Thiyl Radical Reactions in the Chemical Degradation of Pharmaceutical Proteins


Abstract
:
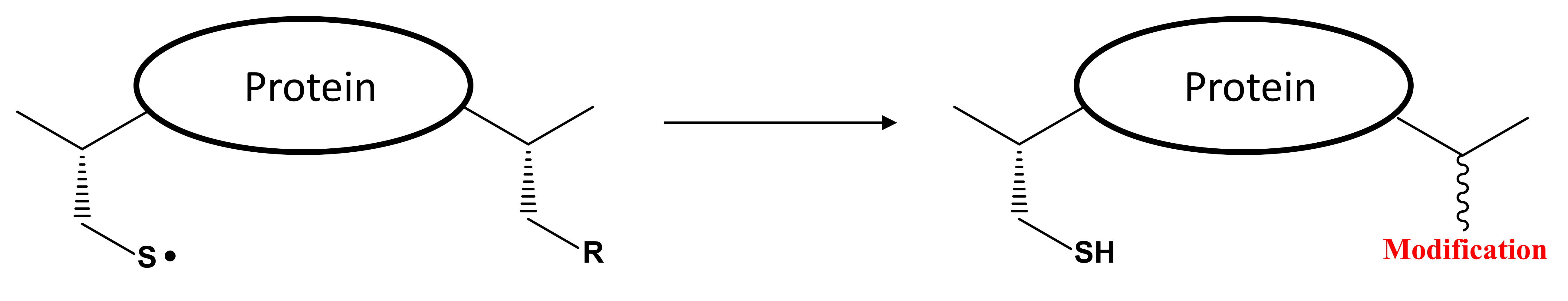{kind=link}
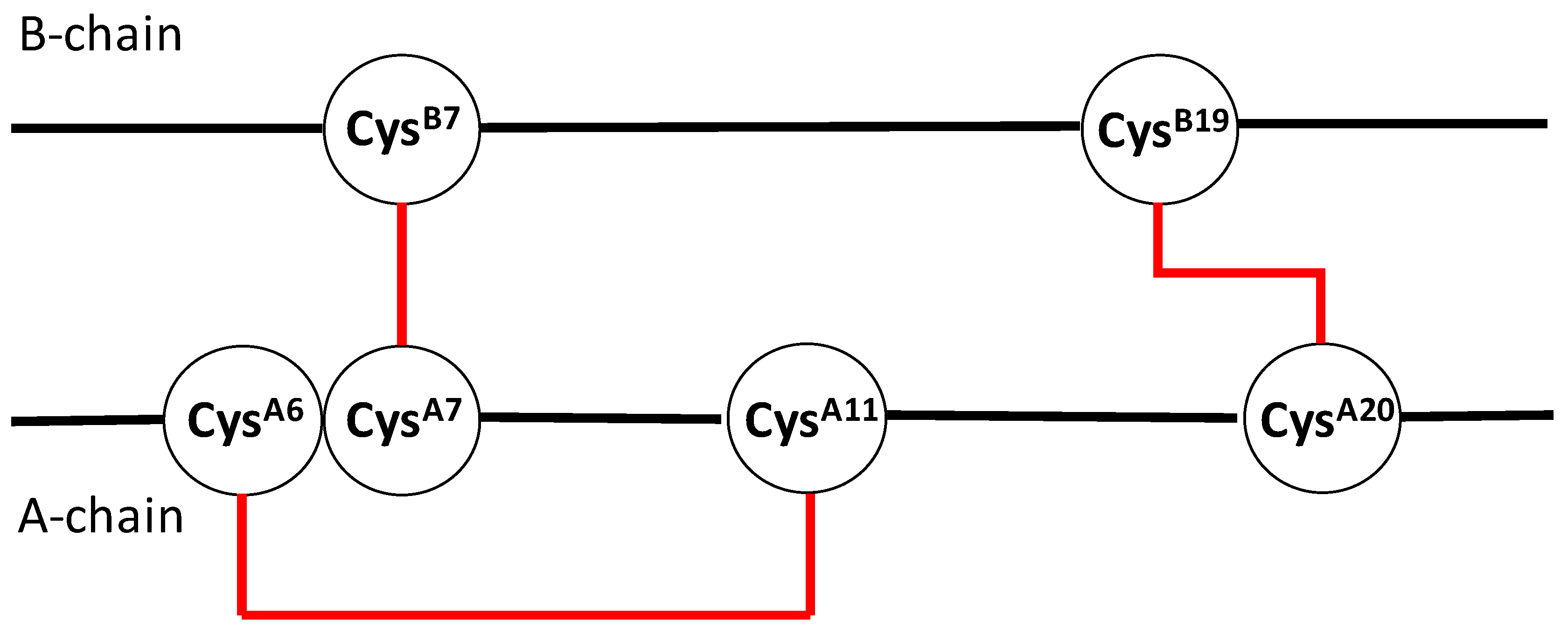{kind=link}
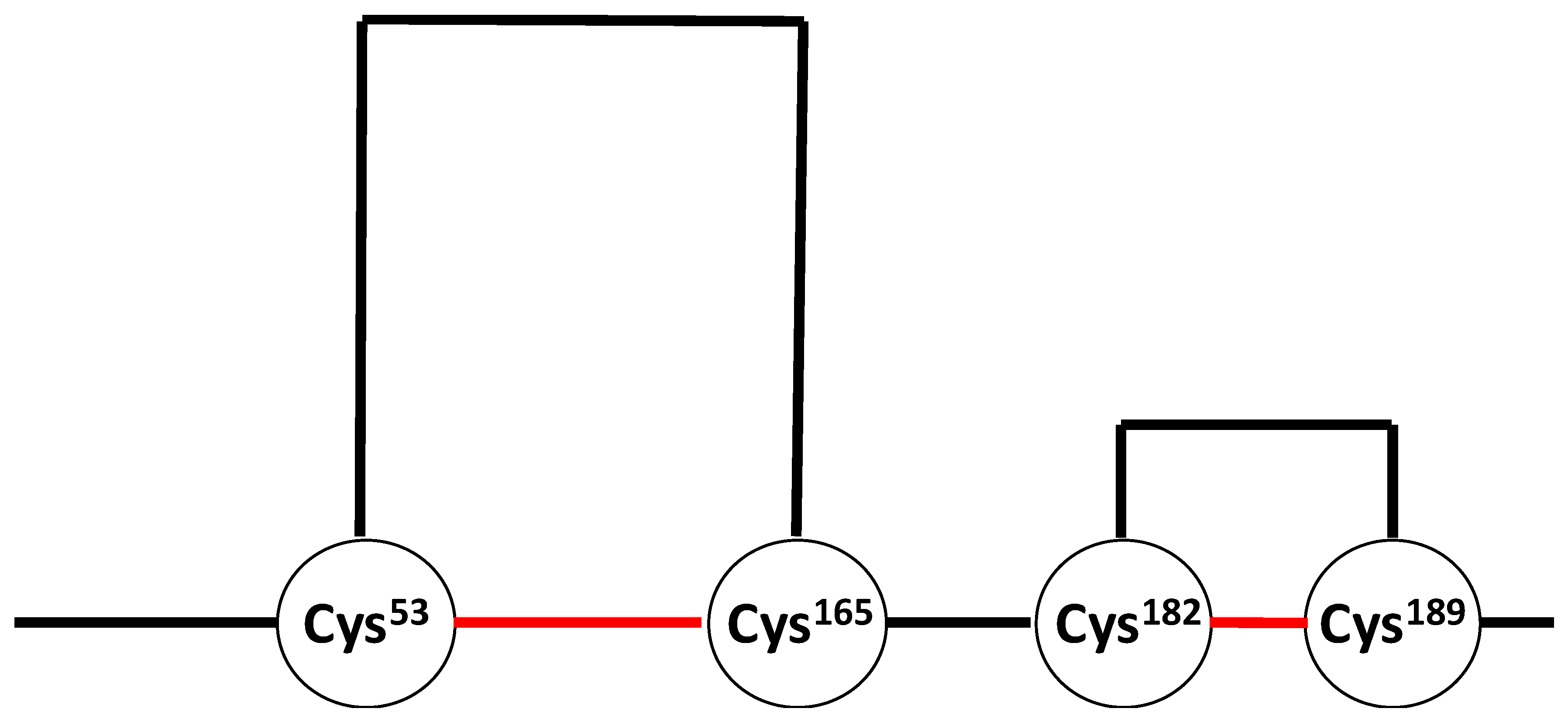{kind=link}
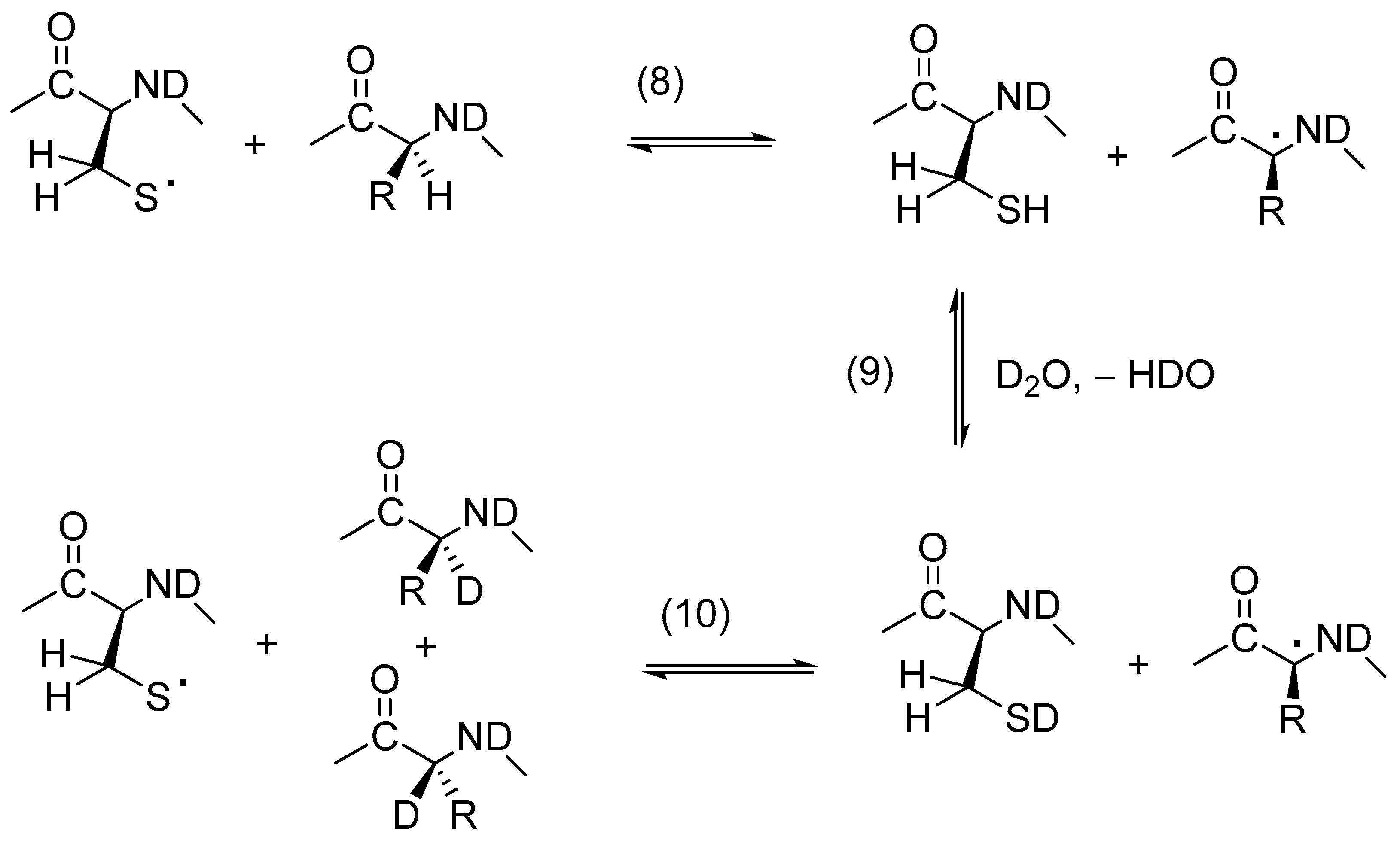{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
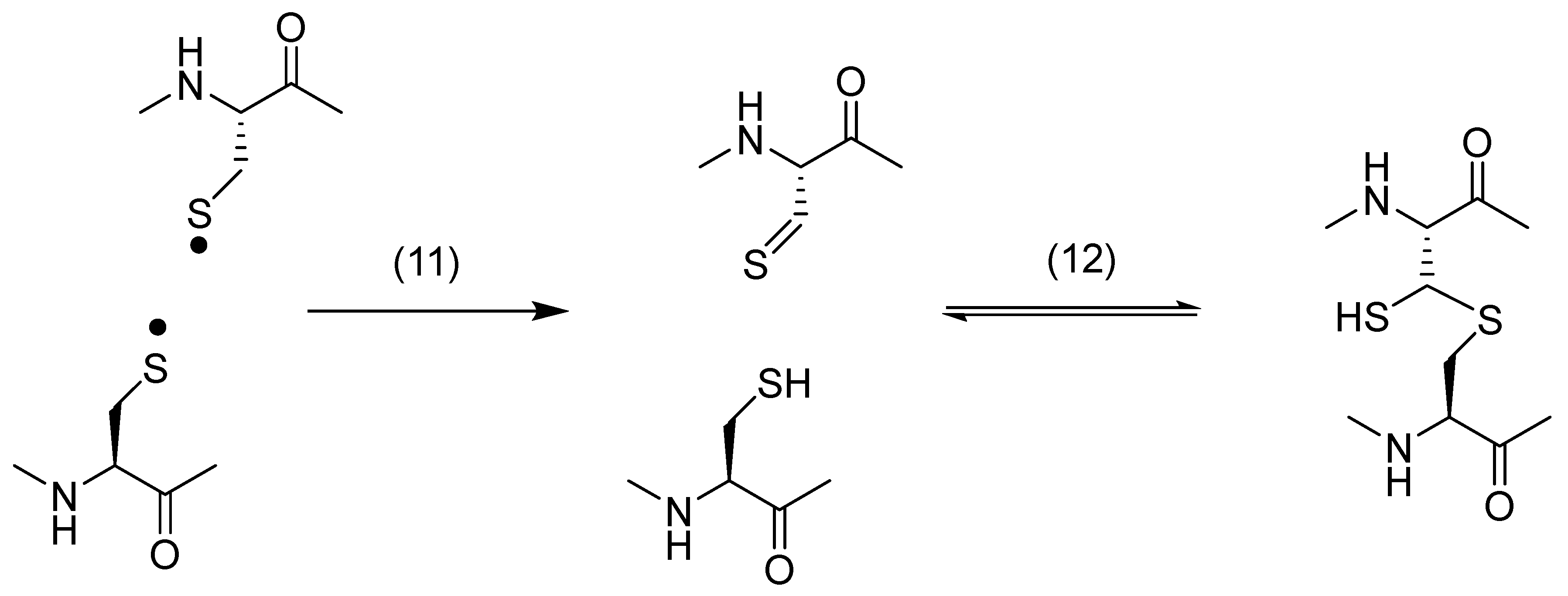
2. Thiyl Radicals in Reversible HAT Reactions




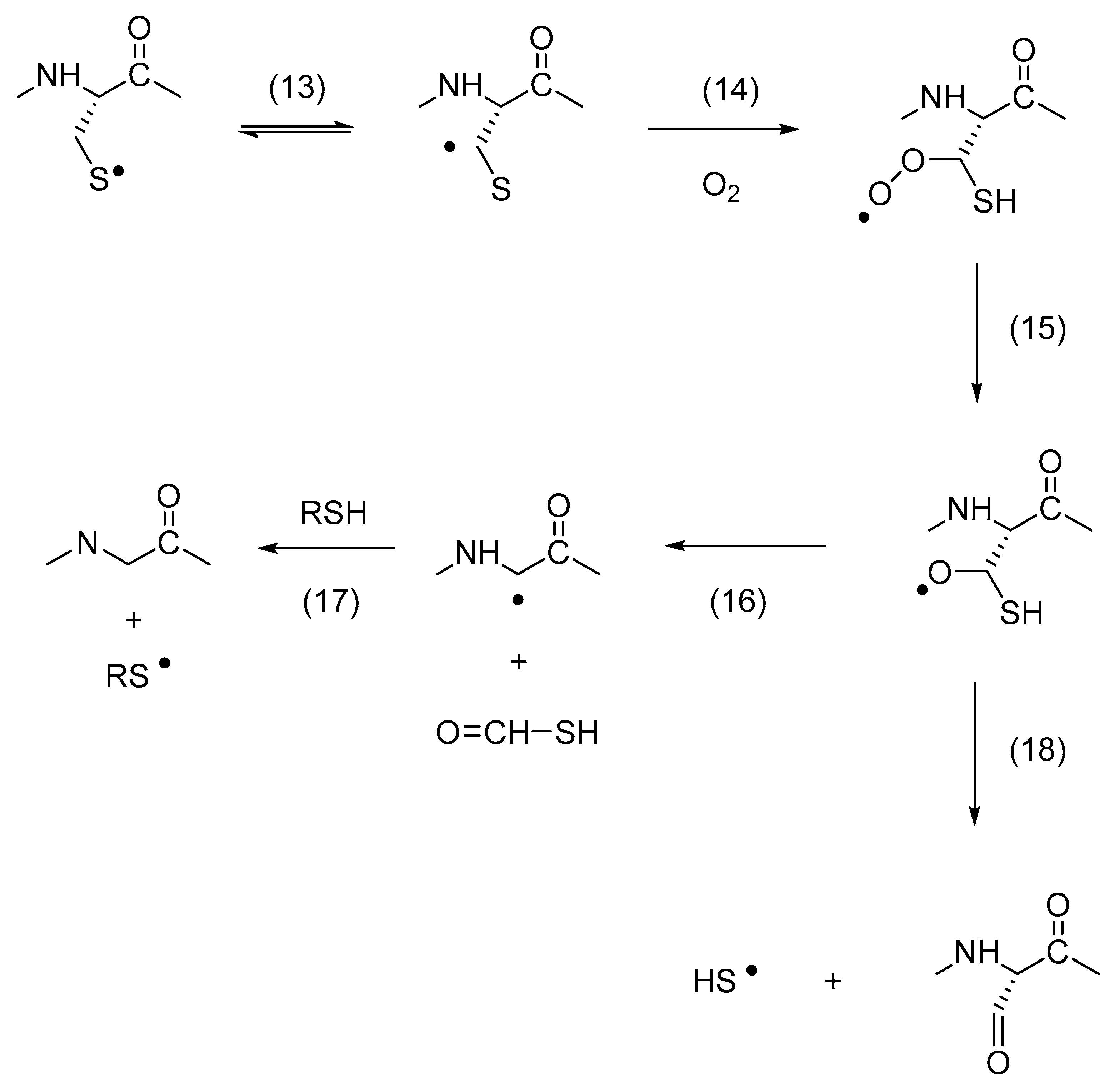
3. Thiyl Radical Reactions with Molecular Oxygen
4. Insulin
4.1. HAT Reactions in Solution
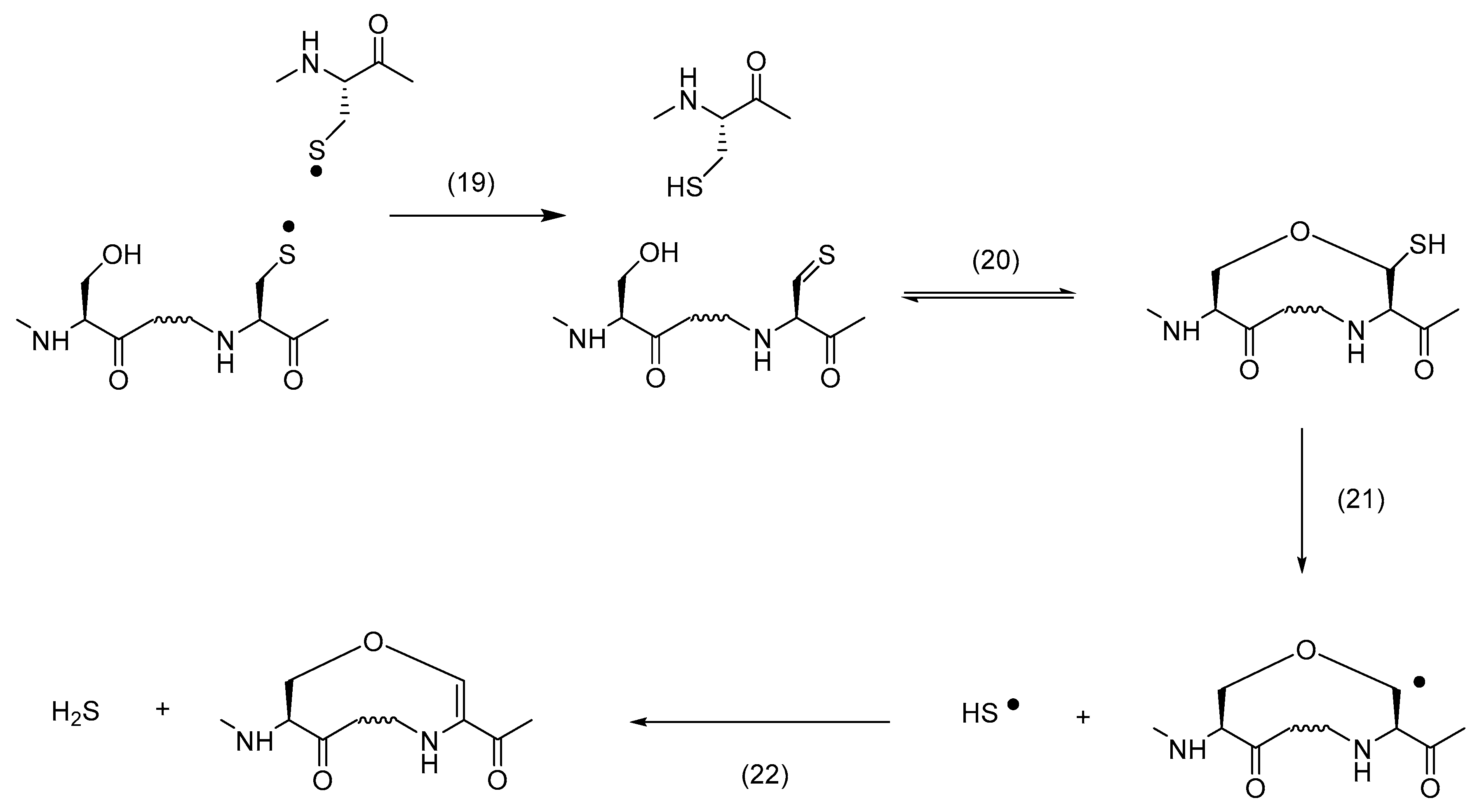
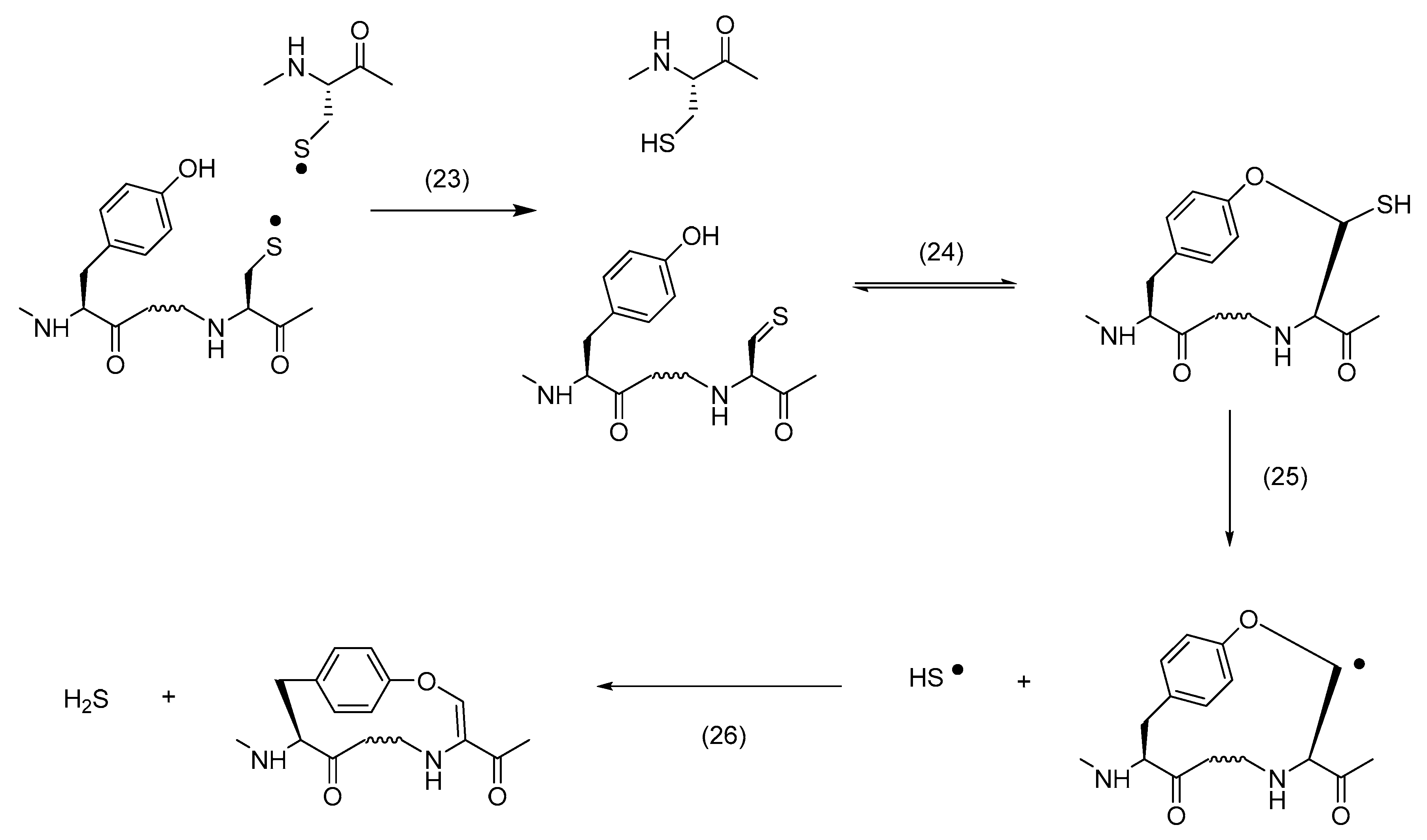
4.2. Additional Reactions of Thiyl Radicals Leading to Cross-Links in Solution
4.3. Thiyl Radical Reactions in Solids
5. Growth Hormone
5.1. The Conversion of Cys to Gly
5.2. The Formation of Ether and Vinyl ether
5.3. Formation of Non-Native Disulfides
5.4. hGH Cleavage Products
6. Monoclonal Antibodies
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Manning, M.C.; Chou, D.K.; Murphy, B.M.; Payne, R.W.; Katayama, D.S. Stability of Protein Pharmaceuticals: An Update. Pharm. Res. 2010, 27, 544–575. [Google Scholar] [CrossRef] [PubMed]
- Grassi, L.; Cabrele, C. Susceptibility of Protein Therapeutics to Spontaneous Chemical Modifications by Oxidation, Cyclization, and Elimination Reactions. Amino Acids 2019, 51, 1409–1431. [Google Scholar] [CrossRef] [PubMed]
- Harmon, P.A.; Kosuda, K.; Nelson, E.; Mowery, M.; Reed, R.A. A Novel Peroxy Radical Based Oxidative Stressing System for Ranking the Oxidizability of Drug Substances. J. Pharm. Sci. 2006, 95, 2014–2028. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, D.; Ji, J.A.; Wang, Y.J.; Schöneich, C. Oxidation of Human Growth Hormone by Oxygen-Centered Radicals: Formation of Leu-101 Hydroperoxide and Tyr-103 Oxidation Products. Mol. Pharm. 2012, 9, 803–814. [Google Scholar] [CrossRef]
- Escobar-Alvarez, E.; Leinisch, F.; Araya, G.; Monasterio, O.; Lorentzen, L.G.; Silva, E.; Davies, M.J.; Lopez-Alarcon, C. The Peroxyl Radical-Induced Oxidation of Escherichia Coli FtsZ and Its Single Tryptophan Mutant (Y222W) Modifies Specific Side-Chains, Generates Protein Cross-Links and Affects Biological Function. Free Radic. Biol. Med. 2017, 112, 60–68. [Google Scholar] [CrossRef]
- Leinisch, F.; Mariotti, M.; Rykaer, M.; Lopez-Alarcon, C.; Hagglund, P.; Davies, M.J. Peroxyl Radical-And Photo-Oxidation of Glucose 6-Phosphate Dehydrogenase Generates Cross-Links and Functional Changes Via Oxidation of Tyrosine and Tryptophan Residues. Free Radic. Biol. Med. 2017, 112, 240–252. [Google Scholar] [CrossRef]
- Randolph, T.W.; Schiltz, E.; Sederstrom, D.; Steinmann, D.; Mozziconacci, O.; Schöneich, C.; Freund, E.; Ricci, M.S.; Carpenter, J.F.; Lengsfeld, C.S. Do Not Drop: Mechanical Shock in Vials Causes Cavitation, Protein Aggregation, and Particle Formation. J. Pharm. Sci. 2015, 104, 602–611. [Google Scholar] [CrossRef]
- Sreedhara, A.; Lau, K.; Li, C.; Hosken, B.; Macchi, F.; Zhan, D.; Shen, A.; Steinmann, D.; Schöneich, C.; Lentz, Y. Role of Surface Exposed Tryptophan as Substrate Generators for the Antibody Catalyzed Water Oxidation Pathway. Mol. Pharm. 2013, 10, 278–288. [Google Scholar] [CrossRef]
- Haywood, J.; Mozziconacci, O.; Allegre, K.M.; Kerwin, B.A.; Schöneich, C. Light-Induced Conversion of Trp to Gly and Gly Hydroperoxide in IgG1. Mol. Pharm. 2013, 10, 1146–1150. [Google Scholar] [CrossRef]
