
Effects of Propolis Extract and Propolis-Derived Compounds on Obesity and Diabetes: Knowledge from Cellular and Animal Models


Abstract
:1. Introduction
2. Advantages and Disadvantages of Animal and Cellular Models
2.1. Animal Models
2.2. Cellular Models
3. Obesity and Adipocity
3.1. Experimental Models
3.1.1. Animal Models and Pathology
3.1.2. Cellular Models
3.2. Effects of Propolis and Propolis-Derived Components on Obesity
3.2.1. Body Weight and Adipose Tissue Weight
3.2.2. Dyslipidemia
3.2.3. Feeding and Leptin Production
3.2.4. Adipogenesis
3.2.5. Adipokine Production
3.2.6. Induction of Brown/Beige Adipocytes
4. Diabetes Mellitus
4.1. Experimental Models
4.1.1. Animal Models and Pathology
4.1.2. Cellular Models
4.2. Effects of Propolis and Propolis-Derived Compounds on Diabetes Mellitus
4.2.1. Blood Glucose, Hb1Ac, and Lipid Profiles
4.2.2. Blood Insulin Level, Insulin Secretion, and Insulin Resistance
4.2.3. Oxidative Stress
4.2.4. Systemic Inflammation and Immune System
4.2.5. Adipose Tissue Inflammation
4.2.6. Vascular Endothelial Cells and Contraction of Vessels
5. Diabetic Complications
5.1. Diabetic Nephropathy
5.1.1. Pathology and Models of Diabetic Nephropathy
5.1.2. Effects of Propolis and Propolis-Derived Compounds on Diabetic Nephropathy
5.2. Diabetic Retinopathy
5.2.1. Pathology and Models of Diabetic Retinopathy
5.2.2. Effects of Propolis and Propolis-Derived Compounds on Diabetic Retinopathy
5.3. Delayed Wound Healing
5.3.1. Pathology and Models of Diabetes-Deteriorated Wound Healing
5.3.2. Effects of Propolis and Propolis-Derived Compounds on Wound Healing
5.4. NAFLD and Hepatic Steatosis
5.4.1. Animal and Cellular Models
5.4.2. Effects of Propolis and Propolis-Derived Compounds on NAFLD
6. Effects of Propolis on Diabetes and Diabetic Complications in Humans
6.1. Blood and Urinary Indices
6.2. Wound Healing
7. Perspectives
Funding
Conflicts of Interest
References
- Bhupathiraju, S.N.; Frank, B.H. Epidemiology of obesity and diabetes and their cardiovascular complications. Circ. Res. 2016, 118, 1723–1735. [Google Scholar] [CrossRef] [PubMed]
- Geiss, L.S.; Wang, J.; Cheng, Y.J.; Thompson, T.J.; Barker, L.; Li, Y.; Albright, A.L.; Gregg, E.W. Prevalence and incidence trends for diagnosed diabetes among adults aged 20 to 79 years, United States, 1980-2012. JAMA 2014, 312, 1218–1226. [Google Scholar] [CrossRef] [PubMed]
- Kayyali, R.; Slater, N.; Sahi, A.; Mepani, D.; Lalji, K.; Abdallah, A. Type 2 Diabetes: how informed are the general public? A cross-sectional study investigating disease awareness and barriers to communicating knowledge in high-risk populations in London. BMC Public Health 2019, 19, 138. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. Pathophysiology of type 2 diabetes. Acta Clin. Belg. 2003, 58, 335–341. [Google Scholar] [CrossRef]
- Volpe, C.M.O.; Villar-Delfino, P.H.; Dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 119. [Google Scholar] [CrossRef]
- Bierhaus, A.; Schiekofer, S.; Schwaninger, M.; Andrassy, M.; Humpert, P.M.; Chen, J.; Hong, M.; Luther, T.; Henle, T.; Klöting, I.; et al. Diabetes-associated sustained activation of the transcription factor nuclear factor-κB. Diabetes 2001, 50, 2792–2808. [Google Scholar] [CrossRef]
