Interaction Effects between Doxorubicin and Hernandezine on the Pharmacokinetics by Liquid Chromatography Coupled with Mass Spectrometry

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
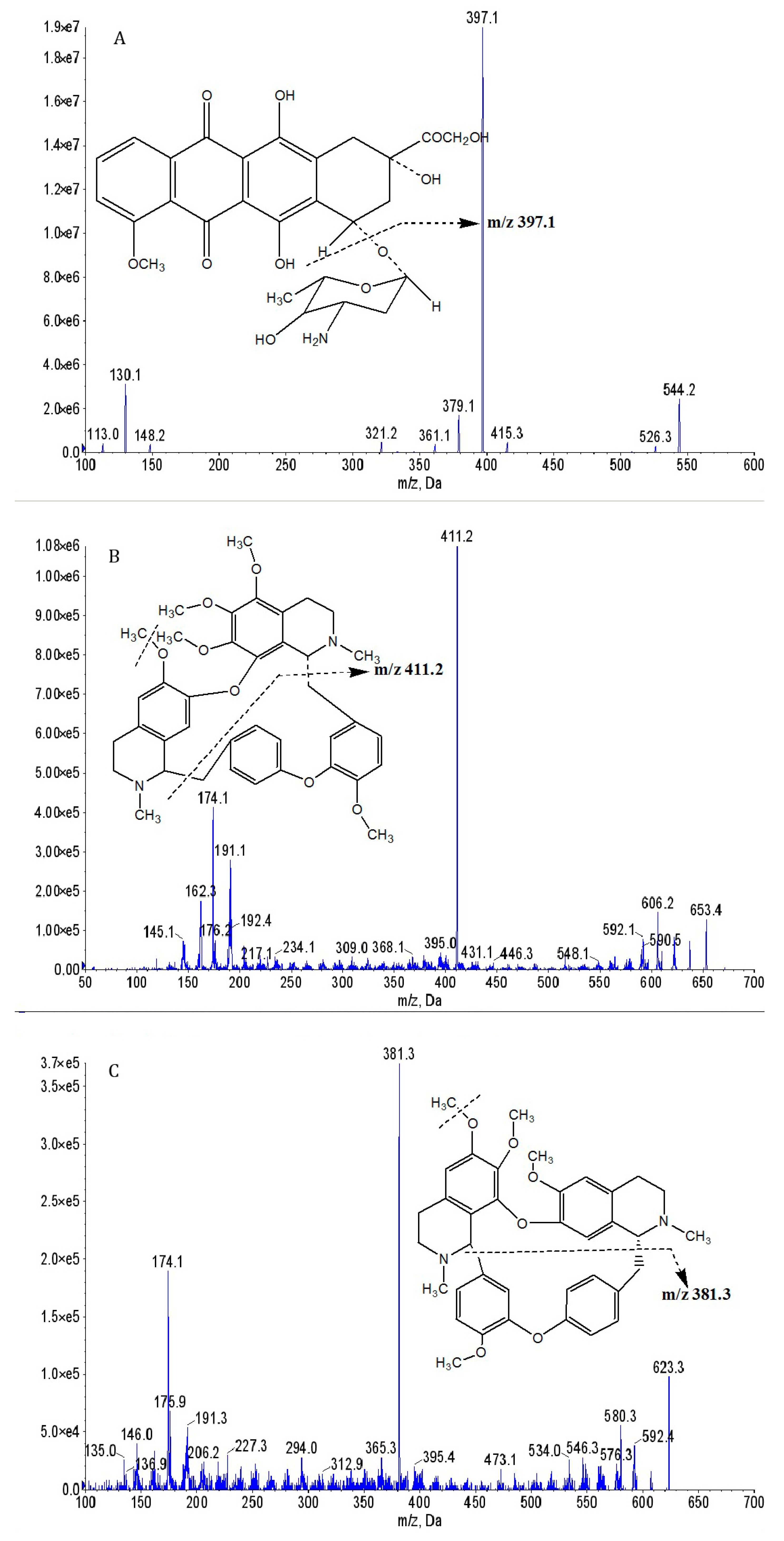
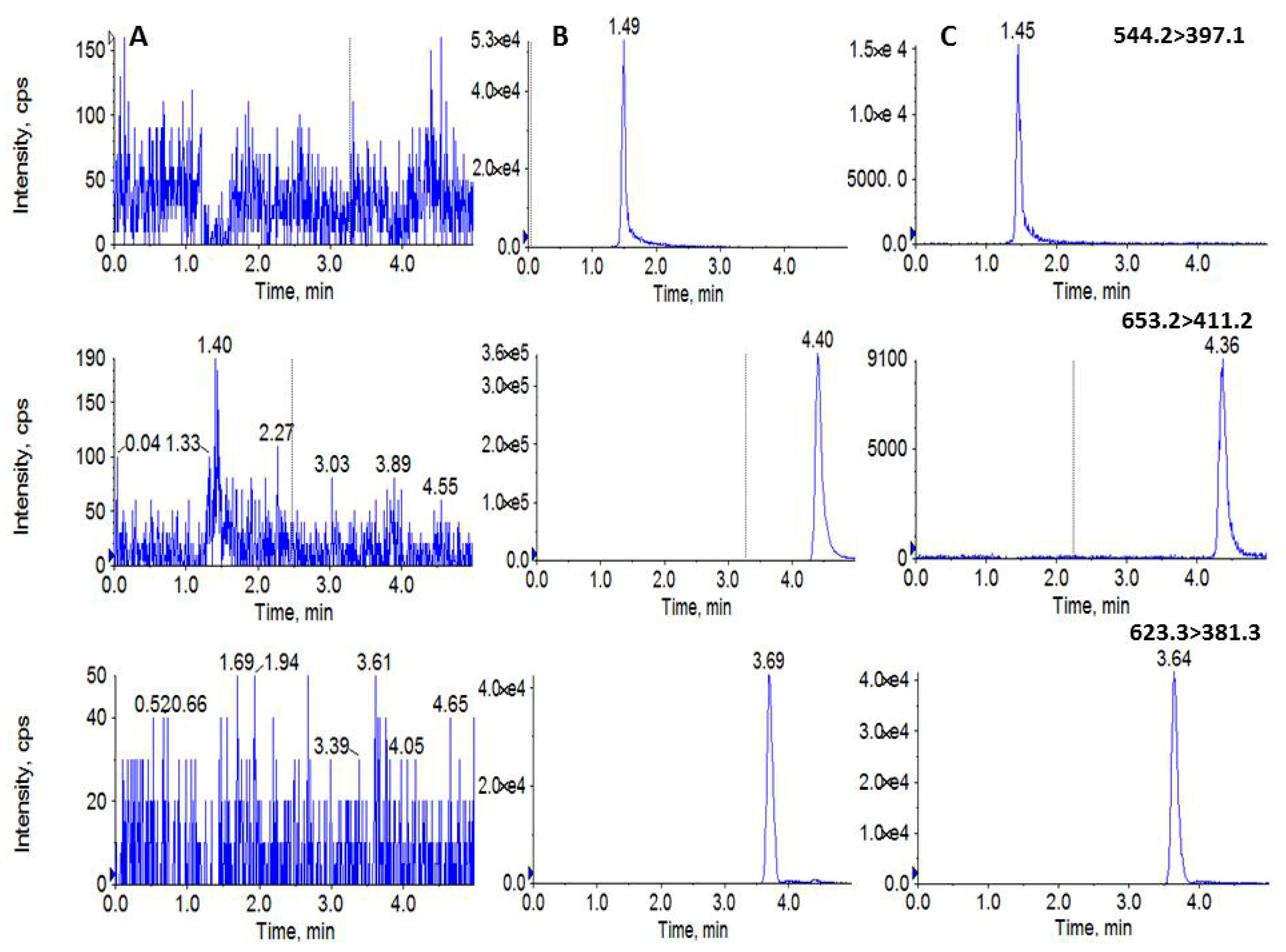
2.1. Method Development
2.2. Method Validation
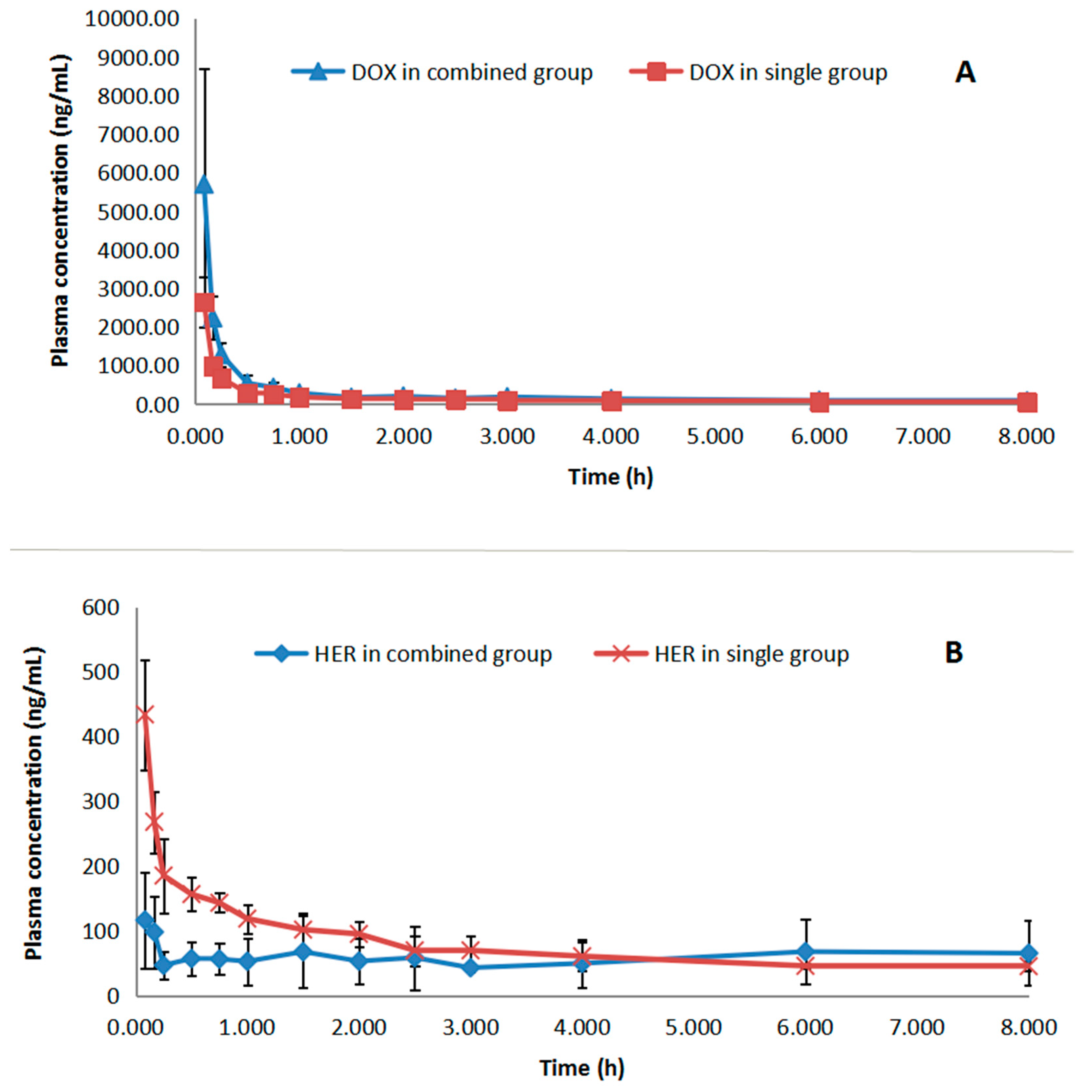
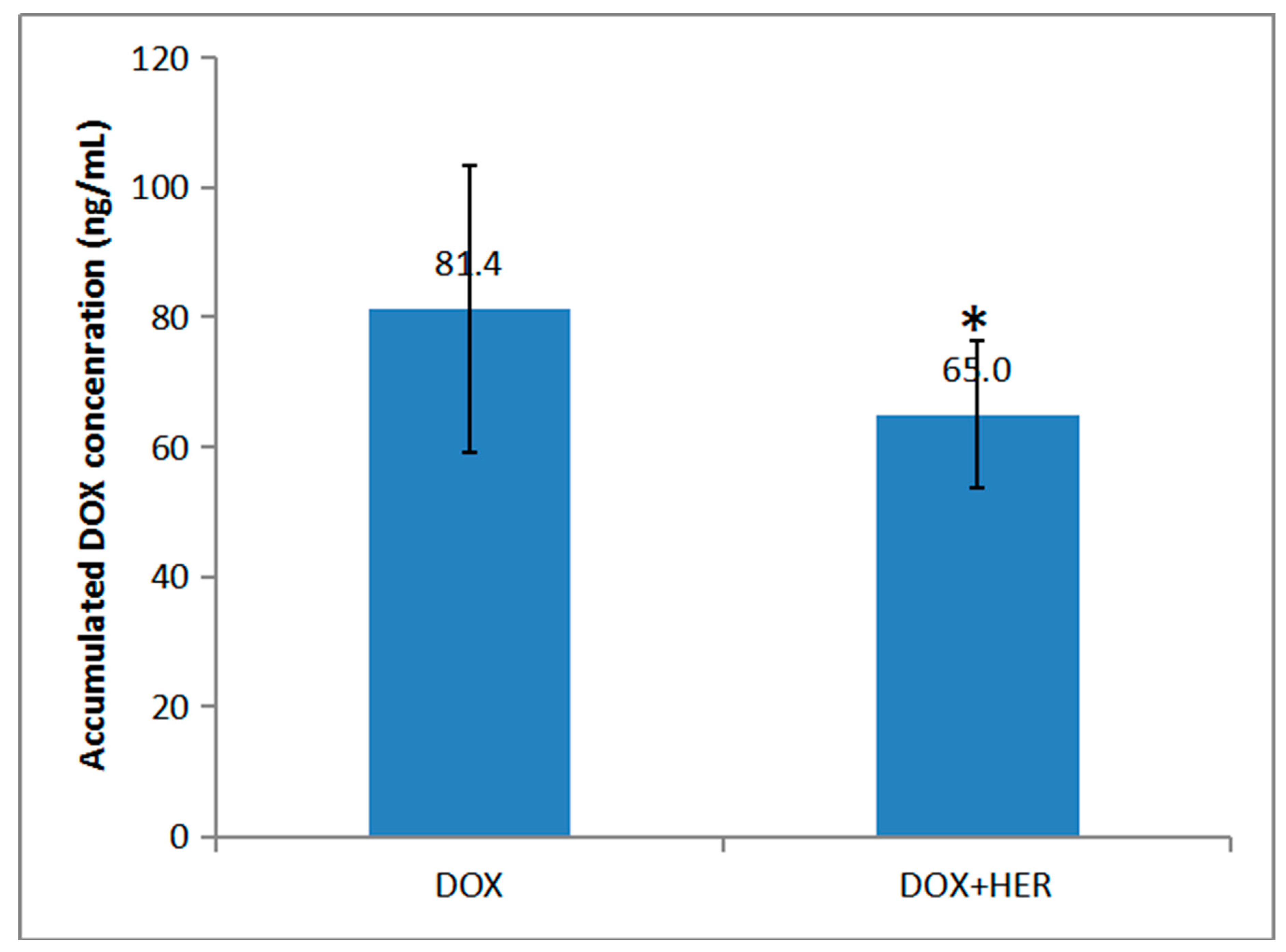
2.3. Pharmacokinetics
3. Experimental
3.1. Chemicals and Reagents
3.2. Animals
3.3. Instrumentation and Conditions
3.4. Preparation of Stock Solutions, Working Solutions, Calibration Standards, and Quality Control Samples
3.5. Sample Preparation
3.6. Pharmacokinetic Study
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Friedrich, K.; Noel, D.; Pinter, T. Propective randomized trial of docetaxel versus doxorubicin in patients with metastatic breast cancer. J. Clin. Oncol. 1999, 17, 2341–2354. [Google Scholar] [CrossRef] [PubMed]
- Duggan, S.T.; Keating, G.M. Pegylated liposomal doxorubicin. Drugs 2011, 71, 2531–2558. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.; Alvarez, J.A.; Scully, R.E.; Miller, T.L.; Lipshultz, S.E. Anthracycline-induced cardiotoxicity: Course, pathophysiology, prevention and management. Expert Opin. Pharmacother. 2007, 8, 1039–1058. [Google Scholar] [CrossRef] [PubMed]
- Gianni, L.; Herman, E.H.; Lipshultz, S.E.; Minotti, G.; Sarvazyan, N.; Sawyer, D.B. Anthracycline cardiotoxicity: From bench to bedside. J. Clin. Oncol. 2008, 26, 3777–3784. [Google Scholar] [CrossRef] [PubMed]
- Menna, P.; Recalcati, S.; Cairo, G.; Minotti, G. An introduction to the metabolic determinants of anthracycline cardiotoxicity. Cardiovasc. Toxicol. 2007, 7, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Takemura, G.; Fujiwara, H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog. Cardiovasc. Dis. 2007, 49, 330–352. [Google Scholar] [CrossRef]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Zhang, Q.L.; Li, Z.; Cong, H.; Li, J.M.; Sheng, H.M. Comparison of effect of hernandezine on the delayed rectifier potassium current of pulmonary artery smooth muscle cell of normal and pulmonary hypertensive rats. Chin. J. Pharmacol. Toxicol. 1998, 12, 235–236. [Google Scholar]