- Bane, J.; Mozziconacci, O.; Yi, L.; Wang, Y.J.; Sreedhara, A.; Schöneich, C. Photo-Oxidation of IgG1 and Model Peptides: Detection and Analysis of Triply Oxidized His and Trp Side Chain Cleavage Products. Pharm. Res. 2017, 34, 229–242. [Google Scholar] [CrossRef]
- Wecksler, A.T.; Yin, J.; Lee Tao, P.; Kabakoff, B.; Sreedhara, A.; Deperalta, G. Photodisruption of the Structurally Conserved Cys-Cys-Trp Triads Leads to Reduction-Resistant Scrambled Intrachain Disulfides in an IgG1 Monoclonal Antibody. Mol. Pharm. 2018, 15, 1598–1606. [Google Scholar] [CrossRef] [PubMed]
- Mohr, D.; Wolff, M.; Kissel, T. Gamma Irradiation for Terminal Sterilization of 17beta-Estradiol Loaded Poly-(D,L-Lactide-Co-Glycolide) Microparticles. J. Control. Release 1999, 61, 203–217. [Google Scholar] [CrossRef]
- Creed, D. The Photophysics and Photochemistry of the Near-Uv Absorbing Amino-Acids. 3. Cystine and Its Simple Derivatives. Photochem. Photobiol. 1984, 39, 577–583. [Google Scholar] [CrossRef]
- Creed, D. The Photophysics and Photochemistry of the Near-Uv Absorbing Amino-Acids. 1. Tryptophan and Its Simple Derivatives. Photochem. Photobiol. 1984, 39, 537–562. [Google Scholar] [CrossRef]
- Dose, K. Photolysis of Free Cystine in Presence of Aromatic Amino Acids. Photochem. Photobiol. 1968, 8, 331. [Google Scholar] [CrossRef]
- Lorenz, C.M.; Wolk, B.M.; Quan, C.P.; Alcala, E.W.; Eng, M.; McDonald, D.J.; Matthews, T.C. The Effect of Low Intensity Ultraviolet-C Light on Monoclonal Antibodies. Biotechnol. Prog. 2009, 25, 476–482. [Google Scholar] [CrossRef]
- Mossoba, M.M.; Makino, K.; Riesz, P. Photo-Ionization of Aromatic-Amino-Acids in Aqueous-Solutions-A Spin-Trapping and Electron-Spin Resonance Study. J. Phys. Chem. 1982, 86, 3478–3483. [Google Scholar] [CrossRef]
- Truong, T.B. Charge-Transfer to a Solvent State. 5. Effect of Solute-Solvent Interaction on the Ionization-Potential of the Solute-Mechanism for Photo-Ionization. J. Phys. Chem. 1980, 84, 964–970. [Google Scholar] [CrossRef]
- Nauser, T.; Schöneich, C. Thiyl Radicals Abstract Hydrogen Atoms from the (alpha)C-H Bonds in Model Peptides: Absolute Rate Constants and Effect of Amino Acid Structure. J. Am. Chem. Soc. 2003, 125, 2042–2043. [Google Scholar] [CrossRef]
- Breslow, R. Biomimetic Chemistry: Biology as An Inspiration. J. Biol. Chem. 2009, 284, 1337–1342. [Google Scholar] [CrossRef]
- Schöneich, C.; Asmus, K.D.; Dillinger, U.; Von Bruchhausen, F. Thiyl Radical Attack on Polyunsaturated Fatty Acids: A Possible Route to Lipid Peroxidation. Biochem. Biophys. Res. Commun. 1989, 161, 113–120. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Ferreri, C.; Guerra, M.; Samadi, A.; Bowry, V.W. The Reaction of Thiyl Radical with Methyl Linoleate: Completing the Picture. J. Am. Chem. Soc. 2017, 139, 4704–4714. [Google Scholar] [CrossRef] [PubMed]
- Chatgilialoglu, C.; Ferreri, C.; Melchiorre, M.; Sansone, A.; Torreggiani, A. Lipid Geometrical Isomerism: From Chemistry to Biology and Diagnostics. Chem. Rev. 2014, 114, 255–284. [Google Scholar] [CrossRef] [PubMed]
- Ferreri, C.; Costantino, C.; Landi, L.; Mulazzani, Q.G.; Chatgilialoglu, C. The Thiyl Radical-Mediated Isomerization of Cis-Monounsaturated Fatty Acid Residues in Phospholipids: A Novel Path of Membrane Damage? Chem. Commun. 1999, 5, 407–408. [Google Scholar] [CrossRef]
- Lykakis, I.N.; Ferreri, C.; Chatgilialoglu, C. The Sulfhydryl Radical (HS Center dot/S Center Dot-): A Contender for the Isomerization of Double Bonds in Membrane Lipids. Angew. Chem. Int. Edit. 2007, 46, 1914–1916. [Google Scholar] [CrossRef]
