
Lignans from the Twigs of Litsea cubeba and Their Bioactivities

 ,
,
Abstract
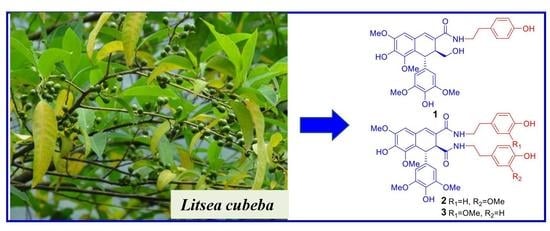
:
1. Introduction
2. Results and Discussions
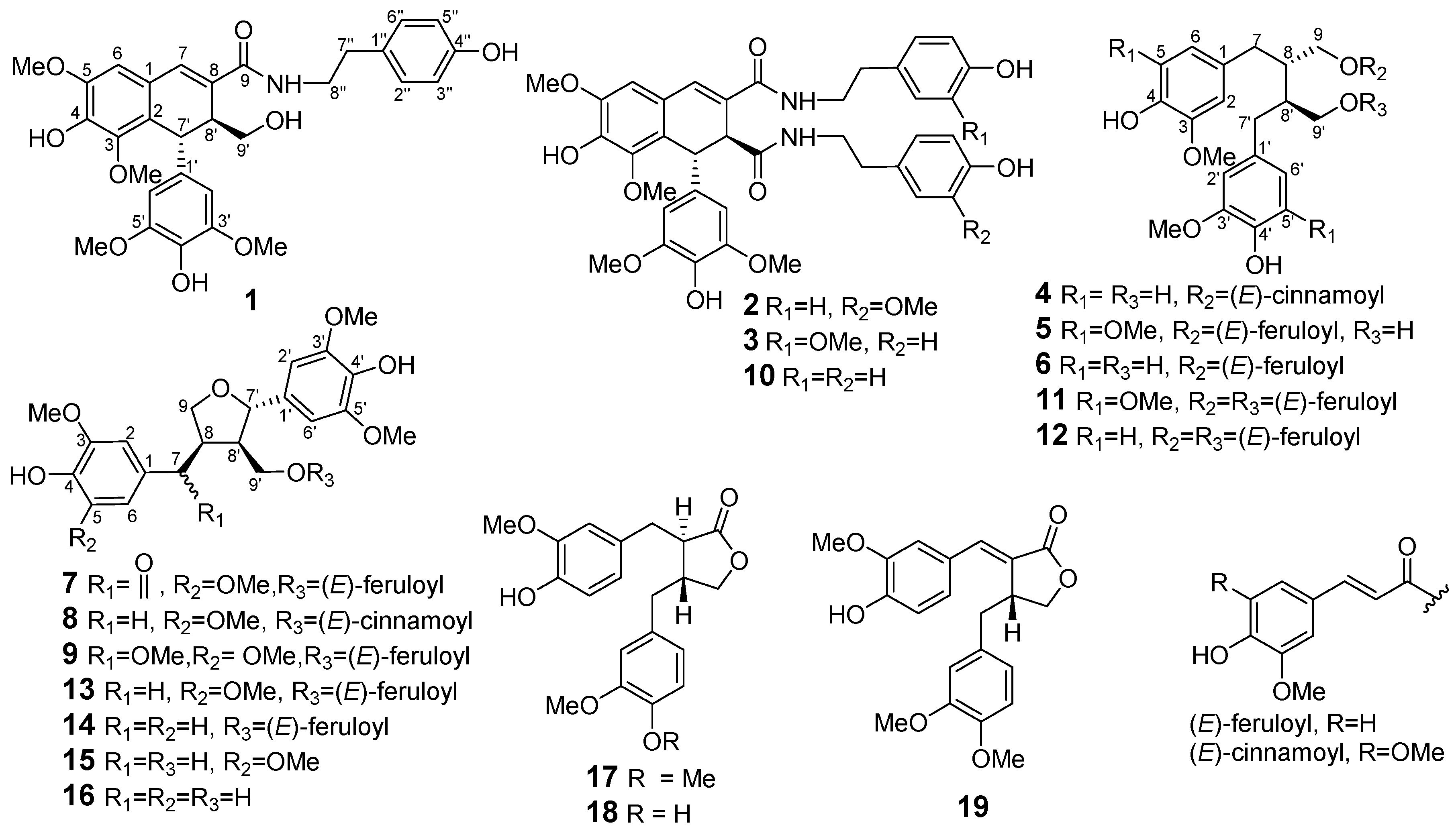
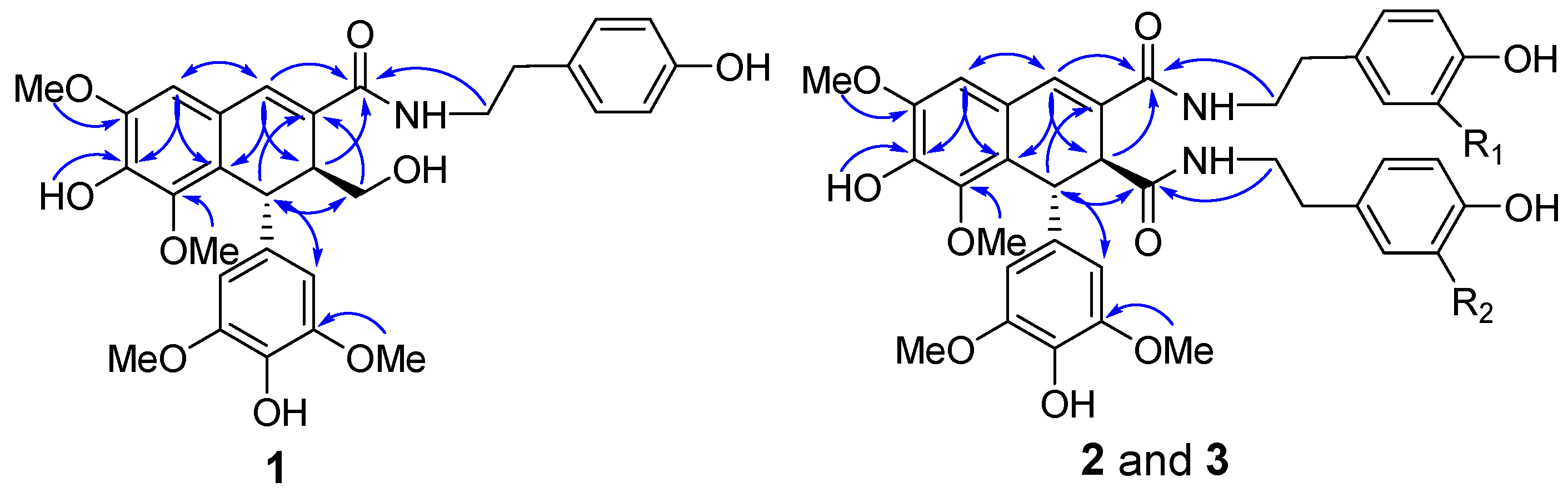
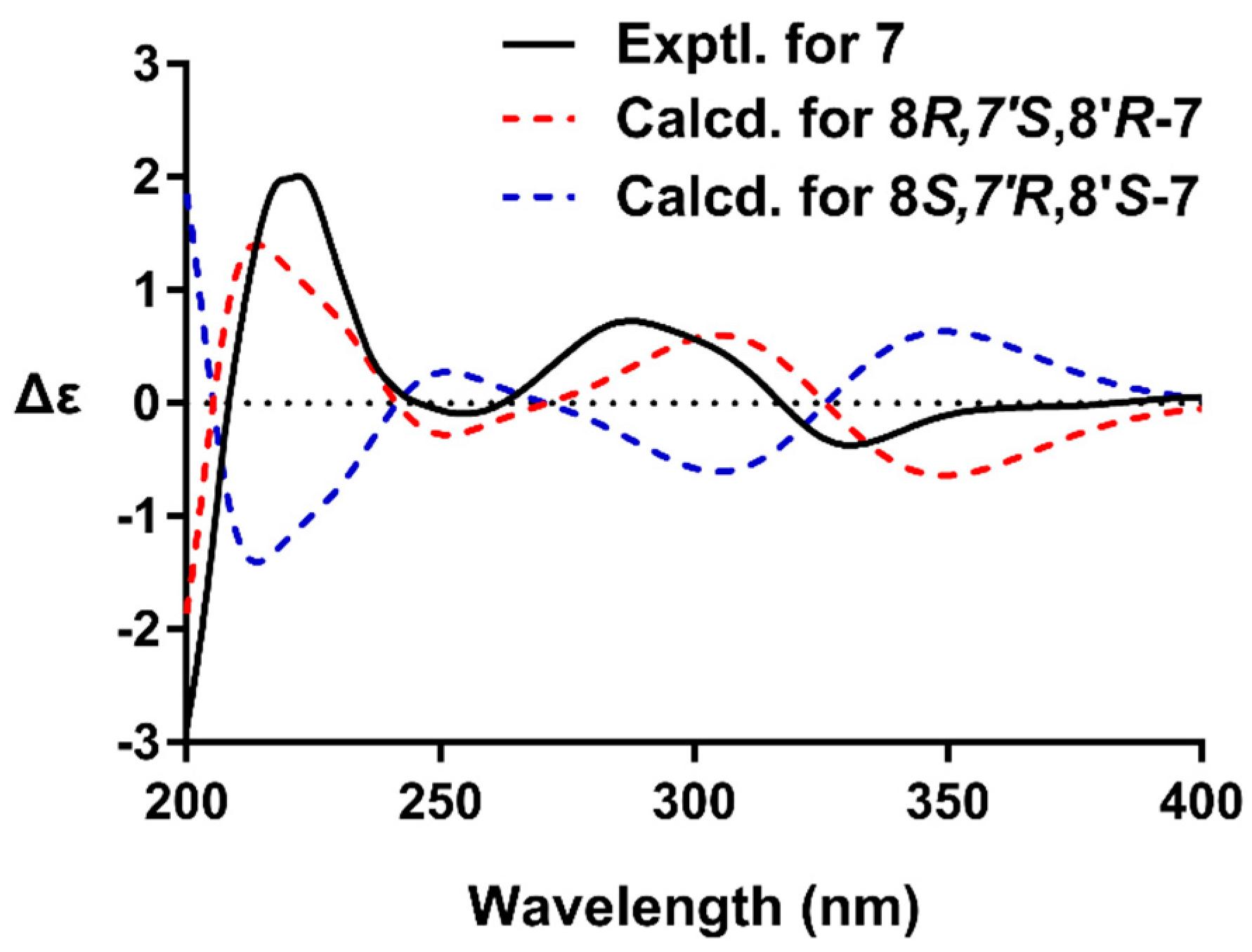
2.1. Structure Elucidation
2.2. Biological Activities of Compounds 1–19
2.2.1. Cytotoxic Activity
2.2.2. Inhibitory Activity of Protein Tyrosine Phosphatase 1B
2.2.3. Anti-Inflammatory Activity
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. (−)-(7′R,8′S)-N-[2-(4-Hydroxyphenyl)-ethyl]-4,4′,9′-trihydroxy-3,5,3′,5′-tetramethoxy-2,7′-cyclo-lignan-7-en-9-amide (1)
3.5. (−)-(7′R,8′S)-N1-[2-(4-Hydroxyphenyl)-ethyl]-N2-[2-(4-hydroxy-3-methoxyphenyl)-ethyl]-4,4′-dihydro-xy-3,5,3′,5′-tetramethoxy-2,7′-cyclolignan-7-en-9,9′-diamide (2)
3.6. (−)-(7′R,8′S)-N1-[2-(4-Hydroxy-3-methoxyphenyl)-ethyl]-N2-[2-(4-hydroxyphenyl)-ethyl]-4,4′-dihydro-xy-3,5,3′,5′-tetramethoxy-2,7′-cyclolignan-7-en-9,9′-diamide (3)
3.7. (+)-(8S,8′S)-9-O-(E)-Cinnamoyl-secoisolariciresinol (4)
3.8. (+)-(8S,8′S)-9-O-(E)-Feruloyl-5,5′-dimethoxysecoisolariciresinol (5)
3.9. (+)-(8S,8′S)-9-O-(E)-Feruloyl-secoisolariciresinol (6)
3.10. (+)-(8R,7′S,8′R)-9′-O-(E)-Feruloyl-5,5′-dimethoxylariciresinol-7-one (7)
3.11. (+)-(8R,7′S,8′R)-9′-O-(E)-Cinnamoyl-5,5′-dimethoxylariciresinol (8)
3.12. 9′-O-(E)-Feruloyl-5,7,5′-trimethoxylariciresinol (9)
3.13. Cytotoxicity Assay
3.14. PTP1B Inhibition Assay
3.15. Nitric Oxide (NO) Production in RAW264.7 Macrophages
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- National Pharmacopoeia Commission. Chinese Pharmacopoeia; China Medical Science and Technology Press: Beijing, China, 2015. [Google Scholar]
- Editorial Committee of Chinese Materia Medica, State Administration Bureau of Traditional Chinese Medicine. Chinese Materia Medica (Zhonghua Bencao); Shanghai Science & Technology Press: Shanghai, China, 1999. [Google Scholar]