- Xu, P.X.; Sun, S.S. Effect of the Hernandezine on contraction of rat aortic strips. Pharmacol. Clin. Chin. Mater. Med. 1998, 5, 15–18. [Google Scholar]
- Kruger, M.; Boney, R.; Ordoobadi, A.J.; Sommers, T.F.; Trapani, J.G.; Coffin, A.B. Natural Bizbenzoquinoline Derivatives Protect Zebrafish Lateral Line Sensory Hair Cells from Aminoglycoside Toxicity. Front. Cell Neurosci. 2016, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Ho, L.J.; Chang, D.M.; Lee, T.C.; Chang, M.L.; Lai, J.H. Plant alkaloid tetrandrine downregulates protein kinase C-dependent signaling pathway in T cells. Eur. J. Pharmacol. 1999, 367, 389–398. [Google Scholar] [CrossRef]
- Virginio, C.; Graziani, F.; Terstappen, G.C. Differential inhibition of rat alpha3* and alpha7 nicotinic acetylcholine receptors by tetrandrine and closely related bis-benzylisoquinoline derivatives. Neurosci. Lett. 2005, 381, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Imoto, K.; Takemura, H.; Kwan, C.Y.; Sakano, S.; Kaneko, M.; Ohshika, H. Inhibitory effects of tetrandrine and hernandezine on Ca2+ mobilization in rat glioma C6 cells. Res. Commun. Mol. Pathol. Pharmacol. 1997, 95, 129–146. [Google Scholar] [PubMed]
