
Dual Geometry Schemes in Tetrel Bonds: Complexes between TF4 (T = Si, Ge, Sn) and Pyridine Derivatives




Abstract
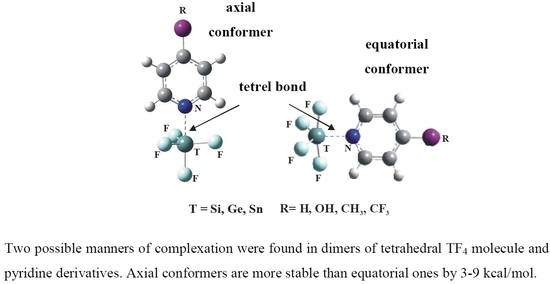
:
1. Introduction
2. Results
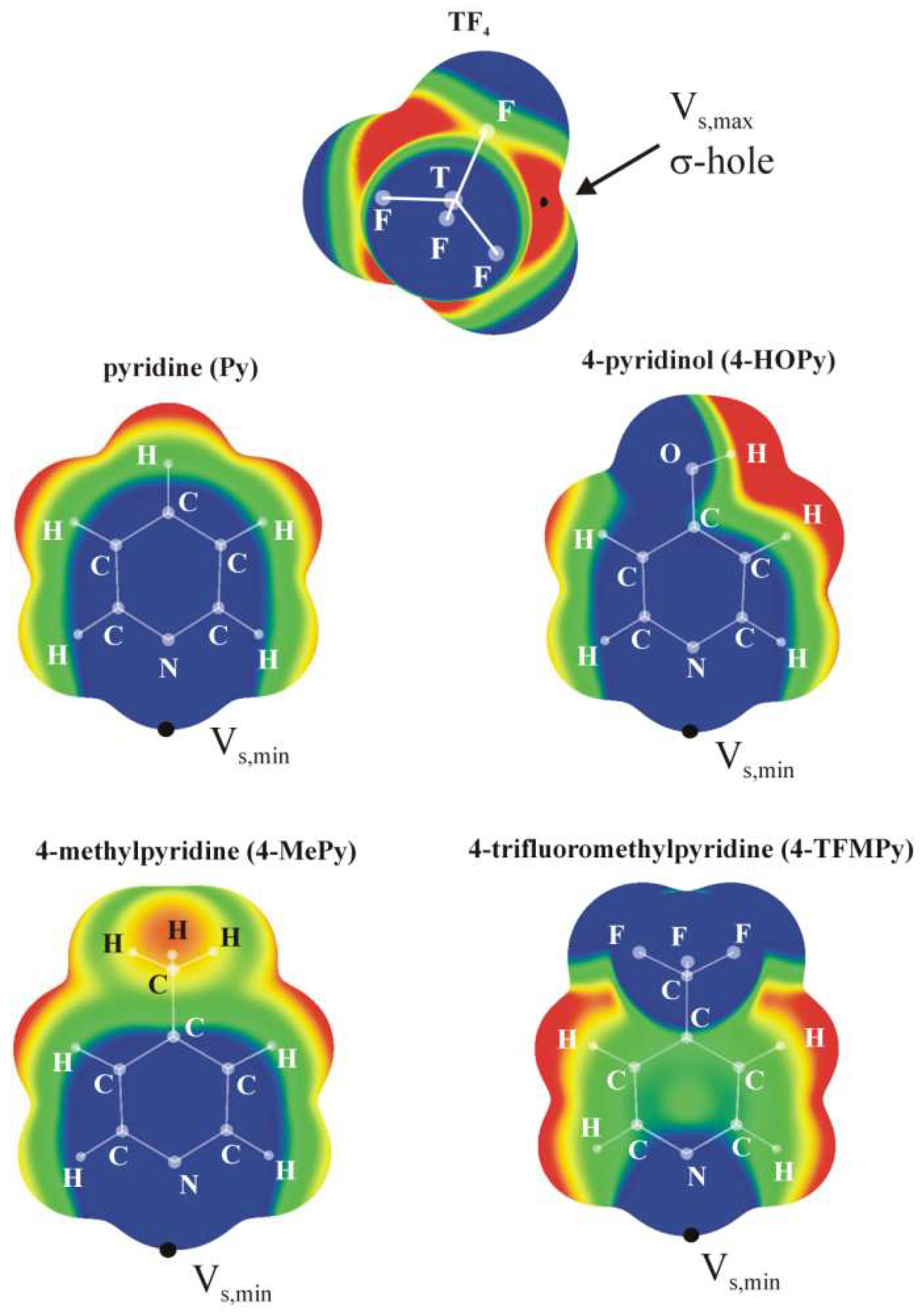
2.1. Electrostatic Potentials of Monomers
2.2. Structures of Complexes
2.3. Energetics
2.4. Analysis of Wave Function
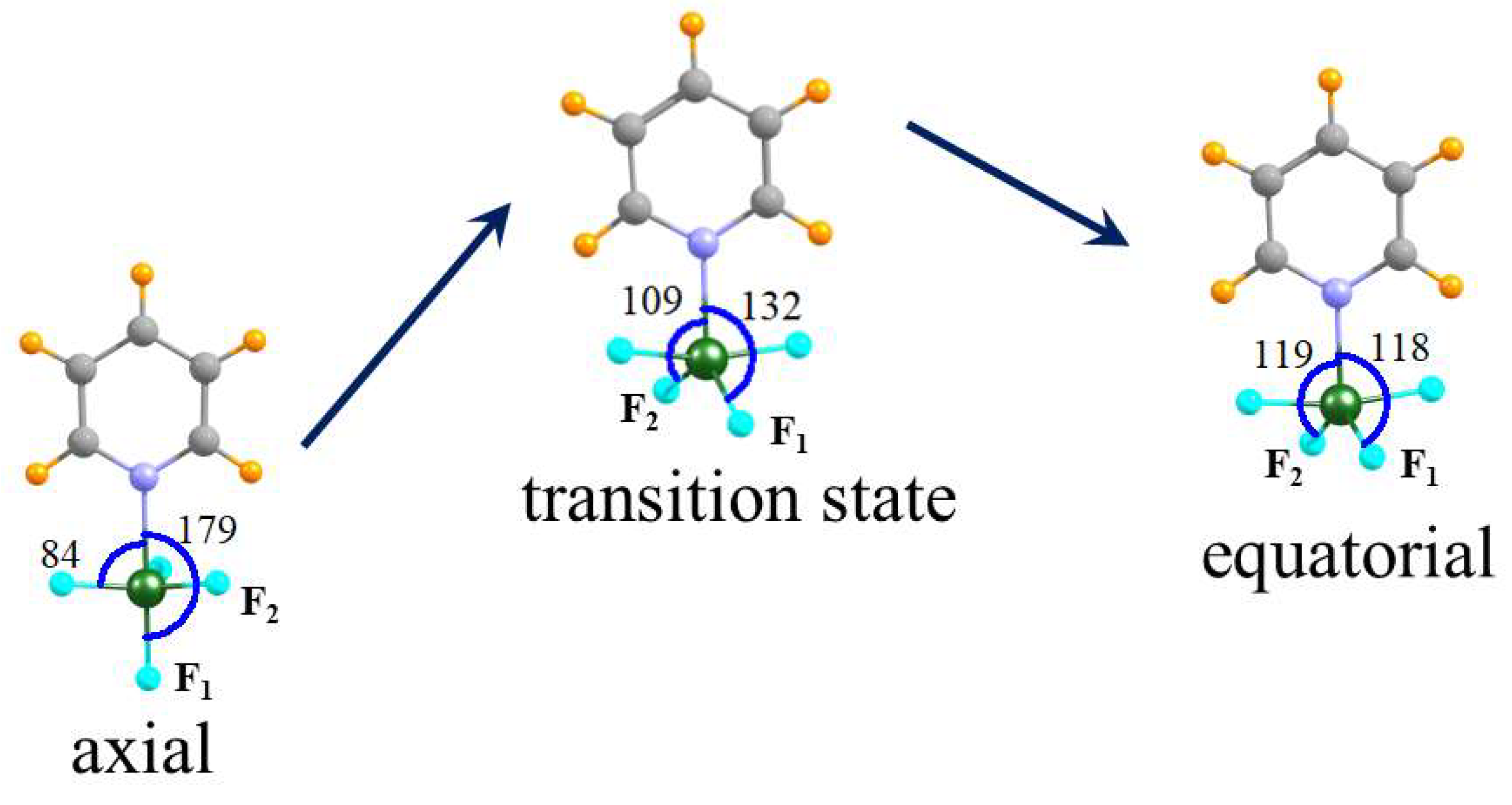
2.5. Conversion between Axial and Equatorial Complexes
2.6. Other Types of Geometry
3. Discussion
4. Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schneider, H.J. Binding mechanisms in supramolecular complexes. Angew. Chem. Int. Ed. 2009, 48, 3924–3977. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.J.; Yatsimirski, A. Principles and Methods in Supramolecular Chemistry; JohnWiley: Chichester, UK, 2000. [Google Scholar]
- Grabowski, S.J. What is the covalency of hydrogen bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef]
- Wash, P.L.; Ma, S.; Obst, U.; Rebek, J. Nitrogen−Halogen Intermolecular Forces in Solution. J. Am. Chem. Soc. 1999, 121, 7973–7974. [Google Scholar] [CrossRef]
- Legon, A.C. Prereactive complexes of dihalogens XY with Lewis bases B in the gas phase: A systematic case for the Halogen analogue B small middle dot small middle dot small middle dotXY of the hydrogen bond B small middle dot small middle dot small middle dotHX. Angew. Chem. Int. Ed. Engl. 1999, 38, 2686–2714. [Google Scholar] [CrossRef]
- Caronna, T.; Liantonio, R.; Logothetis, T.A.; Metrangolo, P.; Pilati, T.; Resnati, G. Halogen bonding and π···π stacking control reactivity in the solid state. J. Am. Chem. Soc. 2004, 126, 4500–4501. [Google Scholar] [CrossRef] [PubMed]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed]
- Glaser, R.; Chen, N.; Wu, H.; Knotts, N.; Kaupp, M. 3C NMR study of halogen bonding of haloarenes: Measurements of solvent effects and theoretical analysis. J. Am. Chem. Soc. 2004, 126, 4412–4419. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Bilewicz, E. Cooperativity halogen bonding effect–Ab initio calculations on H2CO⋯(ClF)n complexes. Chem. Phys. Lett. 2006, 427, 51–55. [Google Scholar] [CrossRef]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the sigma-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. Halogen bonding: An interim discussion. Chem. Phys. Chem. 2013, 14, 278–294. [Google Scholar] [CrossRef] [PubMed]
- Bundhun, A.; Ramasami, P.; Murray, J.S.; Politzer, P. Trends in σ-hole strengths and interactions of F3MX molecules (M = C, Si, Ge and X = F., Cl, Br, I). J. Mol. Model. 2013, 19, 2739–2746. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S. σ-Hole interactions: Perspectives and misconceptions. Crystals 2017, 7, 212. [Google Scholar] [CrossRef]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen bond: A sister noncovalent bond to halogen bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. A new noncovalent force: Comparison of P···N interaction with hydrogen and halogen bonds. J. Chem. Phys. 2011, 134, 094315–094319. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Aerogen bonding interaction: A new supramolecular force? Angew. Chem. Int. Ed. 2015, 54, 7340–7343. [Google Scholar] [CrossRef]
