Design and Synthesis of Novel Breast Cancer Therapeutic Drug Candidates Based upon the Hydrophobic Feedback Approach of Antiestrogens

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Evaluation
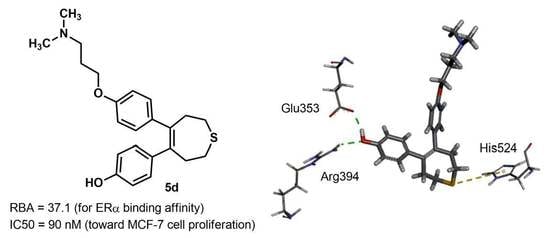
2.3. Docking Study
3. Materials and Methods
3.1. General Consideration
3.2. Synthesis
3.3. Competitive Binding Assay Using Human ER
3.4. MCF-7 Cell Proliferation Assay
3.5. Docking Simulation Study
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Umetani, M.; Domoto, H.; Gormley, A.K.; Yuhanna, I.S.; Cummins, C.L.; Javitt, N.B.; Korach, K.S.; Shaul, P.W.; Mangelsdorf, D.J. 27-Hydroxycholesterol is an endogenous SERM that inhibits the cardiovascular effects of estrogen. Nat. Med. 2007, 13, 1185. [Google Scholar] [CrossRef] [PubMed]
- Takano-Yamamoto, T.; Rodan, G.A. Direct effects of 17 beta-estradiol on trabecular bone in ovariectomized rats. Proc. Natl. Acad. Sci. USA 1990, 87, 2172. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, V.D.; Kipp, J.; Joe, I. Estradiol, in the CNS, targets several physiologically relevant membrane-associated proteins. Brain Res. Rev. 2001, 37, 141. [Google Scholar] [CrossRef]
- Green, S.; Walter, P.; Kumar, V.; Krust, A.; Bornert, J.M.; Argos, P.; Chambon, P. Human oestrogen receptor cDNA: Sequence, expression and homology to v-erb-A. Nature 1986, 320, 134. [Google Scholar] [CrossRef] [PubMed]
- Leng, X.H.; Bray, P.F. Hormone therapy and platelet function. Drug Discov. Today Disease Mechanisms 2005, 2, 85. [Google Scholar] [CrossRef]
- Motivala, A.; Pitt, B. Drospirenone for Oral Contraception and Hormone Replacement Therapy. Drugs 2007, 67, 647. [Google Scholar] [CrossRef]
- Labrie, F.; Labrie, C.; Belanger, A.; Simard, J. Selective Estrogen Receptor Modulators; Manni, A., Verderame, M., Eds.; Humana Press: Totowa, NJ, USA, 2002. [Google Scholar]
- Jameera, B.A.; Jubie, S.; Nanjan, M.J. Estrogen receptor agonists/antagonists in breast cancer therapy: A critical review. Bioorg. Chem. 2017, 71, 257. [Google Scholar] [CrossRef]
- Patel, H.K.; Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther. 2018, 186, 1. [Google Scholar] [CrossRef]
- Valera, M.C.; Fontaine, C.; Dupuis, M.; Noirrit-Esclassan, E.; Vinel, A.; Guillaume, M.; Gourdy, P.; Lenfant, F.; Arnal, J.F. Towards optimization of estrogen receptor modulation in medicine. Pharmacol. Ther. 2018, 189, 123. [Google Scholar] [CrossRef]
- Shiau, A.K.; Barstad, D.; Loria, P.M.; Cheng, L.; Kushner, P.J.; Agard, D.A.; Greene, G.L. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell 1998, 95, 927. [Google Scholar] [CrossRef]
- Jordan, V.C. Tamoxifen: A most unlikely pioneering medicine. Nat. Rev. Drug Discov. 2003, 2, 205. [Google Scholar] [CrossRef] [PubMed]
- Shagufta; Ahmad, I. Tamoxifen a pioneering drug: An update on the therapeutic potential of tamoxifen derivatives. Eur. J. Med. Chem. 2018, 143, 515. [Google Scholar] [CrossRef] [PubMed]
- McCague, R.; Jarman, M.; Leung, O.T.; Foster, A.B.; Leclercq, G.; Stoessel, S.J. Inhibitors of steroid hormone biosynthesis and action. Steroid Biochem. 1988, 31, 545. [Google Scholar] [CrossRef]
- Katzenellenbogen, B.S.; Norman, M.J.; Eckert, R.L.; Peltz, S.W.; Mangel, W.F. Bioactivities, estrogen receptor interactions, and plasminogen activator-inducing activities of tamoxifen and hydroxy-tamoxifen isomers in MCF-7 human breast cancer cells. Cancer Res. 1984, 44, 112. [Google Scholar]
- Gauthier, S.; Mailhot, J.; Labrie, F. New Highly Stereoselective Synthesis of (Z)-4-Hydroxytamoxifen and (Z)-4-Hydroxytoremifene via McMurry Reaction. J. Org. Chem. 1996, 61, 3890. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.D.; Forman, B.M. Simple and efficient production of (Z)-4-hydroxytamoxifen, a potent estrogen receptor modulator. J. Org. Chem. 2003, 68, 9489. [Google Scholar] [CrossRef]
- Mitlak, B.H.; Cohen, F.J. Selective estrogen receptor modulators: A look ahead. Drugs 1999, 57, 653. [Google Scholar] [CrossRef]
- Jones, C.D.; Blaszczak, L.C.; Goettel, M.E.; Suarez, T.; Crowell, T.A.; Mabry, T.E.; Ruenitz, P.C.; Srivatsan, V. Antiestrogens. 3. Estrogen receptor affinities and antiproliferative effects in MCF-7 cells of phenolic analogs of trioxifene, [3,4-dihydro-2-(4-methoxyphenyl)-1-naphthalenyl][4-[2-(1-pyrrolidinyl)ethoxy]phenyl] methanone. J. Med. Chem. 1992, 35, 931. [Google Scholar] [CrossRef]
