
S-Acyl-2-Thioethyl: A Convenient Base-Labile Protecting Group for the Synthesis of siRNAs Containing 5′-Vinylphosphonate

Abstract
:1. Introduction
2. Results and Discussion
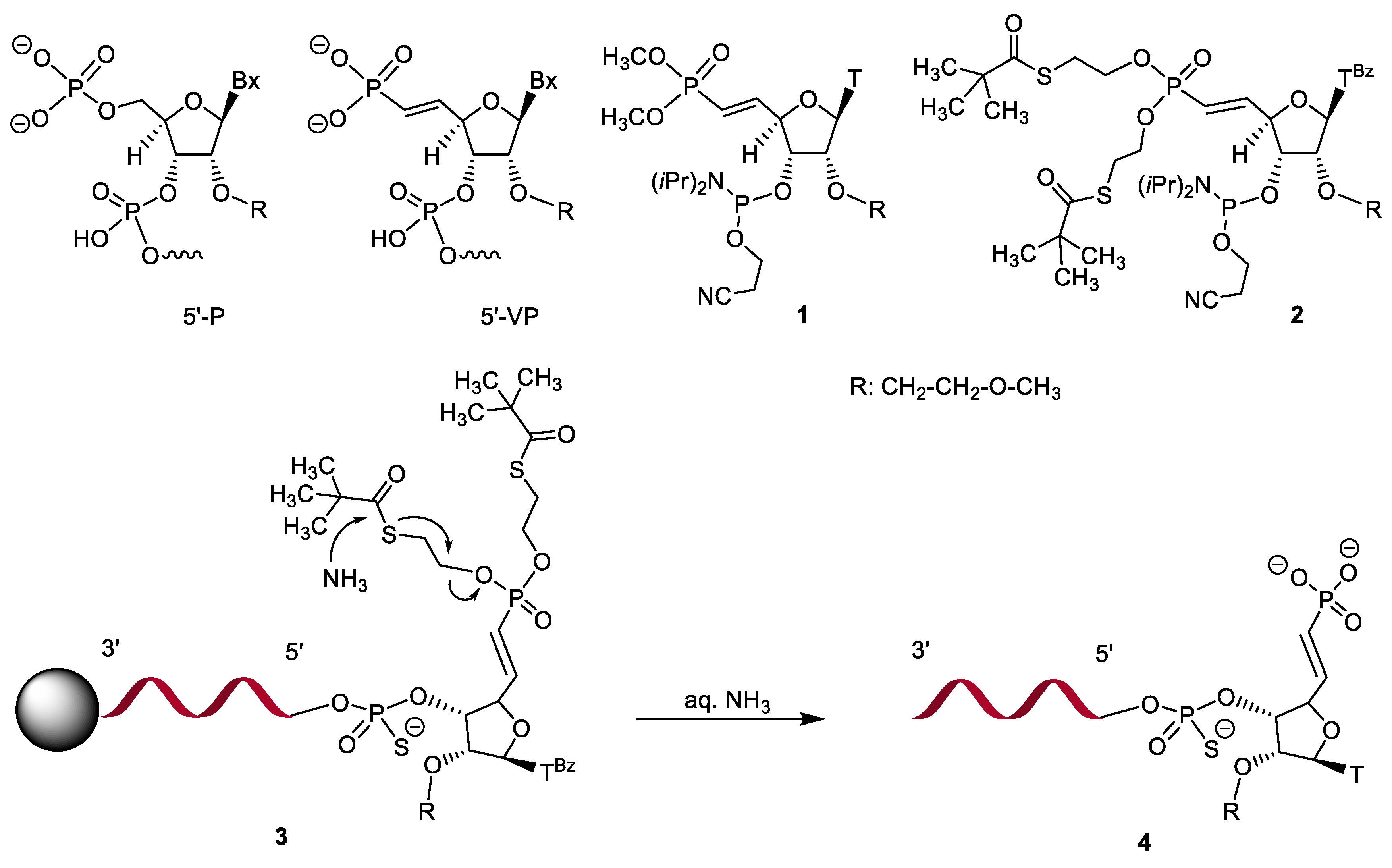
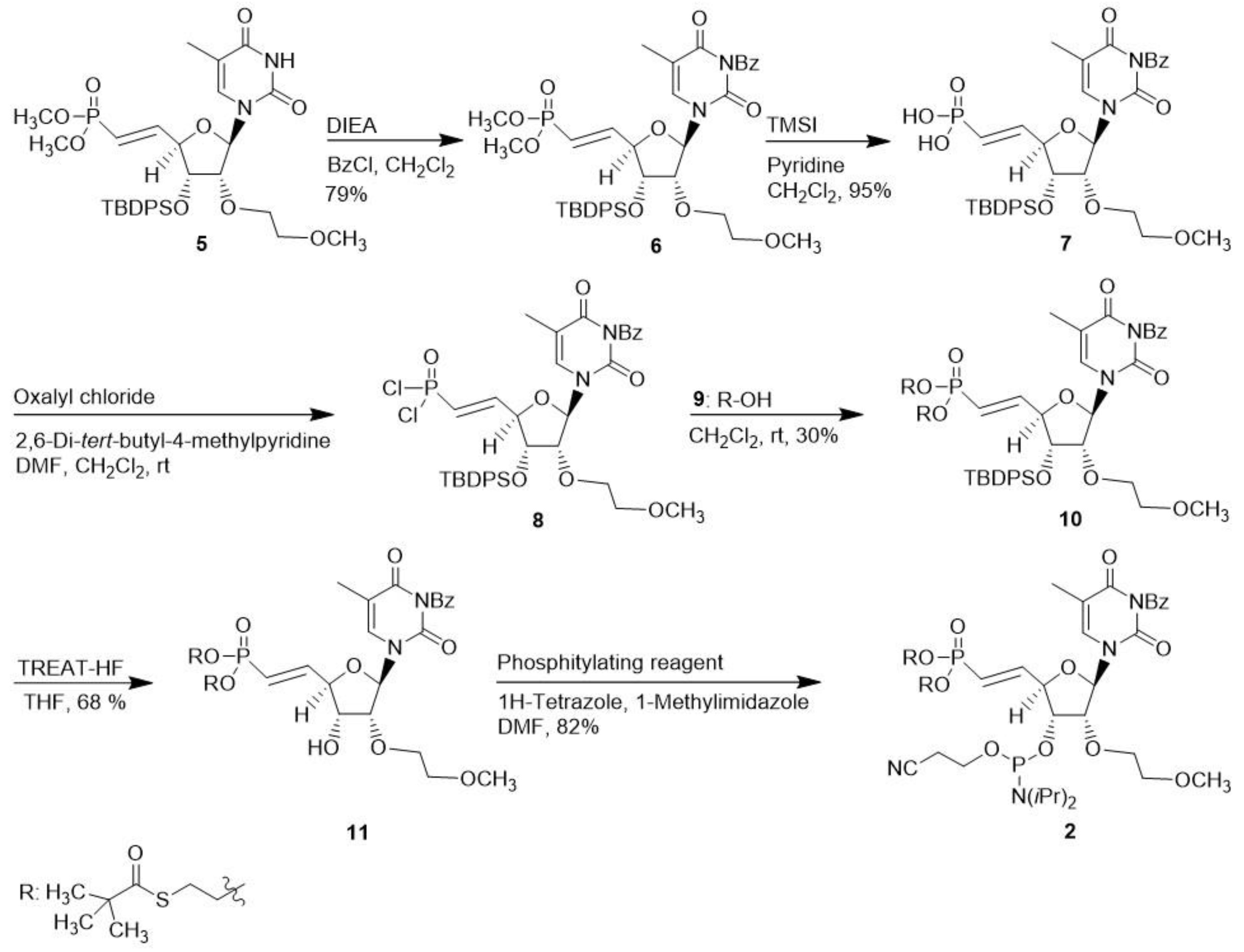
2.1. Synthesis of 5′-deoxy-(E)-5′-vinyl-bis(tBu-SATE)phosphonate)-2′-O-(2-methoxyethyl)-thymidine-3′-phosphoramidite 2
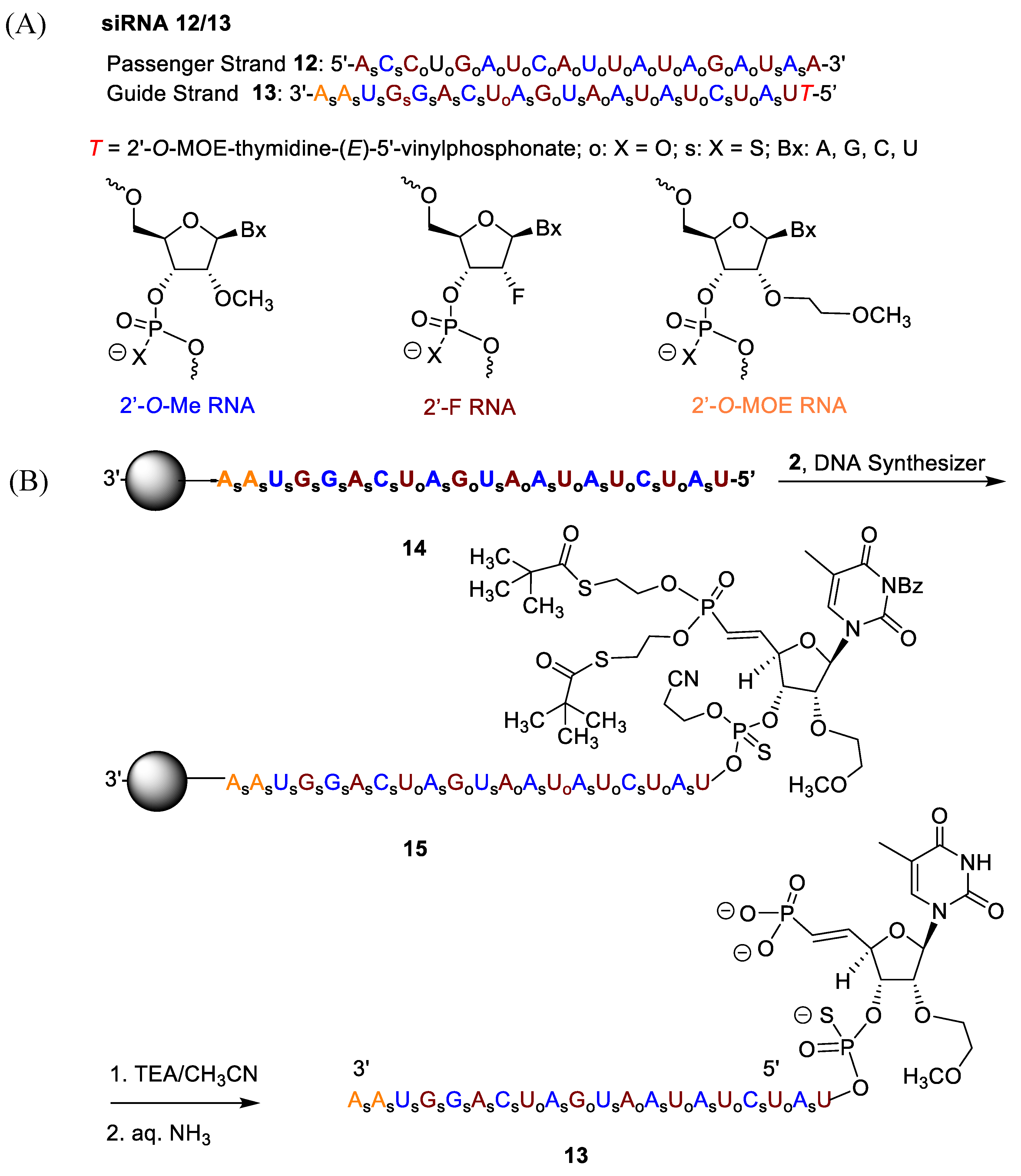
2.2. Synthesis of Oligonucleotides Using 5′-deoxy-(E)-5′-vinyl-bis(tBu-SATE)phosphonate)-2′-O-(2-methoxyethyl)-thymidine-3′-phosphoramidite 2
3. Materials and Methods
General
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.; Adams, D.; Silva, A.; Lozeron, P.; Hawkins, P.N.; Mant, T.; Perez, J.; Chiesa, J.; Warrington, S.; Tranter, E.; et al. Safety and Efficacy of RNAi Therapy for Transthyretin Amyloidosis. New England J. Med. 2013, 369, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, A.; Barros, S.; Ivanciu, L.; Cooley, B.; Qin, J.; Racie, T.; Hettinger, J.; Carioto, M.; Jiang, Y.; Brodsky, J.; et al. An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat. Med. 2015, 21, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef] [PubMed]