- Zhang, S.Y.; Guo, Q.; Gao, X.L.; Guo, Z.Q.; Zhao, Y.F.; Chai, X.Y.; Tu, P.F. A phytochemical and pharmacological advance on medicinal plant Litsea cubeba (Lauraceae). Chin. J. Chin. Mater. Med. 2014, 39, 769–776. [Google Scholar]
- Li, W.R.; Shi, Q.S.; Liang, Q.; Xie, X.B.; Huang, X.M.; Chen, Y.B. Antibacterial activity and kinetics of Litsea cubeba oil on Escherichia coli. PLoS ONE 2014, 9, e110983. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Chen, C.K.; Huang, F.M.; Chen, C.H. Two dibenzopyrrocoline alkaloids from Litsea cubeba. J. Nat. Prod. 1996, 59, 80–82. [Google Scholar] [CrossRef]
- Lee, S.S.; Lin, Y.J.; Chen, C.K.; Liu, K.C.S.; Chen, C.H. Quaternary alkaloids from Litsea cubeba and Cryptocarya konishii. J. Nat. Prod. 1993, 56, 1971–1976. [Google Scholar] [CrossRef]
- Wu, Y.C.; Liou, J.Y.; Duh, C.Y.; Lee, S.S.; Lu, S.T. Litebamine, a novel phenanthrene alkalord from Quaternary alkaloids from Litsea cubeba. Tetrahedron Lett. 1991, 32, 4169–4170. [Google Scholar] [CrossRef]
- Feng, T.; Xu, Y.; Cai, X.H.; Du, Z.Z.; Luo, X.D. Antimicrobially active isoquinoline alkaloids from Litsea cubeba. Planta Med. 2009, 75, 76–79. [Google Scholar] [CrossRef]
- Zhang, S.Y.; Guo, Q.; Cao, Y.; Zhang, Y.; Gao, X.L.; Tu, P.F.; Chai, X.Y. Alkaloids from roots and stems of Litsea cubeba. Chin. J. Chin. Mater. Med. 2014, 39, 3964–3968. [Google Scholar]
- Guo, Q.; Bai, R.F.; Su, G.Z.; Zhu, Z.X.; Tu, P.F.; Chai, X.Y. Chemical constituents from the roots and stems of Litsea cubeba. J. Asian Nat. Prod. Res. 2015, 1, 51–58. [Google Scholar]
- Zhang, S.Y.; Zhang, Q.; Guo, Q.; Zhao, Y.F.; Gao, X.L.; Chai, X.Y.; Tu, P.F. Characterization and simultaneous quantification of biological aporphine alkaloids in Litsea cubeba by HPLC with hybrid ion trap time-of-flight mass spectrometry and HPLC with diode array detection. J. Sep. Sci. 2015, 38, 2614–2624. [Google Scholar] [CrossRef]