- Hu, H.; Jiang, H.; Ren, H.; Hu, X.; Wang, X.; Han, C. AGEs and chronic subclinical inflammation in diabetes: disorders of immune system. Diabetes Metab. Res. Rev. 2015, 31, 127–137. [Google Scholar] [CrossRef]
- Newsholme, P.; Keane, K.N.; Carlessi, R.; Cruzat, V. Oxidative stress pathways in pancreatic β-cells and insulin-sensitive cells and tissue: importance to cell metabolism, function, and dysfunction. Am. J. Physiol. 2019, 317, C420–C433. [Google Scholar] [CrossRef]
- Nishikawa, T.; Kukidome, D.; Sonoda, K.; Fujisawa, K.; Matsuhisa, T.; Motoshima, H.; Matsumura, T.; Araki, E. Impact of mitochondrial ROS production on diabetic vascular complications. Diabetes Res. Clin. Pract. 2007, 77S, S41–S45. [Google Scholar] [CrossRef]
- Ho, E.; Bray, T.M. Antioxidants, NFκB activation, and diabetogenesis. Proc. Soc. Exp. Biol. Med. 1999, 222, 205–213. [Google Scholar] [CrossRef]
- Jha, J.C.; Ho, F.; Dan, C.; Jandeleit-Dahm, K. A causal link between oxidative stress and inflammation in cardiovascular and renal complications of diabetes. Clin. Sci. 2018, 132, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Santiago, A.R.; Boia, R.; Aires, I.D.; Ambrósio, A.F.; Fernandes, R. Sweet stress: coping with vascular dysfunction in diabetic retinopathy. Front. Physiol. 2018, 9, 820. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Yiang, T.; Lai, T.T.; Li, C.J. The oxidative stress and mitochondrial dysfunction during the pathogenesis of diabetic retinopathy. Oxid. Med. Cell Longev. 2018, 2018, 3420187. [Google Scholar] [CrossRef] [PubMed]
- Spahis, S.; Delvin, E.; Borys, J.M.; Levy, E. Oxidative stress as a critical factor in nonalcoholic fatty liver disease pathogenesis. Antioxid. Redox Signal. 2017, 26, 519–541. [Google Scholar] [CrossRef] [PubMed]
- Rudel, L.L.; Parks, J.S.; Johnson, F.L.; Babiak, J. Low density lipoproteins in atherosclerosis. J. Lipid. Res. 1986, 27, 465–474. [Google Scholar] [PubMed]
- Liu, Q.; Bengmark, S.; Qu, S. The role of hepatic fat accumulation in pathogenesis of non-alcoholic fatty liver disease (NAFLD). Lipids Health Dis. 2010, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.; Schattenberg, J.M.; Leclercq, I.; Yeh, M.M.; Goldin, R.; Teoh, N.; Schuppan, D. Mouse models of nonalcoholic steatohepatitis: toward optimization of their relevance to human nonalcoholic steatohepatitis. Hepatology 2019, 69, 2241–2257. [Google Scholar] [CrossRef]
- Walenbergh, S.M.; Koek, G.H.; Bieghs, V.; Shiri-Sverdlov, R. Non-alcoholic steatohepatitis: The role of oxidized low-density lipoproteins. J. Hepatol. 2013, 58, 801–810. [Google Scholar] [CrossRef]
- Bellentani, S.; Scaglioni, F.; Marino, M.; Bedogni, G. Epidemiology of non-alcholoc fatty liver disease. Dig. Dis. 2010, 28, 155–161. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Younossi, Y.; Golabi, P.; Mishra, A.; Rafiq, N.; Henry, L. Epidemiology of chronic liver diseases in the USA in the past three decades. Gut 2019, in press. [Google Scholar] [CrossRef]
- Huang, S.; Zhang, C.P.; Wang, K.; Li, G.Q.; Hu, F.L. Recent advances in the chemical composition of propolis. Molecules 2014, 19, 19610–19632. [Google Scholar] [CrossRef] [PubMed]
- Lemos, M.; de Barros, M.P.; Sousa, J.P.; da Silva Filho, A.A.; Bastos, J.K.; de Andrade, S.F. Baccharis dracunculifolia, the main botanical source of Brazilian green propolis, displays antiulcer activity. J. Pharm. Pharmacol. 2007, 59, 603–608. [Google Scholar] [CrossRef] [PubMed]