- Deleavey, G.F.; Damha, M.J. Designing Chemically Modified Oligonucleotides for Targeted Gene Silencing. Chem. Biol. (Oxford, U.K.) 2012, 19, 937–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, T.P.; Allerson, C.R.; Dande, P.; Vickers, T.A.; Sioufi, N.; Jarres, R.; Baker, B.F.; Swayze, E.E.; Griffey, R.H.; Bhat, B. Positional Effect of Chemical Modifications on Short Interference RNA Activity in Mammalian Cells. J. Med. Chem. 2005, 48, 4247–4253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allerson, C.R.; Sioufi, N.; Jarres, R.; Prakash, T.P.; Naik, N.; Berdeja, A.; Wanders, L.; Griffey, R.H.; Swayze, E.E.; Bhat, B. Fully 2′-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J. Med. Chem 2005, 48, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Juranek, S.; Li, H.; Sheng, G.; Tuschl, T.; Patel, D.J. Structure of an argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature 2008, 456, 921–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, K.; Weinberg, D.E.; Bartel, D.P.; Patel, D.J. Structure of yeast Argonaute with guide RNA. Nature 2012, 486, 368–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirle, N.T.; MacRae, I.J. The crystal structure of human Argonaute2. Science 2012, 336, 1037–1040. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Kinberger, G.A.; Murray, H.M.; Chappell, A.; Riney, S.; Graham, M.J.; Lima, W.F.; Swayze, E.E.; Seth, P.P. Synergistic effect of phosphorothioate, 5′-vinylphosphonate and GalNAc modifications for enhancing activity of synthetic siRNA. Bioorg. Med. Chem. Lett. 2016, 26, 2817–2820. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Lima, W.F.; Murray, H.M.; Li, W.; Kinberger, G.A.; Chappell, A.E.; Gaus, H.; Seth, P.P.; Bhat, B.; Crooke, S.T.; et al. Identification of metabolically stable 5′-phosphate analogs that support single-stranded siRNA activity. Nucleic Acids Res. 2015, 43, 2993–3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, W.F.; Prakash, T.P.; Murray, H.M.; Kinberger, G.A.; Li, W.; Chappell, A.E.; Li, C.S.; Murray, S.F.; Gaus, H.; Seth, P.P.; et al. Single-stranded siRNAs activate RNAi in animals. Cell 2012, 150, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Schirle, N.T.; Kinberger, G.A.; Murray, H.F.; Lima, W.F.; Prakash, T.P.; MacRae, I.J. Structural Analysis of Human Argonaute-2 Bound to a Modified siRNA Guide. J. Am. Chem Soc. 2016, 138, 8694–8697. [Google Scholar] [CrossRef] [PubMed]
- Parmar, R.G.; Brown, C.R.; Matsuda, S.; Willoughby, J.L.S.; Theile, C.S.; Charissé, K.; Foster, D.J.; Zlatev, I.; Jadhav, V.; Maier, M.A.; et al. Facile Synthesis, Geometry, and 2′-Substituent-Dependent in vivo Activity of 5′-(E)- and 5′-(Z)-Vinylphosphonate-Modified siRNA Conjugates. J. Med. Chem. 2018, 61, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Peyrottes, S.; Egron, D.; Lefebvre, I.; Gosselin, G.; Imbach, J.L.; Perigaud, C. SATE pronucleotide approaches: An overview. Mini