- Leung, Y.M.; Berdik, M.; Kwan, C.Y.; Loh, T.T. Effects of tetrandrine and closely related bis-benzylisoquinoline derivatives on cytosolic Ca2+ in human leukaemic HL-60 cells: A structure-activity relationship study. Clin. Exp. Pharmacol. Physiol. 1996, 23, 653–659. [Google Scholar] [CrossRef]
- Law, B.Y.; Mok, S.W.; Chan, W.K.; Xu, S.W.; Wu, A.G.; Yao, X.J.; Wang, J.R.; Liu, L.; Wong, V.K. Hernandezine, a novel AMPK activator induces autophagic cell death in drug-resistant cancers. Oncotarget 2016, 7, 8090–8104. [Google Scholar] [CrossRef] [Green Version]
- HsiaoS, H.; Lu, Y.J.; Yang, C.C.; Tuo, W.C.; Li, Y.Q.; Huang, Y.H.; Hsieh, C.H.; Hung, T.H.; Wu, C.P. Hernandezine, a Bisbenzylisoquinoline Alkaloid with Selective Inhibitory Activity against Multidrug-Resistance-Linked ATP-Binding Cassette Drug Transporter ABCB1. J. Nat. Prod. 2016, 79, 2135–2142. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Liu, X.; Jiang, T. Quantification of pegylated liposomal doxorubicin and doxorubicinol in rat plasma by liquid chromatography/electrospray tandem mass spectroscopy: Application to preclinical pharmacokinetic studies. Talanta 2008, 74, 887–895. [Google Scholar] [CrossRef]
- Chin, D.L.; Lum, B.L.; Sikic, B.I.J. Rapid determination of PEGylated liposomal doxorubicin and its major metabolite in human plasma by ultraviolet-visible high-performance liquid chromatography. Chromatogr. B Analyt.Technol. Biomed. Life Sci. 2002, 779, 259–269. [Google Scholar] [CrossRef]
- Song, Y.; Wang, Z.B.; Zhang, B.Z.; Zhang, W.P.; Zhang, Y.J.; Yang, C.J.; Meng, F.H.; Feng, X.S. Determination of a novel anticancer AMPK activator Hernandezine in rat plasma and tissues with a validated UHPLC-MS/MS method: Application to pharmacokinetics and tissue distribution study. J. Pharm. Biomed. Anal. 2017, 141, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.N.; Moyer, M.D. API-ionspray MS and MS/MS study on the structural characterization of bisbenzylisoquinoline alkaloids. J. Pharm. Biomed. Anal. 2004, 34, 53–56. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Veterinary Medicine (CVM). Bioanalytical Method Validation Guidance for Industry; Food and Drug Administration: Rockville, MD, USA, 2018.