- Trusheva, B.; Popova, M.; Bankova, V.; Simova, S.; Marcucci, M.C.; Miorin, P.L.; Pasin, F.R.; Tsvetkova, I. Bioactive constituents of Brazilian red propolis. Evid. Based Complement. Altern. Med. 2006, 3, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Bankova, V.S.; de Castro, S.L.; Marcucci, M.C. Propolis: Recent advances in chemistry and plant origin. Apidologie 2000, 31, 3–15. [Google Scholar] [CrossRef]
- Sforcin, J.M.; Bankova, V. Propolis: Is there a potential for the development of new drugs? J. Ethinopharmacol. 2011, 133, 253–260. [Google Scholar] [CrossRef]
- Zabaiou, N.; Fouache, A.; Trousson, A.; Baron, S.; Zellagui, A.; Lahouel, M.; Lobaccaro, J.A. Biological properties of propolis extracts: Something new from an ancient product. Chem. Phys. Lipids 2017, 207, 214–222. [Google Scholar] [CrossRef]
- De Figueiredo, S.M.; Nogueira-Machado, J.A.; Almeida Bde, M.; Abreu, S.R.; de Abreu, J.A.; Filho, S.A.; Binda, N.S.; Caligiorne, R.B. Immunomodulatory properties of green propolis. Recent Pat. Endocr. Metab. Immune Drug Discov. 2014, 8, 85–94. [Google Scholar] [CrossRef]
- Franchin, M.; Freires, I.A.; Lazarini, J.G.; Nani, B.D.; da Cunha, M.G.; Colón, D.F.; de Alencar, S.M.; Rosalen, P.L. The use of Brazilian propolis for discovery and development of novel anti-inflammatory drugs. Eur. J. Med. Chem. 2018, 153, 49–55. [Google Scholar] [CrossRef]
- Kleinert, M.; Clemmensen, C.; Hofmann, S.M.; Moore, M.C.; Renner, S.; Woods, S.C.; Huypens, P.; Beckers, J.; de Angelis, M.H.; Schürmann, A.; et al. Animal models of obesity and diabetes mellitus. Nat. Rev. Endocrinol. 2018, 14, 140–162. [Google Scholar] [CrossRef]
- Chahoud, I.; Paumgartten, F.J. Influence of litter size on the postnatal growth of rat pups: Is there a rationale for litter-size standardization in toxicity studies? Environ. Res. 2009, 109, 1021–1027. [Google Scholar] [CrossRef]
- Da Silva Xavier, G.; Hodson, D.J. Mouse models of peripheral metabolic disease. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Mistry, S.B.; Omana, J.J.; Kini, S. Rat models for bariatric surgery and surgery for type 2 diabetes mellitus. Obes. Surg. 2009, 19, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Liao, J.K. A mouse model of diet-induced obesity and insulin resistance. Meth. Mol. Biol. 2012, 821, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Ayala, J.E.; Samuel, V.T.; Morton, G.J.; Obici, S.; Croniger, C.M.; Shulman, G.I.; Wasserman, D.H.; McGuinness, O.P. NIH Mouse Metabolic Phenotyping Center Consortium. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis. Model Mech. 2010, 3, 525–534. [Google Scholar] [CrossRef] [Green Version]
- Reitman, M.L. Of mice and men-environmental temperature, body temperature, and treatment of obesity. FEBS Lett. 2018, 592, 2098–2107. [Google Scholar] [CrossRef] [Green Version]
- Like, A.A.; Rossini, A.A. Streptozotocin-induced pancreatic insulitis: New model of diabetes mellitus. Science 1976, 193, 415–417. [Google Scholar] [CrossRef]
- Cassidy, A.; Minihane, A.M. The role of metabolism (and the microbiome) in defining the clinical efficacy of dietry flavonoids. Am. J. Clin. Nutr. 2017, 105, 10–22. [Google Scholar] [CrossRef] [Green Version]