- Stenlid, J.H.; Johansson, A.J.; Brinck, T. σ-Holes and σ-lumps direct the Lewis basic and acidic interactions of noble metal nanoparticles: Introducing regium bonds. Phys. Chem. Chem. Phys. 2018, 20, 2676–2692. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Clark, T.; Riley, K.E.; Politzer, P. Σ-holes, π-holes and electrostatically-driven interactions. J. Mol. Model. 2012, 18, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. Directionality of π-holes in nitro compounds. Chem. Commun. 2015, 51, 1491–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauza, A.; Mooibroek, T.J.; Frontera, A. The bright future of unconventional σ/π-hole interactions. ChemPhysChem 2015, 16, 2496–2517. [Google Scholar] [CrossRef] [PubMed]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Comparison between tetrel bonded complexes stabilized by σ and π hole interactions. Molecules 2018, 23, 1416. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cheng, J.; Yang, X.; Liu, Z.; Li, W.; Li, Q. Comparison of σ-hole and π-hole tetrel bonds formed by pyrazine and 1,4-dicyanobenzene: The interplay between anion-π and tetrel bonds. Chem. Phys. Chem. 2017, 18, 2442–2450. [Google Scholar] [CrossRef] [PubMed]
- Wenbo, D.; Xin, Y.; Jianbo, C.; Wenzuo, L.; Qingzhong, L. Comparison for σ-hole and π-hole tetrel-bonded complexes involving F2C=CFTF3 (T = C, Si, and Ge): Substitution, hybridization, and solvation effects. J. Fluorine Chem. 2018, 207, 38–44. [Google Scholar]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel Bonding Interactions. Chem. Rec. 2016, 16, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Kubelka, J.; Bickelhaupt, F.M. Activation strain analysis of SN2 reactions at C, N, O, and F centers. J. Phys.Chem. A 2017, 121, 885–891. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Dronskowski, R. Tetrel bonds in infinite molecular chains by electronic structure theory and their role for crystal stabilization. J. Phys. Chem. A 2017, 121, 1381–1387. [Google Scholar] [CrossRef]
- Scheiner, S. Steric crowding in tetrel bonds. J. Phys. Chem. A 2018, 122, 2550–2562. [Google Scholar] [CrossRef]
- Scheiner, S. Tetrel Bonding as a vehicle for strong and selective anion binding. Molecules 2018, 23, 1147. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Elguero, J.; Alkorta, I. Complexes of CO2 with the azoles: Tetrel bonds, hydrogen bonds and other secondary interactions. Molecules 2018, 23, 906. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. Tetrel bonding interactions in perchlorinated cyclopenta- and cyclohexatetrelanes: A combined DFT and CSD study. Molecules 2018, 23, 1770. [Google Scholar] [CrossRef] [PubMed]
- García-Llinás, X.; Bauzá, A.; Seth, S.K.; Frontera, A. Importance of R–CF3···O tetrel bonding interactions in biological systems. J. Phys. Chem. A 2017, 121, 5371–5376. [Google Scholar] [CrossRef]
- Mundlapati, V.R.; Sahoo, D.K.; Bhaumik, S.; Jena, S.; Chandrakar, A.; Biswal, H.S. Noncovalent carbon-bonding interactions in proteins. Angew. Chem. Int. Ed. 2018, 57, 16496–16500. [Google Scholar] [CrossRef] [PubMed]