- Bains, O.S.; Grigliatti, T.A.; Reid, R.E.; Riggs, K.W. Naturally occurring variants of human aldo-keto reductases with reduced in vitro metabolism of daunorubicin and doxorubicin. J. Pharmacol. Exp. Ther. 2010, 335, 533–545. [Google Scholar] [CrossRef]
- Bains, O.S.; Karkling, M.J.; Grigliatti, T.A.; Reid, R.E.; Riggs, K.W. Two nonsynonymous single nucleotide polymorphisms of human carbonyl reductase 1 demonstrate reduced in vitro metabolism of daunorubicin and doxorubicin. Drug Metab. Dispos. 2009, 37, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Salvatorelli, E.; Menna, P.; Gonzalez Paz, O.; Surapaneni, S.; Aukerman, S.L.; Chello, M.; Covino, E.; Sung, V.; Minotti, G.J. Pharmacokinetic characterization of amrubicin cardiac safety in an ex vivo human myocardial strip model. II. Amrubicin shows metabolic advantages over doxorubicin and epirubicin. Pharmacol. Exp. Ther. 2012, 341, 474–483. [Google Scholar] [CrossRef]
- Center of Clinical Drug Research, Shanghai University of Traditional Chinese Medicine, Shanghai, People’s Republic of China. Drug Analysis System [CP/DK]. Available online: http://www.drugchina.net/# (accessed on 5 September 2019).
Sample Availability: Samples of the compounds doxorubicin, hernandezine and tetrandrine are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Samples | Calibration Curves | Correlation Coefficients (r) | Linear Ranges (ng/mL) | LOQs (ng/mL) |
|---|---|---|---|---|---|
| DOX | Plasma | Y = 0.0047 + 0.00015x | 0.994 | 32–8000 | 32 |
| Heart | Y = −0.0028 + 0.00019x | 0.992 | 32–8000 | 32 | |
| HER | Plasma | Y = −0.0038 + 0.00298x | 0.998 | 20–4000 | 20 |
| Heart | Y = −0.0197 + 0.00373x | 0.994 | 20–4000 | 20 |
| Analytes | QC Conc. (ng/mL) | Intraday | Interday | ||
|---|---|---|---|---|---|
| Precision (RSD, %) | Accurary (mean %) | Precision (RSD, %) | Accurary (mean %) | ||
| DOX | 80 | 1.6 | −5.7 | 4.3 | −6.0 |
| 800 | 6.4 | 5.5 | 6.8 | 5.3 | |
| 4000 | 4.0 | −14.0 | 9.3 | −3.6 | |
| HER | 40 | 5.6 | −5.2 | 4.6 | −3.3 |
| 400 | 5.6 | −0.8 | 3.1 | −1.4 | |
| 3200 | 1.4 | −7.8 | 1.9 | −9.0 | |
| Analytes | QC Conc. (ng/mL) | Matrix Effect | Recovery | ||
|---|---|---|---|---|---|
| Mean ± SD (%) | RSD (%) | Mean ± SD (%) | RSD (%) | ||
| DOX | 80 | 112.8 ± 1.8 | 1.6 | 88.7 ± 6.2 | 7.0 |
| 800 | 95.3 ± 11.1 | 11.6 | 103.2 ± 2.6 | 2.5 | |
| 4000 | 92.9 ± 4.3 | 4.6 | 95.1 ± 2.1 | 2.2 | |
| HER | 40 | 104.0 ± 1.7 | 1.7 | 91.7 ± 4.3 | 4.7 |