- Ferreri, C.; Kratzsch, S.; Brede, O.; Marciniak, B.; Chatgilialoglu, C. Trans Lipid Formation Induced by Thiols in Human Monocytic Leukemia Cells. Free Radical. Biol. Med. 2005, 38, 1180–1187. [Google Scholar] [CrossRef]
- Koppaka, V.; Axelsen, P.H. Accelerated Accumulation of Amyloid Beta Proteins on Oxidatively Damaged Lipid Membranes. Biochemistry 2000, 39, 10011–10016. [Google Scholar] [CrossRef]
- Musiek, E.S.; Holtzman, D.M. Three Dimensions of the Amyloid Hypothesis: Time, Space and ‘Wingmen’. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef]
- Gesslbauer, B.; Kuerzl, D.; Valpatic, N.; Bochkov, V.N. Unbiased Identification of Proteins Covalently Modified by Complex Mixtures of Peroxidized Lipids Using a Combination of Electrophoretic Mobility Band Shift with Mass Spectrometry. Antioxidants 2018, 7, 116. [Google Scholar] [CrossRef]
- Grimm, M.O.; Rothhaar, T.L.; Grosgen, S.; Burg, V.K.; Hundsdorfer, B.; Haupenthal, V.J.; Friess, P.; Kins, S.; Grimm, H.S.; Hartmann, T. Trans Fatty Acids Enhance Amyloidogenic Processing of the Alzheimer Amyloid Precursor Protein (APP). J. Nutr. Biochem. 2012, 23, 1214–1223. [Google Scholar] [CrossRef]
- Walling, C.; Rabinowitz, R. The Photolysis of Isobutyl Disulfide in Cumene. J. Am. Chem. Soc. 1959, 81, 1137–1143. [Google Scholar] [CrossRef]
- Pryor, W.A.; Gojon, G.; Church, D.F. Relative Rate Constants for Hydrogen-Atom Abstraction by Cyclohexanethiyl and Benzenethiyl Radicals. J. Org. Chem. 1978, 43, 793–800. [Google Scholar] [CrossRef]
- Pryor, W.A.; Gojon, G.; Stanley, J.P. Hydrogen Abstraction by Thiyl Radicals. J. Am. Chem. Soc. 1973, 95, 945–946. [Google Scholar] [CrossRef]
- Akhlaq, M.S.; Schuchmann, H.P.; Von Sonntag, C. The Reverse of the ‘Repair’ Reaction of Thiols: H-Abstraction at Carbon by Thiyl Radicals. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1987, 51, 91–102. [Google Scholar] [CrossRef]
- Schöneich, C.; Bonifacic, M.; Asmus, K.D. Reversible H-Atom Abstraction from Alcohols by Thiyl Radicals-Determination of Absolute Rate Constants by Pulse-Radiolysis. Free Radical. Res. Com. 1989, 6, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Schöneich, C.; Asmus, K.D.; Bonifacic, M. Determination of Absolute Rate Constants for the Reversible Hydrogen-Atom Transfer Between Thiyl Radicals and Alcohols or Ethers. J. Chem. Soc. Faraday T. 1995, 91, 1923–1930. [Google Scholar] [CrossRef]
- Pogocki, D.; Schöneich, C. Thiyl Radicals Abstract Hydrogen Atoms from Carbohydrates: Reactivity and Selectivity. Free Radic. Biol. Med. 2001, 31, 98–107. [Google Scholar] [CrossRef]
- Dang, H.S.; Roberts, B.P.; Tocher, D.A. Selective Radical-Chain Epimerisation at Electron-Rich Chiral Tertiary C-H Centres Using Thiols as Protic Polarity-Reversal Catalysts. J. Chem. Soc. Perk. T. 2001, 1, 2452–2461. [Google Scholar] [CrossRef]
- Roberts, B.P. Polarity-Reversal Catalysis of Hydrogen-Atom Abstraction Reactions: Concepts and Applications in Organic Chemistry. Chem. Soc. Rev. 1999, 28, 25–35. [Google Scholar] [CrossRef]
- Backman, L.R.F.; Funk, M.A.; Dawson, C.D.; Drennan, C.L. New Tricks for the Glycyl Radical Enzyme Family. Crit. Rev. Biochem. Mol. 2017, 52, 674–695. [Google Scholar] [CrossRef] [Green Version]
- Parast, C.V.; Wong, K.K.; Lewisch, S.A.; Kozarich, J.W.; Peisach, J.; Magliozzo, R.S. Hydrogen-Exchange of the Glycyl Radical of Pyruvate Formate-Lyase Is Catalyzed by Cysteine-419. Biochemistry 1995, 34, 2393–2399. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.D.; Himo, F. Catalytic Mechanism of Pyruvate-Formate Lyase Revisited. J. Phys. Chem. B 2004, 108, 15347–15354. [Google Scholar] [CrossRef]