- Pasinetti, G.M.; Singh, R.; Westfall, S.; Herman, F.; Faith, J.; Ho, L. The role of the gut microbiota in the metabolism of polyphenols as characterized by gnotbiotic mice. J. Alzheimers Dis. 2018, 63, 409–421. [Google Scholar] [CrossRef]
- Martignoni, M.; Groothuis, G.M.M.; de Kanter, R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin. Drug Metab. Toxicol. 2006, 2, 875–894. [Google Scholar] [CrossRef]
- Breinholt, V.M.; Offord, E.A.; Brouwer, C.; Nielsen, S.E.; Brøsen, K.; Friedberg, T. In vitro investigation of cytochrome P450-mediated metabolism of dietry flavonoids. Food Chem. Toxicol. 2002, 40, 609–616. [Google Scholar] [CrossRef]
- Carrão, D.B.; de Albuquerque, N.C.P.; Marques, L.M.M.; Crotti, A.E.M.; Pilon, A.C.; Bolzani, V.D.S.; Berretta, A.A.; de Oliveira, A.R.M. In vitro metabolism of artepillin C by rat and human liver microsomes. Plant Med. 2017, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.W.; Jiang, X.L.; Gonzalez, F.J.; Yu, A.M. Humanized transgenic mouse models for drug metabolism and pharmacokinetics research. Curr. Drug Metab. 2011, 12, 997–1006. [Google Scholar] [CrossRef] [PubMed]
- Kilkenny, C.; Browne, W.J.; Cuthill, I.C.; Emerson, M.; Altman, D.G. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.C.; Lilley, E. Implementing guidelines on reporting research using animals (ARRIVE etc.): New requirements for publication in BJP. Br. J. Pharmacol. 2015, 172, 3189–3193. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.K.; Park, S.H.; Choi, Y.J.; Shin, D.; Kang, Y.H. Chrysin inhibits diabetic renal tubulointerstitial fibrosis through blocking epithelial to mesenchymal transition. J. Mol. Med. 2015, 93, 759–772. [Google Scholar] [CrossRef]
- Lee, E.J.; Kang, M.K.; Kim, D.Y.; Kim, Y.H.; Oh, H.; Kang, Y.H. Chrysin inhibits advanced glycation end products-induced kidney fibrosis in renal mesangial cells and diabetic kidneys. Nutrients 2018, 10, 882. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.J.; Kang, M.K.; Kim, Y.H.; Kim, D.Y.; Oh, H.; Kim, S.I.; Oh, S.Y.; Kang, Y.H. Dietary chrysin suppresses formation of actin cytoskeleton and focal adhesion in AGE-exposed mesangial cells and diabetic kidney: Role of autophagy. Nutrients 2019, 11, 127. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.K.; Park, S.H.; Kim, Y.H.; Lee, E.J.; Antika, L.D.; Kim, D.Y.; Choi, Y.J.; Kang, Y.H. Chrysin ameliorates podocyte injury and slit diaphragm protein loss via inhibition of the PERK-eIF2α-ATF-CHOP pathway in diabetic mice. Acta Pharmacol. Sin. 2017, 38, 1129–1140. [Google Scholar] [CrossRef]
- Choi, S.S.; Cha, B.Y.; Iida, K.; Lee, Y.S.; Yonezawa, T.; Teruya, T.; Nagai, K.; Woo, J.T. Artepillin C, as a PPARγ ligand, enhances adipocyte differentiation and glucose uptake in 3T3-L1 cells. Biochem. Pharmacol. 2011, 81, 925–933. [Google Scholar] [CrossRef]
- Nakashima, K.; Murakami, T.; Tanabe, H.; Inoue, M. Identification of a naturally occurring retinoid X receptor agonist from Brazilian green propolis. Biochim. Biophys. Acta 2014, 1840, 3034–3041. [Google Scholar] [CrossRef]
- Ahn, S.; Kim, J.; An, S.; Pyo, J.J.; Jung, D.; Lee, J.; Hwang, S.Y.; Gong, J.; Shin, I.; Kim, H.P.; et al. 2-Phenyl-8-(1-phenylallyl)-chromenone compounds have a pan-PPAR modulator pharmacophore. Bioorg. Med. Chem. 2019, 27, 2948–2958. [Google Scholar] [CrossRef] [PubMed]