- Guo, Q.; Zeng, K.W.; Gao, X.L.; Zhu, Z.X.; Zhang, S.Y.; Chai, X.Y.; Tu, P.F. Chemical constituents with NO production inhibitory and cytotoxic activities from Litsea cubeba. J. Nat. Med. 2015, 69, 94–99. [Google Scholar] [CrossRef]
- Lin, B.; Zhang, H.; Zhao, X.X.; Rahman, K.; Wang, Y.; Ma, X.Q.; Zheng, C.J.; Zhang, Q.Y.; Han, T.; Qin, L. Inhibitory effects of the root extract of Litsea cubeba (lour.) pers. on adjuvant arthritis in rats. J. Ethnopharmacol. 2013, 147, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.H.; Zhang, F.X.; Wei, X.Y.; Liu, M.F. A review of the studies on Litsea alkaloids. J. Trop. Subtrop. Bot. 1999, 7, 87–92. [Google Scholar]
- Zhang, W.; Hu, J.F.; Lv, W.W.; Zhao, Q.C.; Shi, G.B. Antibacterial, antifungal and cytotoxic isoquinoline alkaloidsfrom Litsea cubeba. Molecules 2012, 17, 12950–12960. [Google Scholar] [CrossRef]
- Wang, L.Y.; Chen, M.H.; Wu, J.; Sun, H.; Liu, W.; Qu, Y.H.; Li, Y.C.; Wu, Y.Z.; Li, R.; Zhang, D.; et al. Bioactive glycosides form the twigs of Litsea cubeba. J. Nat. Prod. 2017, 80, 1808–1818. [Google Scholar] [CrossRef]
- Wang, L.Y.; Qu, Y.H.; Li, Y.C.; Wu, Y.Z.; Li, R.; Guo, Q.L.; Wang, S.J.; Wang, Y.N.; Yang, Y.C.; Lin, S. Water soluble constituents from the twigs of Litsea cubeba. Chin. J. Chin. Mater. Med. 2017, 42, 2704–2713. [Google Scholar]
- Wallis, A.F.A. Stereochemistry of cyclolignan—A revised structure of thomasic acid. Tetrahedron Lett. 1968, 9, 5287–5288. [Google Scholar] [CrossRef]
- Assoumatine, T.; Datta, P.K.; Hooper, T.S.; Yvon, B.L.; Charlton, J.L. A short asymmetric synthesis of (+)-lyoniresinol dimethyl ether. J. Org. Chem. 2004, 69, 4140–4144. [Google Scholar] [CrossRef]
- Chaves, M.H.; Roque, N.F. Amides and lignanamides from Porcelia macrocarpa. Phytochemistry 1997, 46, 879–881. [Google Scholar] [CrossRef]