- Rauk, A.; Yu, D.; Armstrong, D.A. Oxidative Damage to and by Cysteine in Proteins: An ab Initio Study of the Radical Structures, C-H, S-H, and C-C Bond Dissociation Energies, and Transition Structures for H Abstraction by Thiyl Radicals. J. Am. Chem. Soc. 1998, 120, 8848–8855. [Google Scholar] [CrossRef]
- Nauser, T.; Casi, G.; Koppenol, W.H.; Schöneich, C. Reversible Intramolecular Hydrogen Transfer Between Cysteine Thiyl Radicals and Glycine and Alanine in Model Peptides: Absolute Rate Constants Derived from Pulse Radiolysis and Laser Flash Photolysis. J. Phys. Chem. B 2008, 112, 15034–15044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauser, T.; Koppenol, W.H.; Schöneich, C. Reversible Hydrogen Transfer Reactions in Thiyl Radicals From Cysteine and Related Molecules: Absolute Kinetics and Equilibrium Constants Determined by Pulse Radiolysis. J. Phys. Chem. B 2012, 116, 5329–5341. [Google Scholar] [CrossRef] [Green Version]
- Lesslie, M.; Lau, J.K.C.; Lawler, J.T.; Siu, K.W.M.; Oomens, J.; Berden, G.; Hopkinson, A.C.; Ryzhov, V. Alkali-Metal-Ion-Assisted Hydrogen Atom Transfer in the Homocysteine Radical. Chem. Eur. J. 2016, 22, 2243–2246. [Google Scholar] [CrossRef]
- Lesslie, M.; Lau, J.K.C.; Pacheco, G.; Lawler, J.T.; Siu, K.W.M.; Hopkinson, A.C.; Ryzhov, V. Hydrogen Atom Transfer in Metal Ion Complexes of the Glutathione Thiyl Radical. Int. J. Mass. Spectrom. 2018, 429, 39–46. [Google Scholar] [CrossRef]
- Hofstetter, D.; Thalmann, B.; Nauser, T.; Koppenol, W.H. Hydrogen Exchange Equilibria in Thiols. Chem. Res. Toxicol. 2012, 25, 1862–1867. [Google Scholar] [CrossRef]
- Mozziconacci, O.; Williams, T.D.; Schöneich, C. Intramolecular Hydrogen Transfer Reactions of Thiyl Radicals from Glutathione: Formation of Carbon-Centered Radical at Glu, Cys, and Gly. Chem. Res. Toxicol. 2012, 25, 1842–1861. [Google Scholar] [CrossRef]
- Reid, D.L.; Armstrong, D.A.; Rauk, A.; Von Sonntag, C. H-Atom Abstraction by Thiyl Radicals from Peptides and Cyclic Dipeptides. A Theoretical Study of Reaction Rates. Phys. Chem. Chem. Phys. 2003, 5, 3994–3999. [Google Scholar] [CrossRef]
- Hao, G.; Gross, S.S. Electrospray Tandem Mass Spectrometry Analysis of S-and N-Nitrosopeptides: Facile Loss of NO and Radical-Induced Fragmentation. J. Am. Soc. Mass. Spectr. 2006, 17, 1725–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauser, T.; Pelling, J.; Schoneich, C. Thiyl Radical Reaction with Amino Acid Side Chains: Rate Constants for Hydrogen Transfer and Relevance for Posttranslational Protein Modification. Chem. Res. Toxicol. 2004, 17, 1323–1328. [Google Scholar] [CrossRef] [PubMed]
- Schöneich, C.; Mozziconacci, O.; Koppenol, W.H.; Nauser, T. Intramolecular 1,2-and 1,3-Hydrogen Transfer Reactions of Thiyl Radicals. Isr. J. Chem. 2014, 54, 265–271. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, N.; Schuchmann, H.P.; Von Sonntag, C. Pulse Radiolysis of 2-Mercaptoethanol in Oxygenated Aqueous Solution. Generation and Reactions of the Thiylperoxyl Radical. J. Phys. Chem. 1994, 98, 6541–6547. [Google Scholar] [CrossRef]
- Tamba, M.; Dajka, K.; Ferreri, C.; Asmus, K.D.; Chatgilialoglu, C. One-Electron Reduction of Methanesulfonyl Chloride. The Fate of MeSO2Cl•− and MeSO2• Intermediates in Oxygenated Solutions and Their Role in the Cis-Trans Isomerization of Mono-Unsaturated Fatty Acids. J. Am. Chem. Soc. 2007, 129, 8716–8723. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.; Swarts, S.; Champagne, M.; Sevilla, M.D. An ESR Investigation of the Reactions of Glutathione, Cysteine and Penicillamine Thiyl Radicals: Competitive Formation of RSO. R. RSSR, and RSS. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1988, 53, 767–786. [Google Scholar] [CrossRef]