- Gara, R.K.; Sundram, V.; Chauhan, S.C.; Jaggi, M. Anti-cancer potential of a novel SERM ormeloxifene. Curr. Med. Chem. 2013, 20, 4177. [Google Scholar] [CrossRef]
- Robertson, J.F.R.; Lindemann, J.; Garnett, S.; Anderson, E.; Nicholson, R.I.; Kuter, I.; Gee, J.M.W. A Good Drug Made Better: The Fulvestrant Dose-Response Story. Clin. Breast Cancer 2014, 14, 381. [Google Scholar] [CrossRef]
- Endo, Y.; Yoshimi, T.; Miyaura, C. Boron clusters for medicinal drug design: Selective estrogen receptor modulators bearing carborane. Pure Appl. Chem. 2003, 75, 1197. [Google Scholar] [CrossRef]
- Endo, Y.; Yoshimi, T.; Ohta, K.; Suzuki, T.; Ohta, S. Potent Estrogen Receptor Ligands Based on Bisphenols with a Globular Hydrophobic Core. J. Med. Chem. 2005, 48, 3941. [Google Scholar] [CrossRef] [PubMed]
- Gharpure, S.J.; Anuradha, D.; Prasad, J.V.K.; Srinivasa, R.P. Stereoselective Synthesis of cis-2,6-Disubstituted Morpholines and 1,4-Oxathianes by Intramolecular Reductive Etherification of 1,5-Diketones. Eur. J. Org. Chem. 2015, 86. [Google Scholar] [CrossRef]
- Schnapperelle, I.; Bach, T. Modular Synthesis of Phenanthro[9,10-c]thiophenes by a Sequence of C-H Activation, Suzuki Cross-Coupling and Photocyclization Reactions. Chem. Eur. J. 2014, 20, 9725. [Google Scholar] [CrossRef]
- Ohta, K.; Chiba, Y.; Kaise, A.; Endo, Y. Novel retinoid X receptor (RXR) antagonists having a dicarba-closo-dodecaborane as a hydrophobic moiety. Bioorg. Med. Chem. 2015, 23, 861. [Google Scholar] [CrossRef]
- Ohta, K.; Ogawa, T.; Kaise, A.; Endo, Y. Enhanced estrogen receptor beta (ERβ) selectivity of fluorinated carborane-containing ER modulators. Bioorg. Med. Chem. Lett. 2013, 23, 6555. [Google Scholar] [CrossRef]
- Ohta, K.; Ogawa, T.; Oda, A.; Kaise, A.; Endo, Y. Aliphatic Substitution of o-Carboranyl Phenols Enhances Estrogen Receptor Beta Selectivity. Chem. Pharm. Bull. 2014, 62, 386. [Google Scholar] [CrossRef]
- Ohta, K.; Ogawa, T.; Oda, A.; Kaise, A.; Endo, Y. Design and synthesis of carborane-containing estrogen receptor-beta (ERb)-selective ligands. Bioorg. Med. Chem. Lett. 2015, 25, 4174. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43. [Google Scholar] [CrossRef]
Sample Availability: Samples of all the compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ring | X | R | RBA (%) a | EC50 (μM) b | Emax (%) c | IC50 (μM) b | CLogP d |
|---|---|---|---|---|---|---|---|---|
| 4a | 5 | S | H | 1.2 | 0.39 | 119 | – | 3.1 |
| 4b | SO | H | 0.003 | NT e | NT e | NT e | 0.8 | |
| 4c | SO2 | H | 0.04 | 0.30 | 130 | – | 0.7 | |
| 4d | S | (CH2)3N(CH3)2 | 4.0 | – | – | 0.41 | 4.2 | |
| 5a | 7 | S | H | 4.9 | 0.048 | 99 | – | 3.9 |
| 5b | SO | H | 0.001 | NT f | NT f | NT f | 1.6 | |
| 5c | SO2 | H | 0.01 | 4.7 | 89 | – | 1.5 | |
| 5d | S | (CH2)3N(CH3)2 | 37.1 | – | – | 0.090 | 4.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohta, K.; Kaise, A.; Taguchi, F.; Aoto, S.; Ogawa, T.; Endo, Y. Design and Synthesis of Novel Breast Cancer Therapeutic Drug Candidates Based upon the Hydrophobic Feedback Approach of Antiestrogens. Molecules 2019, 24, 3966. https://doi.org/10.3390/molecules24213966
Ohta K, Kaise A, Taguchi F, Aoto S, Ogawa T, Endo Y. Design and Synthesis of Novel Breast Cancer Therapeutic Drug Candidates Based upon the Hydrophobic Feedback Approach of Antiestrogens. Molecules. 2019; 24(21):3966. https://doi.org/10.3390/molecules24213966
Chicago/Turabian StyleOhta, Kiminori, Asako Kaise, Fumi Taguchi, Sayaka Aoto, Takumi Ogawa, and Yasuyuki Endo. 2019. "Design and Synthesis of Novel Breast Cancer Therapeutic Drug Candidates Based upon the Hydrophobic Feedback Approach of Antiestrogens" Molecules 24, no. 21: 3966. https://doi.org/10.3390/molecules24213966
APA StyleOhta, K., Kaise, A., Taguchi, F., Aoto, S., Ogawa, T., & Endo, Y. (2019). Design and Synthesis of Novel Breast Cancer Therapeutic Drug Candidates Based upon the Hydrophobic Feedback Approach of Antiestrogens. Molecules, 24(21), 3966. https://doi.org/10.3390/molecules24213966