- Frontera, A.; Bauzá, A. S⋅⋅⋅Sn tetrel bonds in the activation of peroxisome proliferator-activated receptors (PPARs) by organotin molecules. Chem. Eur. J. 2018, 24, 16582–16587. [Google Scholar] [CrossRef]
- Trievel, R.C.; Scheiner, S. Crystallographic and computational characterization of methyl tetrel bonding in S-adenosylmethionine-dependent methyltransferases. Molecules 2018, 23, 2965. [Google Scholar] [CrossRef]
- Marin-Luna, M.; Alkorta, I.; Elguero, J. Cooperativity in tetrel bonds. J. Phys. Chem. A 2016, 120, 648–656. [Google Scholar] [CrossRef]
- Shukla, R.; Chopra, D. Characterization of the short O=C⋯O=C π-hole tetrel bond in the solid state. CrystEngComm 2018, 20, 3308–3312. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. RCH3···O Interactions in biological systems: Are they trifurcated H-bonds or noncovalent carbon bonds? Crystals 2016, 6, 26. [Google Scholar] [CrossRef]
- Scheiner, S.; Lu, J. Chalcogen, and pnicogen bonding involving hypervalent atoms. Chem. Eur. J. 2018, 24, 8167–8177. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Tetrel bonds with π-electrons acting as lewis bases—Theoretical results and experimental evidences. Molecules 2018, 23, 1183. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms In Molecules: A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Bader, R.F.W. A bond path: A universal indicator of bonded interactions. J. Phys. Chem. A 1998, 102, 7314. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Essen, H. The characterization of atomic interactions. J. Chem. Phys. 1984, 80, 1943. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Slee, T.S.; Cremer, D.; Kraka, E. Description of conjugation and hyperconjugation in terms of electron distributions. J. Am. Chem. Soc. 1983, 105, 5061. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. A description of the chemical-bond in terms of local properties of electron density and energy. Croat. Chem. Acta. 1984, 57, 1259. [Google Scholar]
- Liu, M.; Li, Q.; Scheiner, S. Comparison of tetrel bonds in neutral and protonated complexes of pyridineTF3 and furan TF3 (T = C, Si, and Ge) with NH3. Phys. Chem. Chem. Phys. 2017, 19, 5550–5559. [Google Scholar] [CrossRef]
- Esrafili, M.; Mousavian, P. Strong tetrel bonds: Theoretical aspects and experimental evidence. Molecules 2018, 23, 2642. [Google Scholar] [CrossRef]
- Baryshnikov, G.V.; Minaev, B.F.; Minaeva, V.A.; Podgornaya, A.T.; Ågren, H. Application of Bader’s atoms in molecules theory to the description of coordination bonds in the complex compounds of Ca2+ and Mg2+ with methylidene rhodanine and its anion. Russ. J. Gen. Chem. 2012, 82, 1254. [Google Scholar] [CrossRef]
- Karaush, N.; Baryshnikov, G.V.; Minaev, B.F. Alkali and alkaline-earth metal complexes with tetraoxa[8]circulene sheet: A computational study by DFT and QTAIM methods. RSC Adv. 2015, 5, 24299–24305. [Google Scholar] [CrossRef]
- De Paul, V.; Nziko, N.; Scheiner, S. Chalcogen bonding between tetravalent SF4 and amines. J. Phys. Chem. A 2014, 118, 10849–10856. [Google Scholar]
- Guo, X.; An, X.; Li, Q. Se···N chalcogen bond and Se···X halogen bond involving F2C=Se: Influence of hybridization, substitution, and cooperativity. J. Phys. Chem. A 2015, 119, 3518–3527. [Google Scholar] [CrossRef] [PubMed]