- Moon, S.S.; Rahman, A.A.; Kim, J.Y.; Kee, S.H. Hanultarin, a cytotoxic lignan as an inhibitor of actin cytoskeleton polymerization from the seeds of Trichosanthes kirilowii. Bioorg. Med. Chem. 2008, 16, 7264–7269. [Google Scholar] [CrossRef]
- Park, H.B.; Lee, K.H.; Kim, K.H.; Lee, I.K.; Noh, H.J.; Choi, S.U.; Lee, K.R. Lignans from the roots of Berberis amurensis. Nat. Prod. Sci. 2009, 15, 17–21. [Google Scholar]
- Zhao, Q.; Liu, J.; Wang, F.N.; Liu, G.F.; Wang, G.Z.; Zhang, K. Lignans from branch of Hypericum petiolulatum. Chin. J. Chin. Mater. Med. 2009, 34, 1373–1376. [Google Scholar]
- Chen, J.J.; Wang, T.Y.; Hwang, T.L. Neolignans, a coumarinolignan, lignan derivatives, and a chromene: Anti-inflammatory constituents from Zanthoxylum avicennae. J. Nat. Prod. 2008, 71, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.H.; Huang, Y.H.; Lin, J.J.; Liau, B.C.; Wang, S.Y.; Wu, Y.C.; Jong, T.T. Cytotoxic lignans esters from Cinnamomum osmophloeum. Planta Med. 2010, 76, 613–619. [Google Scholar] [CrossRef]
- Hsiao, J.J.; Chiang, H.C. Lignans from the wood of Aralia bipinnata. Phytochemistry 1995, 39, 899–902. [Google Scholar] [CrossRef]
- Achenbach, H.; Stöcker, M.; Constenla, M.A. Flavonoid and other constituents of Bauhinia manca. Phytochemistry 1988, 27, 1835–1841. [Google Scholar] [CrossRef]
- Duh, C.Y.; Phoebe, C.H., Jr.; Pezzuto, J.M.; Kinghorn, A.D.; Farnsworth, N.R. Plant anticancer agents, XLII. Cytotoxic constituents from Wikstroemia elliptica. J. Nat. Prod. 1986, 49, 706–709. [Google Scholar] [CrossRef]
- Umehara, K.; Sugawa, A.; Kuroyanagi, M.; Ueno, A.; Taki, T. Studies on differentiation-inducers from Arctium Fructus. Chem. Pharm. Bull. 1993, 41, 1774–1779. [Google Scholar] [CrossRef]
- Wang, H.Y.; Yang, J.S. Chemical components from Arctimu lappa. Acta Pharm. Sin. 1993, 28, 911–917. [Google Scholar]