- Sevilla, M.D.; Yan, M.Y.; Becker, D. Thiol Peroxyl Radical Formation from the Reaction of Cysteine Thiyl Radical with Molecular Oxygen: An ESR Investigation. Biochem. Biophys. Res. Commun. 1988, 155, 405–410. [Google Scholar] [CrossRef]
- Brange, J.; Langkjoer, L. Insulin Structure and Stability. In Stability and Characterization of Protein and Peptide Drugs; Wang, Y.J., Pearlman, R., Eds.; Plenum Press: New York, NJ, USA, 1993; pp. 315–350. [Google Scholar]
- Mozziconacci, O.; Williams, T.D.; Kerwin, B.A.; Schöneich, C. Reversible Intramolecular Hydrogen Transfer Between Protein Cysteine Thiyl Radicals and Alpha C-H Bonds in Insulin: Control of Selectivity by Secondary Structure. J. Phys. Chem. B 2008, 112, 15921–15932. [Google Scholar] [CrossRef]
- Correia, M.; Neves-Petersen, M.T.; Jeppesen, P.B.; Gregersen, S.; Petersen, S.B. UV-Light Exposure of Insulin: Pharmaceutical Implications Upon Covalent Insulin Dityrosine Dimerization and Disulphide Bond Photolysis. PLoS ONE 2012, 7, e50733. [Google Scholar] [CrossRef] [Green Version]
- Darrington, R.T.; Anderson, B.D. Effects of Insulin Concentration and Self-Association on the Partitioning of its A-21 Cyclic Anhydride Intermediate to Desamido Insulin and Covalent Dimer. Pharm. Res. 1995, 12, 1077–1084. [Google Scholar] [CrossRef]
- Darrington, R.T.; Anderson, B.D. Evidence for a Common Intermediate in Insulin Deamidation and Covalent Dimer Formation: Effects of pH and Aniline Trapping in Dilute Acidic Solutions. J. Pharm. Sci. 1995, 84, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.I.; Sievers, S.A.; Sawaya, M.R.; Wall, J.S.; Eisenberg, D. Molecular Basis for Insulin Fibril Assembly. Proc. Natl. Acad. Sci. USA 2009, 106, 18990–18995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanova, M.I.; Thompson, M.J.; Eisenberg, D. A Systematic Screen of Beta(2)-Microglobulin And Insulin for Amyloid-Like Segments. Proc. Natl. Acad. Sci. USA 2006, 103, 4079–4082. [Google Scholar] [CrossRef] [Green Version]
- Brange, J.; Ribel, U.; Hansen, J.F.; Dodson, G.; Hansen, M.T.; Havelund, S.; Melberg, S.G.; Norris, F.; Norris, K.; Snel, L.; et al. Monomeric Insulins Obtained by Protein Engineering and Their Medical Implications. Nature 1988, 333, 679–682. [Google Scholar] [CrossRef]
- Weiss, M.A.; Lawrence, M.C. A Thing of Beauty: Structure and Function of Insulin’s “Aromatic Triplet”. Diabetes Obes. Metab. 2018, 20, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Ohgaku, S.; Iwasaki, M.; Maegawa, H.; Shigeta, Y.; Inouye, K. Supernormal Insulin: [D-PheB24]-Insulin with Increased Affinity for Insulin Receptors. Biochem. Biophys. Res. Commun. 1982, 107, 329–336. [Google Scholar] [CrossRef]
- Wei, Y.; Mathies, G.; Yokoyama, K.; Chen, J.; Griffin, R.G.; Stubbe, J. A Chemically Competent Thiosulfuranyl Radical on the Escherichia Coli Class III Ribonucleotide Reductase. J. Am. Chem. Soc. 2014, 136, 9001–9013. [Google Scholar] [CrossRef] [Green Version]
- Nauser, T.; Carreras, A. Carbon-Centered Radicals Add Reversibly to Histidine-Implications. Chem. Commun. 2014, 50, 14349–14351. [Google Scholar] [CrossRef] [Green Version]
- Santschi, N.; Nauser, T. An Experimental Radical Electrophilicity Index. Chemphyschem 2017, 18, 2973–2976. [Google Scholar] [CrossRef]
- Mozziconacci, O.; Haywood, J.; Gorman, E.M.; Munson, E.; Schöneich, C. Photolysis of Recombinant Human Insulin in the Solid State: Formation of a Dithiohemiacetal Product at the C-Terminal Disulfide Bond. Pharm. Res. 2012, 29, 121–133. [Google Scholar] [CrossRef]