- Esrafili, M.; Mohammadirad, N. Substituent effects in cooperativity of chalcogen bonds. Mol. Phys. 2015, 113, 3282–3290. [Google Scholar] [CrossRef]
- Esrafili, M.; Nurazar, R. Chalcogen bonds formed through π-holes: SO3 complexes with nitrogen and phosphorus bases. Mol. Phys. 2016, 114, 276–282. [Google Scholar] [CrossRef]
- Azofra, L.; Alkorta, I.; Scheiner, S. Chalcogen bonds in complexes of SOXY (X, Y = F, Cl) with nitrogen bases. J. Phys. Chem. A 2015, 119, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, U.; Scheiner, S. Effects of charge and substituent on the S···N chalcogen bond. J. Phys. Chem. A 2014, 118, 3183–3192. [Google Scholar] [CrossRef] [PubMed]
- Ramasami, P.; Ford, T. Chalcogen-bonded complexes of some carbon dioxide analogues. J. Mol. Struct. 2014, 1072, 28–31. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen bonds, and σ-hole and π-hole bonds-mechanisms protecting doublet and octet electron structures. Phys. Chem. Chem. Phys. 2017, 19, 29742–29759. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Asadollahi, S.; Mousavian, P. Anionic tetrel bonds: An Ab initio study. Chem. Phys. Lett. 2018, 691, 394–400. [Google Scholar] [CrossRef]
- Grabowski, S.J.; Sokalski, W.A. Are various Σ-hole bonds steered by the same mechanisms? ChemPhysChem. 2017, 18, 1569–1577. [Google Scholar] [CrossRef]
- Azofra, L.M.; Scheiner, S. Chalcogen, and CH··O Hydrogen bonds in complexes pairing carbonyl-Containing molecules with 1, 2, and 3 molecules of CO2. J. Chem. Phys. 2015, 142, 034307–034317. [Google Scholar] [CrossRef] [PubMed]
- Nziko, V.d.P.N.; Scheiner, S. Comparison of p-hole tetrel bonding with s-hole halogen bonds in complexes of XCN (X = F, Cl, Br, I) and NH3. Phys. Chem. Chem. Phys. 2016, 18, 3581–3590. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, Q.; Scheiner, S. The Π-tetrel bond and its influence on hydrogen bonding and proton transfer. ChemPhysChem. 2018, 19, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, Q. Comparison for Σ-hole and π-hole tetrel-bonded complexes involving cyanoacetaldehyde. Mol. Phys. 2018, 116, 222–230. [Google Scholar] [CrossRef]
- Shen, S.; Zeng, Y.; Li, X.; Meng, L.; Zhang, X. Insight into the π-Hole··· π-electrons tetrel bonds between F2ZO (Z = C, Si, Ge) and unsaturated hydrocarbons. Int. J. Quantum Chem. 2018, 118, e25521–25533. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Implications of monomer deformation for tetrel and pnicogen bonds. Phys. Chem. Chem. Phys. 2018, 20, 8832–8841. [Google Scholar] [CrossRef] [PubMed]
- Solel, E.; Kozuch, S. On the power of geometry over tetrel bonds. Molecules 2018, 23, 2742. [Google Scholar] [CrossRef]
- Scheiner, S. Systematic elucidation of factors that influence the strength of tetrel bonds. J. Phys. Chem. A 2017, 121, 5561–5568. [Google Scholar] [CrossRef]
- Sethio, D.; Oliveira, V.; Kraka, E. Quantitative assessment of tetrel bonding utilizing vibrational spectroscopy. Molecules 2018, 23, 2763. [Google Scholar] [CrossRef]