Sample Availability: Samples of the compounds 1–19 are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| 2 | 6.71 d (1.5) | 6.42 s | 6.70 d (1.8) | 7.39 s | 6.57 s | 6.67 s | |||
| 5 | 6.71 d (7.5) | 6.71 d (7.8) | |||||||
| 6 | 6.74 s | 6.69 s | 6.60 s | 6.61 dd (7.5, 1.5) | 6.42 s | 6.61 dd (7.8, 1.8) | 7.39 s | 6.57 s | 6.67 s |
| 7 | 7.19 s | 7.18 s | 7.21 s | 2.80 dd (13.5, 7.0); 2.62 dd (13.5, 8.0) | 2.79 dd (14.2, 7.2); 2.62 dd (14.2, 8.4) | 2.80 dd (13.8, 6.6); 2.62 dd (13.8, 8.4) | 2.91 dd (13.2, 5.4); 2.59 dd (13.2, 10.2) | 4.35 d (6.5) | |
| 8 | 2.32 m | 2.31 m | 2.31 m | 4.57 m | 2.82 m | 2.84 m | |||
| 9 | 4.36 dd (11.5, 6.5); 4.11 dd (11.5, 6.0) | 4.42 dd (10.8, 6.0); 4.10 dd (10.8, 6.0) | 4.36 dd (11.4, 6.6); 4.11 dd (11.4, 6.0) | 4.35 t (8.0); 4.22 t (8.0) | 4.04 dd (8.4, 6.6); 3.74 dd (8.4, 6.6) | 4.14 t (8.5); 4.04 t (8.5) | |||
| 2′ | 6.39 s | 6.38 s | 6.38 s | 6.73 d (1.5) | 6.44 s | 6.73 d (1.8) | 6.78 s | 6.68 s | 6.63 s |
| 5′ | 6.69 d (7.5) | 6.69 d (7.8) | |||||||
| 6′ | 6.39 s | 6.38 s | 6.38 s | 6.61 dd (7.5, 1.5) | 6.44 s | 6.61 dd (7.8, 1.8) | 6.78 s | 6.68 s | 6.63 s |
| 7′ | 4.62 s | 5.03 s | 5.03 s | 2.70 dd (13.5, 7.0); 2.63 dd (13.5, 8.0) | 2.70 dd (14.2, 7.2); 2.63 dd (14.2, 8.4) | 2.70 dd (13.8, 6.6); 2.63 dd (13.8, 8.4) | 4.74 d (7.5) | 4.82 d (5.5) | |
| 8′ | 3.14 dd (7.5, 7.5) | 3.66 s | 3.67 s | 1.99 m | 1.99 m | 1.99 m | 3.01 m | 2.61 m | |
| 9′ | 3.59 m 3.28 m | 3.67 m; 3.59 m | 3.67 m; 3.61 m | 3.69 m; 3.59 m | 4.16 d (6.5) | 4.53 dd (11.4, 6.6); 4.30 dd (11.4, 7.8) | |||
| 2′′ | 6.98 d (8.5) | 6.98 d (8.5) | 6.79 d (1.8) | 7.00 s | 7.32 d (1.8) | 7.32 d (1.8) | 7.06 d (1.5) | 6.98 s | 7.27 d (2.0) |
| 3′′ | 6.71 d (8.5) | 6.72 d (8.5) | |||||||
| 5′′ | 6.71 d (8.5) | 6.72 d (8.5) | 6.71 d (7.8) | 6.86 d (8.4) | 6.81 d (8.4) | 6.82 d (8.5) | 6.85 d (8.0) | ||
| 6′′ | 6.98 d (8.5) | 6.98 d (8.5) | 6.61 dd (7.8, 1.8) | 7.00 s | 7.13 dd (8.4, 1.8) | 7.13 dd (8.4, 1.8) | 6.96 dd (8.5, 1.5) | 6.98 s | 7.11 dd (8.0, 2.0) |