- De Vos, A.M.; Ultsch, M.; Kossiakoff, A.A. Human Growth Hormone and Extracellular Domain of its Receptor: Crystal Structure of the Complex. Science 1992, 255, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Pearlman, R.; Bewley, T.A. Stability and Characterization of Human Growth Hormone. In Stability and Characterization of Protein and Peptide Drugs; Wang, Y.J., Pearlman, R., Eds.; Plenum Press: New York, NJ, USA, 1993; pp. 1–58. [Google Scholar]
- Williams, B.R.; Cho, J.S. Hormone Replacement: The Fountain of Youth? Prim. Care 2017, 44, 481–498. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wu, S.L.; Karger, B.L.; Hancock, W.S. Mass Spectrometric Analysis of Innovator, Counterfeit, and Follow-On Recombinant Human Growth Hormone. Biotechnol. Prog. 2009, 25, 207–218. [Google Scholar] [CrossRef]
- Karlsson, G.; Eriksson, K.; Persson, A.; Mansson, H.; Soderholm, S. The Separation of Recombinant Human Growth Hormone Variants by UHPLC. J. Chromatogr. Sci. 2013, 51, 943–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battersby, J.E.; Hancock, W.S.; Canova-Davis, E.; Oeswein, J.; O’Connor, B. Diketopiperazine Formation and N-Terminal Degradation in Recombinant Human Growth Hormone. Int. J. Pept. Protein. Res. 1994, 44, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Ghezzo-Schöneich, E.; Aced, G.I.; Hong, J.; Milby, T.; Schöneich, C. Metal-Catalyzed Oxidation of Histidine in Human Growth Hormone. Mechanism, Isotope Effects, and Inhibition by a Mild Denaturing Alcohol. J. Biol. Chem. 1997, 272, 9019–9029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovorka, S.W.; Hong, J.; Cleland, J.L.; Schöneich, C. Metal-Catalyzed Oxidation of Human Growth Hormone: Modulation by Solvent-Induced Changes of Protein Conformation. J. Pharm. Sci. 2001, 90, 58–69. [Google Scholar] [CrossRef]
- Mulinacci, F.; Capelle, M.A.; Gurny, R.; Drake, A.F.; Arvinte, T. Stability of Human Growth Hormone: Influence of Methionine Oxidation on Thermal Folding. J. Pharm. Sci. 2011, 100, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Mulinacci, F.; Poirier, E.; Capelle, M.A.; Gurny, R.; Arvinte, T. Influence of Methionine Oxidation on the Aggregation of Recombinant Human Growth Hormone. Eur. J. Pharm. Biopharm. 2013, 85, 42–52. [Google Scholar] [CrossRef]
- Chang, S.H.; Teshima, G.M.; Milby, T.; Gillece-Castro, B.; Canova-Davis, E. Metal-Catalyzed Photooxidation of Histidine in Human Growth Hormone. Anal. Biochem. 1997, 244, 221–227. [Google Scholar] [CrossRef]
- Steinmann, D.; Ji, J.A.; Wang, Y.J.; Schöneich, C. Photodegradation of Human Growth Hormone: A Novel Backbone Cleavage Between Glu-88 and Pro-89. Mol. Pharm. 2013, 10, 2693–2706. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, D.; Mozziconacci, O.; Bommana, R.; Stobaugh, J.F.; Wang, Y.J.; Schöneich, C. Photodegradation Pathways of Protein Disulfides: Human Growth Hormone. Pharm. Res. 2017, 34, 2756–2778. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, A.M.; Christensen, T.; Klausen, N.K.; Nielsen, F.; Sorensen, H.H. Characterisation of a Trisulphide Derivative of Biosynthetic Human Growth Hormone Produced in Escherichia Coli. Eur. J. Biochem. 1994, 219, 365–373. [Google Scholar] [CrossRef]