- Legon, A.C. Tetrel, Pnictogen and chalcogen bonds identified in the gas phase before they had names: A systematic look at non-covalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 14884–14896. [Google Scholar] [CrossRef]
- Scheiner, S. Highly selective halide receptors based on chalcogen, pnicogen, and tetrel bonds. Chem. Eur. J. 2016, 22, 18850–18858. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Assembly of effective halide receptors from components. Comparing hydrogen, halogen, and tetrel bonds. J. Phys. Chem. A 2017, 121, 3606–3615. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Legon, A. An Ab initio investigation of the geometries and binding strengths of tetrel-, pnictogen-, and chalcogen-bonded complexes of Co2, N2o, and Cs2 with simple Lewis bases: Some generalizations. Molecules 2018, 23, 2250. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Li, Q.; Scheiner, S. Comparative strengths of tetrel, pnicogen, chalcogen, and halogen bonds and contributing factors. Molecules 2018, 23, 1681. [Google Scholar] [CrossRef] [PubMed]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–623. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Head-Gordon, M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical hybrid density functional with perturbative second-order correlation. J. Chem. Phys. 2006, 124, e034108. [Google Scholar] [CrossRef] [PubMed]
- Feller, D. The role of databases in support of computational chemistry calculations. J. Comp. Chem. 1996, 17, 1571–1587. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis set exchange: A community database for computational sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–968. [Google Scholar] [CrossRef]
- Fonseca Guerra, C.; Snijders, J.G.; te Velde, G.; Baerends, E.J. Towards an order-N DFT method. Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar] [CrossRef]
- ADF2014, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: http://www.scm.com (accessed on 19 January 2019).
- Bulat, F.; Toro-Labbe, A.; Brinck, T.; Murray, J.S.; Politzer, P. Quantitative analysis of molecular surfaces: Areas, volumes, electrostatic potentials and average local ionization energies. J. Mol. Model. 2010, 16, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- AIMAll; Version 14.11.23; Todd A. Keith (TK Gristmill Software): Overland Park, KS, USA, 2014.
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 6.0: Natural bond orbital analysis program. J. Comput. Chem. 2013, 34, 1429–1437. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lewis Acid | Vs,max (σ-hole) |
|---|---|
| SiF4 | +41.3 |
| GeF4 | +50.9 |
| SnF4 | +70.1 |
| Lewis base | Vs,min a |
| Py | −36.3 |
| 4-HOPy | −37.3 |
| 4-MePy | −37.8 |
| 4-TFMPy | −29.9 |
| Axial Complexes | Equatorial Complexes | ||||||
|---|---|---|---|---|---|---|---|
| R (T···N) | θ (N··TFax) | θ (N··TFeq) | R (N∙∙∙T) | θ (N··TFeq) | θ (N··TFax) | ∆E a | |
| Py∙∙∙SiF4 | 2.143 | 179.6 | 82.9 | 1.973 | 119.1 | 83.7 | 9.42 |
| Py∙∙∙GeF4 | 2.120 | 179.5 | 84.3 | 2.024 | 118.4 | 84.5 | 5.91 |
| Py∙∙∙SnF4 | 2.244 | 178.2 | 83.1 | 2.218 | 118.6 | 83.3 | 3.27 |
| 4-HOPy∙∙∙SiF4 | 2.121 | 179.9 | 83.2 | 1.960 | 118.9 | 84.3 | 8.59 |