Rev. Med. Chem. 2004, 4, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Meade, B.R.; Gogoi, K.; Hamil, A.S.; Palm-Apergi, C.; van den Berg, A.; Hagopian, J.C.; Springer, A.D.; Eguchi, A.; Kacsinta, A.D.; Dowdy, C.F.; et al. Efficient delivery of RNAi prodrugs containing reversible charge-neutralizing phosphotriester backbone modifications. Nat. Biotechnol 2014, 32, 1256–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakash, T.P.; Prhavc, M.; Eldrup, A.B.; Cook, P.D.; Carroll, S.S.; Olsen, D.B.; Stahlhut, M.W.; Tomassini, J.E.; MacCoss, M.; Galloway, S.M.; et al. Synthesis and Evaluation of S-Acyl-2-thioethyl Esters of Modified Nucleoside 5′-Monophosphates as Inhibitors of Hepatitis C Virus RNA Replication. J. Medi. Chem. 2005, 48, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, I.; Perigaud, C.; Pompon, A.; Aubertin, A.M.; Girardet, J.L.; Kirn, A.; Gosselin, G.; Imbach, J.L. Mononucleoside phosphotriester derivatives with S-acyl-2-thioethyl bioreversible phosphate-protecting groups: intracellular delivery of 3′-azido-2′,3′-dideoxythymidine 5′-monophosphate. J. Med. Chem 1995, 38, 3941–3950. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds 1, 5 and 13 are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
| Comp. | Protecting Group | Deprotection Temperature | Mol. Wt. Calculated | Mol. Wt. Found | Scale μmol | %UV Purity |