| 7′′ | 2.69 t (7.5) | 2.70 t (7.0) | 2.72 t (7.2) | 7.58 d (15.6) | 7.57 d (15.6) | 7.16 d (16.0) | 7.47 d (16.2) | 7.49 d (15.5) | |
| 8′′ | 3.39 dt (7.4, 4.5) | 3.41 t (6.0) | 3.47 m, 3.39 m | 6.42 d (15.6) | 6.41 d (15.6) | 5.89 d (16.0) | 6.39 d (16.2) | 6.34 d (15.5) | |
| 2′′′ | 6.79 d (1.5) | 6.93 d (8.4) | |||||||
| 3′′′ | 6.70 d (8.4) | ||||||||
| 5′′′ | 6.69 d (8.0) | 6.70 d (8.4) | |||||||
| 6′′′ | 6.55 dd (8.0, 1.5) | 6.93 d (8.4) | |||||||
| 7′′′ | 2.58 t (7.0) | 2.56 t (7.2) | |||||||
| 8′′′ | 3.28 t (7.0) | 3.29 m, 3.21 m | |||||||
| OMe-3 | 3.58 s | 3.69 s | 3.69 s | 3.75 s | 3.73 s | 3.75 s | 3.84 s | 3.79 s | 3.80 s |
| OMe-5 | 3.86 s | 3.85 s | 3.85 s | 3.73 s | 3.84 s | 3.79 s | 3.80 s | ||
| OMe-7 | 3.17 s | ||||||||
| OMe-3′ | 3.67 s | 3.67 s | 3,67 s | 3.75 s | 3.73 s | 3.75 s | 3.83 s | 3.88 s | 3.77 s |
| OMe-5′ | 3.67 s | 3.67 s | 3.67 s | 3.73 s | 3.83 s | 3.88 s | 3.77 s | ||
| OMe-3′′ | 3.78 s | 3.88 s | 3.91 s | 3.90 s | 3.91 s | 3.79 s | 3.91 s | ||
| OMe-5′′ | 3.88 s | 3.79 s | |||||||
| OMe-3′′′ | 3.80 s | ||||||||
| OH-4 | 7.76 s | 7.78 s | 7.79 s | 7.29 s | 6.91 s | 7.27 s | 7.09 s | ||
| OH-4′ | 6.90 s | 6.91 s | 6.91 s | 7.26 s | 6.89 s | 7.24 s | 6.98 s | ||
| OH-4′′ | 8.08 s | 7.21 s | 7.75 s | 8.15 s | 8.12 s | 7.77 s | |||
| OH-4′′′ | 7.26 s | 8.07 s | |||||||
| NH | 7.45 t (4.5) | 7.81 t (4.5), 7.59 t (4.5) | 7.72 t (4.5), 7.59 t (4.5) |
| No. | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 132.0 | 123.8 | 123.8 | 132.9 | 132.4 | 132.9 | 129.6 | 131.8 | 134.5 |
| 2 | 124.6 | 126.5 | 126.5 | 113.2 | 107.2 | 113.2 | 107.2 | 106.9 | 106.9 |
| 3 | 147.0 | 146.4 | 146.4 | 148.1 | 148.5 | 148.1 | 148.4 | 148.9 | 148.5 |
| 4 | 141.8 | 142.4 | 142.4 | 145.5 | 134.9 | 145.5 | 142.2 | 135.2 | 136.0 |
| 5 | 148.2 | 148.1 | 148.1 | 115.5 | 148.5 | 115.5 | 148.4 | 148.9 | 148.5 |