- Andersson, C.; Edlund, P.O.; Gellerfors, P.; Hansson, Y.; Holmberg, E.; Hult, C.; Johansson, S.; Kordel, J.; Lundin, R.; Mendel-Hartvig, I.B.; et al. Isolation and Characterization of a Trisulfide Variant of Recombinant Human Growth Hormone Formed During Expression in Escherichia Coli. Int. J. Pept. Protein. Res. 1996, 47, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Canova-Davis, E.; Baldonado, I.P.; Chloupek, R.C.; Ling, V.T.; Gehant, R.; Olson, K.; Gillece-Castro, B.L. Confirmation by Mass Spectrometry of a Trisulfide Variant in Methionyl Human Growth Hormone Biosynthesized in Escherichia Coli. Anal. Chem. 1996, 68, 4044–4051. [Google Scholar] [CrossRef]
- Datola, A.; Richert, S.; Bierau, H.; Agugiaro, D.; Izzo, A.; Rossi, M.; Cregut, D.; Diemer, H.; Schaeffer, C.; Van Dorsselaer, A.; et al. Characterisation of a Novel Growth Hormone Variant Comprising a Thioether Link Between Cys182 and Cys189. ChemMedChem 2007, 2, 1181–1189. [Google Scholar] [CrossRef]
- Junnila, R.K.; Wu, Z.; Strasburger, C.J. The Role of Human Growth Hormone’s C-Terminal Disulfide Bridge. Growth Horm. IGF Res. 2013, 23, 62–67. [Google Scholar] [CrossRef]
- Wang, Z.; Rejtar, T.; Zhou, Z.S.; Karger, B.L. Desulfurization of Cysteine-Containing Peptides Resulting from Sample Preparation for Protein Characterization by Mass Spectrometry. Rapid Commun. Mass Spectrom. 2010, 24, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Fradkin, A.H.; Mozziconacci, O.; Schöneich, C.; Carpenter, J.F.; Randolph, T.W. UV Photodegradation of Murine Growth Hormone: Chemical Analysis and Immunogenicity Consequences. Eur. J. Pharm. Biopharm. 2014, 87, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Asmus, K.D.; Hug, G.L.; Bobrowski, K.; Mulazzani, Q.G.; Marciniak, B. Transients in the Oxidative and H-Atom-Induced Degradation of 1,3,5-Trithiane. Time-Resolved Studies in Aqueous Solution. J. Phys. Chem. A 2006, 110, 9292–9300. [Google Scholar] [CrossRef]
- Hug, G.L.; Janeba-Bartoszewicz, E.; Filipiak, P.; Pedzinski, T.; Kozubek, H.; Marciniak, B. Evidence for Heterolytic Cleavage of C-S Bonds in the Photolysis of 1,3,5-Trithianes. Pol. J. Chem. 2008, 82, 883–892. [Google Scholar]
- Janeba-Bartoszewicz, E.; Hug, G.L.; Andrzejewska, E.; Marciniak, B. Photochemistry of 1,3,5-Trithianes in Solution-Steady-State and Laser Flash Photolysis Studies. J. Photoch. Photobio. A Chem. 2006, 177, 17–23. [Google Scholar] [CrossRef]
- Mozziconacci, O.; Kerwin, B.A.; Schöneich, C. Photolysis of An Intrachain Peptide Disulfide Bond: Primary and Secondary Processes, Formation of H2S, and Hydrogen Transfer Reactions. J. Phys. Chem. B 2010, 114, 3668–3688. [Google Scholar] [CrossRef] [PubMed]
- Strausberg, R.L.; Feingold, E.A.; Grouse, L.H.; Derge, J.G.; Klausner, R.D.; Collins, F.S.; Wagner, L.; Shenmen, C.M.; Schuler, G.D.; Altschul, S.F.; et al. Generation and Initial Analysis of More Than 15,000 Full-Length Human and Mouse cDNA Sequences. Proc. Natl. Acad. Sci. USA 2002, 99, 16899–16903. [Google Scholar] [PubMed] [Green Version]
- Mozziconacci, O.; Stobaugh, J.T.; Bommana, R.; Woods, J.; Franklin, E.; Jorgenson, J.W.; Forrest, M.L.; Schöneich, C.; Stobaugh, J.F. Profiling the Photochemical-Induced Degradation of Rat Growth Hormone with Extreme Ultra-pressure Chromatography-Mass Spectrometry Utilizing Meter-Long Microcapillary Columns Packed with Sub-2-mum Particles. Chromatographia 2017, 80, 1299–1318. [Google Scholar] [CrossRef] [PubMed]
- Mozziconacci, O.; Sharov, V.; Williams, T.D.; Kerwin, B.A.; Schöneich, C. Peptide Cysteine Thiyl Radicals Abstract Hydrogen Atoms from Surrounding Amino Acids: The Photolysis of a Cystine Containing Model Peptide. J. Phys. Chem. B. 2008, 112, 9250–9257. [Google Scholar] [CrossRef] [PubMed]