|---|---|---|---|---|---|---|
| 13 | SATE | 55 °C | 7300.4 | 7299.3 | 2 | 99.3 |
| 13 | SATE | 25 °C | 7300.4 | 7299.1 | 2 | 98.6 |
| 13 | Me | 55 °C | 7300.4 | 7299.1 | 2 | 99.1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikan, M.; Li, W.; Kinberger, G.A.; Seth, P.P.; Swayze, E.E.; Prakash, T.P. S-Acyl-2-Thioethyl: A Convenient Base-Labile Protecting Group for the Synthesis of siRNAs Containing 5′-Vinylphosphonate. Molecules 2019, 24, 225. https://doi.org/10.3390/molecules24020225
Nikan M, Li W, Kinberger GA, Seth PP, Swayze EE, Prakash TP. S-Acyl-2-Thioethyl: A Convenient Base-Labile Protecting Group for the Synthesis of siRNAs Containing 5′-Vinylphosphonate. Molecules. 2019; 24(2):225. https://doi.org/10.3390/molecules24020225
Chicago/Turabian StyleNikan, Mehran, Wenyu Li, Garth A. Kinberger, Punit P. Seth, Eric E. Swayze, and Thazha P. Prakash. 2019. "S-Acyl-2-Thioethyl: A Convenient Base-Labile Protecting Group for the Synthesis of siRNAs Containing 5′-Vinylphosphonate" Molecules 24, no. 2: 225. https://doi.org/10.3390/molecules24020225
APA StyleNikan, M., Li, W., Kinberger, G. A., Seth, P. P., Swayze, E. E., & Prakash, T. P. (2019). S-Acyl-2-Thioethyl: A Convenient Base-Labile Protecting Group for the Synthesis of siRNAs Containing 5′-Vinylphosphonate. Molecules, 24(2), 225. https://doi.org/10.3390/molecules24020225