| 6 | 108.0 | 108.3 | 108.2 | 122.3 | 107.2 | 122.3 | 107.2 | 106.9 | 106.9 |
| 7 | 131.5 | 132.5 | 133.6 | 35.4 | 35.9 | 35.4 | 198.2 | 34.2 | 82.6 |
| 8 | 124.4 | 128.3 | 128.4 | 40.7 | 40.6 | 40.8 | 47.6 | 43.6 | 48.1 |
| 9 | 169.1 | 169.8 | 169.6 | 65.2 | 65.2 | 65.2 | 71.1 | 73.3 | 70.3 |
| 1′ | 135.9 | 135.1 | 135.1 | 133.4 | 131.8 | 133.4 | 132.9 | 134.6 | 131.7 |
| 2′ | 106.4 | 106.4 | 106.4 | 113.2 | 107.3 | 113.2 | 104.7 | 104.3 | 104.4 |
| 3′ | 148.3 | 148.4 | 148.4 | 148.1 | 148.5 | 148.1 | 148.6 | 148.7 | 148.8 |
| 4′ | 135.3 | 135.5 | 135.5 | 145.6 | 135.0 | 145.5 | 136.3 | 136.0 | 136.5 |
| 5′ | 148.3 | 148.4 | 148.4 | 115.4 | 148.5 | 115.4 | 148.6 | 148.7 | 148.8 |
| 6′ | 106.4 | 106.4 | 106.4 | 122.3 | 107.3 | 122.3 | 104.7 | 104.3 | 104.4 |
| 7′ | 39.0 | 39.5 | 39.6 | 34.9 | 35.4 | 35.0 | 84.9 | 84.5 | 85.1 |
| 8′ | 46.1 | 49.1 | 49.1 | 44.1 | 44.1 | 44.2 | 51.5 | 50.3 | 49.4 |
| 9′ | 64.6 | 171.4 | 171.4 | 62.1 | 62.1 | 62.1 | 62.8 | 63.4 | 63.6 |
| 1′′ | 131.2 | 131.1 | 131.7 | 126.1 | 127.4 | 127.5 | 127.2 | 126.0 | 127.3 |
| 2′′ | 130.5 | 130.6 | 113.1 | 106.8 | 111.3 | 11.3 | 111.0 | 106.7 | 111.3 |
| 3′′ | 116.0 | 116.1 | 148.2 | 148.9 | 148.7 | 148.8 | 148.6 | 148.6 | 148.7 |
| 4′′ | 156.6 | 156.7 | 145.9 | 139.4 | 150.1 | 150.1 | 149.9 | 139.5 | 150.1 |
| 5′′ | 116.0 | 116.1 | 115.7 | 148.9 | 116.1 | 116.0 | 115.9 | 148.6 | 116.1 |
| 6′′ | 130.5 | 130.6 | 122.0 | 106.8 | 123.9 | 124.0 | 123.7 | 106.7 | 123.8 |
| 7′′ | 35.6 | 35.5 | 36.0 | 145.9 | 145.6 | 145.6 | 145.6 | 146.2 | 145.8 |
| 8′′ | 42.2 | 42.4 | 42.3 | 116.2 | 116.0 | 116.0 | 115.1 | 115.9 | 114.8 |
| 9′′ | 167.5 | 167.6 | 167.5 | 166.7 | 167.3 | 167.3 | |||
| 1′′′ | 131.8 | 131.2 | |||||||
| 2′′′ | 113.0 | 130.6 | |||||||
| 3′′′ | 148.2 | 116.0 | |||||||
| 4′′′ | 145.8 | 156.6 | |||||||
| 5′′′ | 115.6 | 116.0 | |||||||
| 6′′′ | 122.0 | 130.6 | |||||||
| 7′′′ | 36.1 | 35.7 | |||||||
| 8′′′ | 41.9 | 42.1 | |||||||