| 4-HOPy∙∙∙GeF4 | 2.104 | 179.9 | 84.4 | 2.012 | 118.4 | 85.2 | 5.15 |
| 4-HOPy∙∙∙SnF4 | 2.232 | 177.6 | 83.2 | 2.203 | 118.8 | 83.5 | 2.69 |
| 4-MePy∙∙∙SiF4 | 2.127 | 179.7 | 83.1 | 1.965 | 119.0 | 84.0 | 9.00 |
| 4-MePy∙∙∙GeF4 | 2.110 | 179.6 | 84.5 | 2.017 | 118.4 | 84.9 | 5.55 |
| 4-MePy∙∙∙SnF4 | 2.237 | 178.1 | 83.3 | 2.210 | 118.7 | 83.9 | 3.02 |
| 4-TFMPy∙∙∙SiF4 | 2.189 | 179.7 | 81.9 | - | - | - | - |
| 4-TFMPy∙∙∙GeF4 | 2.142 | 179.5 | 83.7 | 2.039 | 118.5 | 83.9 | 6.63 |
| 4-TFMPy∙∙∙SnF4 | 2.260 | 178.3 | 82.6 | 2.233 | 118.5 | 82.6 | 3.70 |
| Monomer | E(eq) − E(ax) | Vs,max, Axial | Vs,max, Equatorial |
|---|---|---|---|
| SiF4 | 22.56 | 120.6 | 126.5 |
| GeF4 | 17.39 | 116.4 | 120.1 |
| SnF4 | 10.68 | 124.3 | 131.4 |
| Axial Complexes | Equatorial Complexes | |||||
|---|---|---|---|---|---|---|
| (I) | (II) | (III) | (I) | (II) | (III) | |
| Py∙∙∙SiF4 | −26.75 | −22.50 | −27.07 | −50.09 | −47.38 | −50.98 |
| Py∙∙∙GeF4 | −34.73 | −34.37 | −34.93 | −52.02 | −48.71 | −52.62 |
| Py∙∙∙SnF4 | −39.68 | −37.87 | −39.85 | −50.66 | −44.97 | −51.13 |
| 4-HOPy∙∙∙SiF4 | −28.79 | −22.50 | −29.35 | −53.52 | −51.29 | −54.78 |
| 4-HOPy∙∙∙GeF4 | −36.62 | −34.37 | −37.10 | −55.30 | −51.93 | −56.26 |
| 4-HOPy∙∙∙SnF4 | −41.54 | −37.87 | −41.98 | −52.86 | −48.67 | −53.63 |
| 4-MePy∙∙∙SiF4 | −28.35 | −25.97 | −28.84 | −52.10 | −49.96 | −53.25 |
| 4-MePy∙∙∙GeF4 | −36.19 | −36.29 | −36.58 | −53.98 | −51.00 | −54.84 |
| 4-MePy∙∙∙SnF4 | −41.07 | −39.70 | −41.44 | −52.22 | −47.69 | −52.93 |
| 4-TFMPy∙∙∙SiF4 | −21.94 | −9.43 | −22.08 | - | - | - |
| 4-TFMPy∙∙∙GeF4 | −30.62 | −29.93 | −30.67 | −47.32 | −42.98 | −47.72 |
| 4-TFMPy∙∙∙SnF4 | −35.81 | −33.78 | −35.83 | −46.34 | −41.53 | −46.65 |
| Axial Complexes | Equatorial Complexes | |||
|---|---|---|---|---|
| (I) | (II) | (I) | (II) | |
| Py∙∙∙SiF4 | −10.39 | −7.85 | −0.98 | 1.67 |
| Py∙∙∙GeF4 | −20.51 | −18.06 | −14.60 | −12.97 |
| Py∙∙∙SnF4 | −32.41 | −27.64 | −29.14 | −24.59 |
| 4-HOPy∙∙∙SiF4 | −11.13 | −8.66 | −2.53 | −0.06 |
| 4-HOPy∙∙∙GeF4 | −21.52 | −19.29 | −16.36 | −14.86 |
| 4-HOPy∙∙∙SnF4 | −33.65 | −29.09 | −30.96 | −26.45 |
| 4-MePy∙∙∙SiF4 | −11.06 | −8.59 | −2.06 | 0.31 |
| 4-MePy∙∙∙GeF4 | −21.37 | −19.15 | −15.81 | −14.44 |
| 4-MePy∙∙∙SnF4 | −33.41 | −28.89 | −30.39 | −26.20 |
| 4-TFMPy∙∙∙SiF4 | −8.38 | −6.36 | - | - |
| 4-TFMPy∙∙∙GeF4 | −17.98 | −15.41 | −11.35 | −9.67 |
| 4-TFMPy∙∙∙SnF4 | −29.50 | −24.66 | −25.85 | −21.16 |
| Axial Complexes | ||||
|---|---|---|---|---|
| Edef of TF4 | Edef of LB | Sum | Vs,max | |
| Py∙∙∙SiF4 | 20.35 | 0.35 | 20.70 | 93.4 |
| Py∙∙∙GeF4 | 19.49 | 0.58 | 20.07 | 100.3 |
| Py∙∙∙SnF4 | 12.64 | 0.74 | 13.38 | 111.3 |
| 4-HOPy∙∙∙SiF4 | 21.72 | 0.41 | 22.13 | 95.0 |
| 4-HOPy∙∙∙GeF4 | 20.43 | 0.67 | 21.10 | 101.0 |
| 4-HOPy∙∙∙SnF4 | 13.28 | 0.85 | 14.13 | 112.1 |
| 4-MePy∙∙∙SiF4 | 21.35 | 0.37 | 21.72 | 94.6 |
| 4-MePy∙∙∙GeF4 | 20.15 | 0.60 | 20.75 | 101.0 |
| 4-MePy∙∙∙SnF4 | 13.09 | 0.76 | 13.85 | 111.9 |
| 4-TFMPy∙∙∙SiF4 | 17.35 | 0.33 | 17.68 | 89.6 |
| 4-TFMPy∙∙∙GeF4 | 17.70 | 0.58 | 18.28 | 98.5 |
| 4-TFMPy∙∙∙SnF4 | 11.49 | 0.75 | 12.24 | 110.0 |
| Equatorial Complexes | ||||
| Py∙∙∙SiF4 | 53.05 | 0.82 | 53.87 | 107.5 |
| Py∙∙∙GeF4 | 43.08 | 1.01 | 44.09 | 107.7 |