- Mozziconacci, O.; Kerwin, B.A.; Schöneich, C. Reversible Hydrogen Transfer Reactions of Cysteine Thiyl Radicals in Peptides: The Conversion of Cysteine into Dehydroalanine and Alanine, and of Alanine Into Dehydroalanine. J. Phys. Chem. B 2011, 115, 12287–12305. [Google Scholar] [CrossRef] [Green Version]
- Bonifacic, M.; Asmus, K.D. Adduct Formation and Absolute Rate Constants in the Displacement Reaction of Thiyl Radicals with Disulfides. J. Phys. Chem. 1984, 88, 6286–6290. [Google Scholar] [CrossRef]
- Mozziconacci, O.; Kerwin, B.A.; Schöneich, C. Exposure of a Monoclonal Antibody, IgG1, to UV-Light Leads to Protein Dithiohemiacetal and Thioether Cross-Links: A Role for Thiyl Radicals? Chem. Res. Toxicol. 2010, 23, 1310–1312. [Google Scholar] [CrossRef]
- Zhou, S.; Mozziconacci, O.; Kerwin, B.A.; Schöneich, C. The Photolysis of Disulfide Bonds in IgG1 and IgG2 Leads to Selective Intramolecular Hydrogen Transfer Reactions of Cysteine Thiyl Radicals, Probed by Covalent H/D Exchange and RPLC-MS/MS Analysis. Pharm. Res. 2013, 30, 1291–1299. [Google Scholar] [CrossRef]
- Bommana, R.; Subelzu, N.; Mozziconacci, O.; Sreedhara, A.; Schöneich, C. Identification of D-Amino Acids in Light Exposed mAb Formulations. Pharm. Res. 2018, 35, 238. [Google Scholar] [CrossRef] [PubMed]
- Mozziconacci, O.; Kerwin, B.A.; Schöneich, C. Reversible Hydrogen Transfer Between Cysteine Thiyl Radical and Glycine and Alanine in Model Peptides: Covalent H/D Exchange, Radical-Radical Reactions, and L-to D-Ala Conversion. J. Phys. Chem. B 2010, 114, 6751–6762. [Google Scholar] [CrossRef] [PubMed]
- Mozziconacci, O.; Schöneich, C. Effect of Conformation on the Photodegradation of Trp-And Cystine-Containing Cyclic Peptides: Octreotide and Somatostatin. Mol. Pharm. 2014, 11, 3537–3546. [Google Scholar] [CrossRef] [PubMed]
- Mason, B.D.; Schöneich, C.; Kerwin, B.A. Effect of pH and Light on Aggregation and Conformation of An IgG1 mAb. Mol. Pharm. 2012, 9, 774–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, D.D.; Zhang, J.; Maity, H.; Mallela, K.M.G. Effect of Photo-Degradation on the Structure, Stability, Aggregation, and Function of an IgG1 Monoclonal Antibody. Int. J. Pharm. 2018, 547, 438–449. [Google Scholar] [CrossRef] [PubMed]
- Bessa, J.; Boeckle, S.; Beck, H.; Buckel, T.; Schlicht, S.; Ebeling, M.; Kiialainen, A.; Koulov, A.; Boll, B.; Weiser, T.; et al. The Immunogenicity of Antibody Aggregates in a Novel Transgenic Mouse model. Pharm. Res. 2015, 32, 2344–2359. [Google Scholar] [CrossRef] [PubMed]
- Boll, B.; Bessa, J.; Folzer, E.; Rios Quiroz, A.; Schmidt, R.; Bulau, P.; Finkler, C.; Mahler, H.C.; Huwyler, J.; Iglesias, A.; et al. Extensive Chemical Modifications in the Primary Protein Structure of IgG1 Subvisible Particles Are Necessary for Breaking Immune Tolerance. Mol. Pharm. 2017, 14, 1292–1299. [Google Scholar] [CrossRef]







© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schöneich, C. Thiyl Radical Reactions in the Chemical Degradation of Pharmaceutical Proteins. Molecules 2019, 24, 4357. https://doi.org/10.3390/molecules24234357
Schöneich C. Thiyl Radical Reactions in the Chemical Degradation of Pharmaceutical Proteins. Molecules. 2019; 24(23):4357. https://doi.org/10.3390/molecules24234357
Chicago/Turabian StyleSchöneich, Christian. 2019. "Thiyl Radical Reactions in the Chemical Degradation of Pharmaceutical Proteins" Molecules 24, no. 23: 4357. https://doi.org/10.3390/molecules24234357
APA StyleSchöneich, C. (2019). Thiyl Radical Reactions in the Chemical Degradation of Pharmaceutical Proteins. Molecules, 24(23), 4357. https://doi.org/10.3390/molecules24234357