| OMe-3 | 60.4 | 60.3 | 60.3 | 56.5 | 56.5 | 56.1 | 56.7 | 56.6 | 56.6 |
| OMe-5 | 56.5 | 56.2 | 56.2 | 56.5 | 56.7 | 56.6 | 56.6 | ||
| OMe-7 | 56.1 | ||||||||
| OMe-3′ | 56.6 | 56.7 | 56.7 | 56.1 | 56.4 | 56.1 | 56.6 | 56.7 | 56.6 |
| OMe-5′ | 56.6 | 56.7 | 56.7 | 56.4 | 56.6 | 56.7 | 56.6 | ||
| OMe-3′′ | 56.5 | 56.7 | 56.3 | 56.3 | 56.3 | 56.6 | |||
| OMe-5′′ | 56.7 | 56.6 | |||||||
| OMe-3′′′ | 56.6 |
| Compound | IC50 (μM) | ||
|---|---|---|---|
| HCT-116 | NCI-H1650 | A2780 | |
| 1 | >20 | >20 | >20 |
| 2 | >20 | >20 | >20 |
| 3 | >20 | >20 | >20 |
| 4 | >20 | >20 | >20 |
| 5 | >20 | >20 | >20 |
| 6 | >20 | >20 | >20 |
| 7 | >20 | 2.47 | >20 |
| 8 | >20 | 11.25 | >20 |
| 9 | >20 | 13.16 | >20 |
| 10 | >20 | >20 | >20 |
| 11 | >20 | >20 | >20 |
| 12 | >20 | >20 | >20 |
| 13 | >20 | 9.68 | >20 |
| 14 | >20 | 10.52 | >20 |
| 15 | >20 | >20 | >20 |
| 16 | >20 | >20 | >20 |
| 17 | 3.25 | >20 | 0.28 |
| 18 | 13.95 | >20 | 1.53 |
| 19 | 18.47 | >20 | 12.8 |
| Taxol a | 0.005 | 1.28 | 0.02 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Xia, H.; Wang, L.; Xia, G.; Qu, Y.; Shang, X.; Lin, S. Lignans from the Twigs of Litsea cubeba and Their Bioactivities. Molecules 2019, 24, 306. https://doi.org/10.3390/molecules24020306
Li X, Xia H, Wang L, Xia G, Qu Y, Shang X, Lin S. Lignans from the Twigs of Litsea cubeba and Their Bioactivities. Molecules. 2019; 24(2):306. https://doi.org/10.3390/molecules24020306
Chicago/Turabian StyleLi, Xiuting, Huan Xia, Lingyan Wang, Guiyang Xia, Yuhong Qu, Xiaoya Shang, and Sheng Lin. 2019. "Lignans from the Twigs of Litsea cubeba and Their Bioactivities" Molecules 24, no. 2: 306. https://doi.org/10.3390/molecules24020306
APA StyleLi, X., Xia, H., Wang, L., Xia, G., Qu, Y., Shang, X., & Lin, S. (2019). Lignans from the Twigs of Litsea cubeba and Their Bioactivities. Molecules, 24(2), 306. https://doi.org/10.3390/molecules24020306