| Py∙∙∙SnF4 | 26.69 | 1.00 | 27.69 | 119.5 |
| 4-HOPy∙∙∙SiF4 | 54.93 | 0.94 | 55.87 | 109.1 |
| 4-HOPy∙∙∙GeF4 | 44.65 | 1.15 | 45.80 | 108.9 |
| 4-HOPy∙∙∙SnF4 | 27.06 | 1.17 | 28.23 | 119.8 |
| 4-MePy∙∙∙SiF4 | 54.02 | 0.84 | 54.86 | 108.3 |
| 4-MePy∙∙∙GeF4 | 43.90 | 1.03 | 44.93 | 108.4 |
| 4-MePy∙∙∙SnF4 | 27.05 | 1.04 | 28.08 | 119.8 |
| 4-TFMPy∙∙∙SiF4 | - | - | - | - |
| 4-TFMPy∙∙∙GeF4 | 41.43 | 1.04 | 42.47 | 106.4 |
| 4-TFMPy∙∙∙SnF4 | 25.52 | 1.04 | 26.56 | 118.2 |
| EPauli | Eelec | Eoi | Edisp | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ax | eq | eq − ax | ax | eq | eq − ax | ax | eq | eq − ax | ax | eq | eq − ax | |
| Py∙∙∙SiF4 | 89.72 | 117.10 | 27.38 | −70.23 | −93.70 | −23.47 | −40.00 | −67.04 | −27.04 | −4.53 | −4.38 | 0.15 |
| Py∙∙∙GeF4 | 108.13 | 110.56 | 2.43 | −84.50 | −90.48 | −5.98 | −49.68 | −64.82 | −15.14 | −4.63 | −4.19 | 0.44 |
| Py∙∙∙SnF4 | 99.74 | 87.36 | −12.38 | −85.48 | −80.05 | 5.43 | −45.43 | −48.91 | −3.48 | −4.52 | −3.98 | 0.54 |
| 4-HOPy∙∙∙SiF4 | 94.13 | 120.82 | 26.69 | −74.10 | −97.86 | −23.76 | −42.67 | −70.50 | −27.83 | −4.53 | −4.33 | 0.20 |
| 4-HOPy∙∙∙GeF4 | 112.02 | 116.15 | 4.13 | −88.08 | −95.58 | −7.50 | −51.93 | −68.40 | −16.47 | −4.63 | −4.20 | 0.43 |
| 4-HOPy∙∙∙SnF4 | 102.86 | 92.22 | −10.64 | −88.83 | −85.27 | 3.56 | −47.15 | −52.27 | −5.12 | −4.52 | −3.93 | 0.59 |
| 4-MePy∙∙∙SiF4 | 92.92 | 117.10 | 24.18 | −73.01 | −93.70 | −20.60 | −42.06 | −67.04 | −24.98 | −4.54 | −4.38 | 0.16 |
| 4-MePy∙∙∙GeF4 | 110.73 | 110.56 | −0.17 | −86.99 | −90.48 | −3.49 | −51.39 | −64.82 | −13.43 | −4.65 | −4.19 | 0.46 |
| 4-MePy∙∙∙SnF4 | 101.57 | 87.36 | −14.21 | −87.63 | −80.05 | 7.58 | −46.71 | −48.91 | −2.20 | −4.54 | −3.98 | 0.56 |
| 4-TFMPy∙∙∙SiF4 | 80.38 | - | - | −61.74 | - | - | −34.59 | - | - | −4.56 | - | - |
| 4-TFMPy∙∙∙GeF4 | 101.43 | 104.42 | 2.99 | −77.67 | −83.12 | −5.45 | −45.96 | −60.09 | −14.13 | −4.66 | −4.26 | 0.40 |
| 4-TFMPy∙∙∙SnF4 | 94.96 | 82.47 | −12.49 | −79.49 | −73.92 | 5.57 | −42.96 | −46.70 | −3.74 | −4.56 | −3.93 | 0.63 |
| Axial Complexes | Equatorial Complexes | |||||||
|---|---|---|---|---|---|---|---|---|
| Interaction | ρ | ∇2ρ | H | Interaction | ρ | ∇2ρ | H | |
| Py∙∙∙SiF4 | Si···N | 0.054 | 0.145 | −0.022 | Si···N | 0.077 | 0.276 | −0.032 |
| F···H | 0.015 | 0.073 | 0.003 | F···H | 0.019 | 0.094 | 0.003 | |
| - | - | - | F···H | 0.019 | 0.094 | |||
| Py∙∙∙GeF4 | Ge···N | 0.077 | 0.166 | −0.030 | Ge···N | 0.094 | 0.216 | −0.042 |
| F···H | 0.015 | 0.068 | 0.003 | F···H | 0.020 | 0.093 | 0.004 | |
| F···H | 0.015 | 0.070 | 0.003 | F···H | 0.020 | 0.093 | 0.004 | |
| Py∙∙∙SnF4 | Sn···N | 0.073 | 0.211 | −0.020 | Sn···N | 0.077 | 0.230 | −0.022 |
| F···H | 0.014 | 0.059 | 0.002 | F···H | 0.021 | 0.095 | 0.004 | |
| F···H | 0.014 | 0.059 | 0.002 | F···H | 0.021 | 0.095 | 0.004 | |
| 4-HOPy∙∙∙SiF4 | Si···N | 0.057 | 0.159 | −0.023 | Si···N | 0.079 | 0.289 | −0.033 |
| F···H | 0.015 | 0.074 | 0.003 | F···H | 0.021 | 0.100 | 0.004 | |
| - | - | - | F···H | 0.020 | 0.099 | |||
| 4-HOPy∙∙∙GeF4 | Ge···N | 0.079 | 0.174 | −0.032 | Ge···N | 0.097 | 0.225 | −0.044 |
| F···H | 0.015 | 0.070 | 0.003 | F···H | 0.021 | 0.100 | 0.004 | |