| 400 | 94.2 ± 1.5 | 1.6 | 108.4 ± 4.9 | 4.6 | |
| 3200 | 94.5 ± 1.7 | 1.8 | 93.2 ± 0.6 | 0.6 | |
| Analytes | QC Conc. (ng/mL) | Bench Top Stability (at Room Temperature for 4 h) | Auto-Sampler Stability (at 4 °C for 24 h) | Freeze/Thraw Stability | Long Term Stability (at −80 °C for 30 days) |
|---|---|---|---|---|---|
| DOX | 80 | 1.6 | 4.3 | 3.4 | 3.1 |
| 800 | 6.4 | 6.8 | 6.1 | 9.6 | |
| 4000 | 4.0 | 0.2 | 8.2 | 5.0 | |
| HER | 40 | 5.5 | 4.7 | 4.1 | 1.7 |
| 400 | 5.5 | 3.1 | 1.6 | 0.8 | |
| 3200 | 1.3 | 1.9 | 1.9 | 1.6 |
| Pharmacokinetic Parameters | Single DOX Group | Single HER Group | Combination Group | |
|---|---|---|---|---|
| DOX | HER | |||
| Cmax (ng/mL) | 2647 ± 650 | 433.6 ± 85.2 | 5703 ± 2980 | 116.6 ± 74.0 |
| Tmax (h) | 0.083 | 0.083 | 0.083 | 0.083 |
| Ke (1/min) | 0.150 ± 0.03 | 0.090 ± 0.01 | 0.164 ± 0.02 | - |
| t1/2 (h) | 4.6 ± 0.8 | 7.7 ± 1.2 | 4.2 ± 0.6 | - |
| AUC0–t (ng h/mL) | 1109 ± 102 | 647.2 ± 54.9 | 1965 ± 142.5 | 49.9 ± 12.5 |
| AUC0–∞ (ng h/mL) | 1412 ± 114 | 1154 ± 85 | 2453 ± 218 | - |
| MRT0–∞ (h) | 4.9 ± 0.9 | 10.1 ± 1.4 | 4.5 ± 0.5 | - |
| CL/F (L/kg/h) | 3.5 ± 0.5 | 4.3 ± 0.5 | 2.0 ± 0.2 | - |
| Vd/F (L/kg) | 23.6 ± 7.1 | 48.3 ± 5.6 | 12.4 ± 2.1 | 81.8 ± 2.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.; Zhang, Y.; Zhang, W.-P.; Zhang, B.-Z.; Wang, K.-F.; Feng, X.-S. Interaction Effects between Doxorubicin and Hernandezine on the Pharmacokinetics by Liquid Chromatography Coupled with Mass Spectrometry. Molecules 2019, 24, 3622. https://doi.org/10.3390/molecules24193622
Song Y, Zhang Y, Zhang W-P, Zhang B-Z, Wang K-F, Feng X-S. Interaction Effects between Doxorubicin and Hernandezine on the Pharmacokinetics by Liquid Chromatography Coupled with Mass Spectrometry. Molecules. 2019; 24(19):3622. https://doi.org/10.3390/molecules24193622
Chicago/Turabian StyleSong, Yang, Yuan Zhang, Wei-Peng Zhang, Bao-Zhen Zhang, Ke-Fei Wang, and Xue-Song Feng. 2019. "Interaction Effects between Doxorubicin and Hernandezine on the Pharmacokinetics by Liquid Chromatography Coupled with Mass Spectrometry" Molecules 24, no. 19: 3622. https://doi.org/10.3390/molecules24193622
APA StyleSong, Y., Zhang, Y., Zhang, W. -P., Zhang, B. -Z., Wang, K. -F., & Feng, X. -S. (2019). Interaction Effects between Doxorubicin and Hernandezine on the Pharmacokinetics by Liquid Chromatography Coupled with Mass Spectrometry. Molecules, 24(19), 3622. https://doi.org/10.3390/molecules24193622