| F···H | 0.015 | 0.068 | 0.003 | F···H | 0.021 | 0.099 | 0.004 | |
| 4-HOPy∙∙∙SnF4 | Sn···N | 0.075 | 0.218 | −0.021 | Sn···N | 0.079 | 0.240 | −0.023 |
| F···H | 0.014 | 0.059 | 0.002 | F···H | 0.021 | 0.095 | 0.004 | |
| F···H | 0.014 | 0.060 | 0.002 | F···H | 0.020 | 0.093 | 0.003 | |
| 4-MePy∙∙∙SiF4 | Si···N | 0.056 | 0.155 | −0.022 | Si···N | 0.079 | 0.284 | −0.032 |
| F···H | 0.015 | 0.073 | 0.003 | F···H | 0.019 | 0.095 | 0.003 | |
| - | - | - | F···H | 0.019 | 0.094 | |||
| 4-MePy∙∙∙GeF4 | Ge···N | 0.079 | 0.170 | −0.032 | Ge···N | 0.096 | 0.221 | −0.043 |
| F···H | 0.015 | 0.069 | 0.003 | F···H | 0.020 | 0.094 | 0.004 | |
| F···H | 0.015 | 0.069 | 0.003 | F···H | 0.020 | 0.094 | 0.004 | |
| 4-MePy∙∙∙SnF4 | Sn···N | 0.075 | 0.215 | −0.021 | Sn···N | 0.078 | 0.235 | −0.022 |
| F···H | 0.013 | 0.058 | 0.002 | F···H | 0.020 | 0.093 | 0.003 | |
| F···H | 0.013 | 0.058 | 0.002 | F···H | 0.020 | 0.093 | 0.003 | |
| 4-TFMPy∙∙∙SiF4 | Si···N | 0.049 | 0.118 | −0.020 | - | - | - | |
| 4-TFMPy∙∙∙GeF4 | Ge···N | 0.073 | 0.158 | −0.027 | Ge···N | 0.091 | 0.208 | −0.040 |
| F···H | 0.015 | 0.068 | 0.003 | F···H | 0.020 | 0.094 | 0.003 | |
| F···H | 0.015 | 0.068 | 0.003 | F···H | 0.020 | 0.094 | 0.003 | |
| 4-TFMPy∙∙∙SnF4 | Sn···N | 0.071 | 0.204 | −0.019 | Sn···N | 0.075 | 0.222 | −0.020 |
| F···H | 0.014 | 0.060 | 0.002 | F···H | 0.021 | 0.096 | 0.004 | |
| F···H | 0.014 | 0.059 | 0.002 | F···H | 0.021 | 0.096 | 0.004 | |
| Axial Complexes | Equatorial Complexes | |||
|---|---|---|---|---|
| ∑ LP (N) → LP* (T) | CT | ∑ LP (N) → LP* (T) | CT | |
| Py∙∙∙SiF4 | 72.8 | 121 | 161.2 | 212 |
| Py∙∙∙GeF4 | 137.7 | 174 | 158.4 | 220 |
| Py∙∙∙SnF4 | 108.2 | 185 | 111.0 | 202 |
| 4-HOPy∙∙∙SiF4 | 98.5 | 139 | 169.0 | 221 |
| 4-HOPy∙∙∙GeF4 | 146.0 | 182 | 166.2 | 227 |
| 4-HOPy∙∙∙SnF4 | 113.5 | 191 | 116.6 | 210 |
| 4-MePy∙∙∙SiF4 | 90.7 | 138 | 166.9 | 218 |
| 4-MePy∙∙∙GeF4 | 143.8 | 180 | 163.6 | 225 |
| 4-MePy∙∙∙SnF4 | 110.4 | 190 | 114.9 | 209 |
| 4-TFMPy∙∙∙SiF4 | 17.9 | 36 | - | - |
| 4-TFMPy∙∙∙GeF4 | 123.4 | 159 | 149.2 | 206 |
| 4-TFMPy∙∙∙SnF4 | 100.6 | 172 | 103.5 | 192 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zierkiewicz, W.; Michalczyk, M.; Wysokiński, R.; Scheiner, S. Dual Geometry Schemes in Tetrel Bonds: Complexes between TF4 (T = Si, Ge, Sn) and Pyridine Derivatives. Molecules 2019, 24, 376. https://doi.org/10.3390/molecules24020376
Zierkiewicz W, Michalczyk M, Wysokiński R, Scheiner S. Dual Geometry Schemes in Tetrel Bonds: Complexes between TF4 (T = Si, Ge, Sn) and Pyridine Derivatives. Molecules. 2019; 24(2):376. https://doi.org/10.3390/molecules24020376
Chicago/Turabian StyleZierkiewicz, Wiktor, Mariusz Michalczyk, Rafał Wysokiński, and Steve Scheiner. 2019. "Dual Geometry Schemes in Tetrel Bonds: Complexes between TF4 (T = Si, Ge, Sn) and Pyridine Derivatives" Molecules 24, no. 2: 376. https://doi.org/10.3390/molecules24020376
APA StyleZierkiewicz, W., Michalczyk, M., Wysokiński, R., & Scheiner, S. (2019). Dual Geometry Schemes in Tetrel Bonds: Complexes between TF4 (T = Si, Ge, Sn) and Pyridine Derivatives. Molecules, 24(2), 376. https://doi.org/10.3390/molecules24020376