- Surwit, R.S.; Kuhn, C.M.; Cochrance, C.; McCubbin, J.A.; Feinglos, M.N. Diet-induced type II diabetes in C57BL/6J mice. Diabetes 1988, 37, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- Hamann, A.; Matthaei, S. Regulation of energy balance by leptin. Exp. Clin. Endocrinol. Diabetes 1996, 104, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Trayhurn, P. The development of obesity in animals: The role of genetic susceptibility. Clin. Endocrinol. Metab. 1984, 13, 451–474. [Google Scholar] [CrossRef]
- Phillips, M.S.; Liu, Q.; Hammond, H.A.; Dugan, V.; Hey, P.J.; Caskey, C.J.; Hess, J.F. Leptin receptor missense mutation in the fatty Zucker rat. Nat. Genet. 1996, 13, 18–19. [Google Scholar] [CrossRef] [PubMed]
- Zucker, L.M.; Zucker, T.F. Fatty, a new mutation in the rat. J. Hered. 1961, 52, 275–278. [Google Scholar] [CrossRef]
- Bays, H.E. “Sick fat,” metabolic disease, and atherosclerosis. Am. J. Med. 2009, 122, S26–S37. [Google Scholar] [CrossRef]
- Luong, Q.; Huang, J.; Lee, K.Y. Deciphering white adipose tissue heterogeneity. Biology 2019, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Ghaben, A.L.; Scherer, P.E. Adipogenesis and metabolic health. Nat. Rev. Mol. Cell. Biol. 2019, 20, 242–258. [Google Scholar] [CrossRef]
- McNelis, J.C.; Olefsky, J.M. Macrophages, immunity, and metabolic disease. Immunity 2014, 41, 36–48. [Google Scholar] [CrossRef] [Green Version]
- Mraz, M.; Haluzik, M. The role of adipose tissue immune cells in obesity and low-grade inflammation. J. Endocrinol. 2014, 222, R113–R127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Gerhold, K.; Mayers, J.R.; Wiest, M.M.; Watkins, S.M.; Hotamisligil, G.S. Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 2008, 134, 933–944. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.W.; Lee, M.; Oh, K.J. Adipose tissue-derived signatures for obesity and type 2 diabetes: Adipokines, batokines and microRNAs. J. Clin. Med. 2019, 8, 854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Yang, Y.; Xiang, L.; Zhao, Z.; Ye, R. Adipose-derived exosomes: A novel adipokine in pbesity-associated diabetes. J. Cell. Physiol. 2019, 234, 16692–16702. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.Y.; Halo, P.; Leibel, R.L.; Zhang, Y. Effects of obesity on the relationship of leptin mRNA expression and adipocyte size in anatomically distinct fat depots in mice. Am. J. Physiol. 2004, 287, R112–R119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jernås, M.; Palming, J.; Sjöholm, K.; Jennische, E.; Svensson, P.A.; Gabrielsson, B.G.; Levin, M.; Sjögren, A.; Rudemo, M.; Lystig, T.C.; et al. Separation of human adipocytes by size: Hypertrophic fat cells display distinct gene expression. FESEB J. 2006, 20, 1540–1542. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Kastin, A.J.; Huang, W.; Jaspan, J.B.; Maness, L.M. Leptin enters the brain by a saturable system independent of insulin. Peptides 1996, 17, 305–311. [Google Scholar] [CrossRef]
- Baskin, D.G.; Breininger, J.F.; Schwartz, M.W. Leptin receptor mRNA identifies a subpopulation of neuropeptide Y neurons activated by fasting in rat hypothalamus. Diabetes 1999, 48, 828–833. [Google Scholar] [CrossRef]
- Zeng, W.; Pirzgalska, R.M.; Pereira, M.M.; Kubasova, N.; Barateiro, A.; Seixas, E.; Lu, Y.H.; Kozlova, A.; Voss, H.; Martins, G.G.; et al. Sympathetic neuro-adipose connections mediate leptin-driven lipolysis. Cell 2015, 163, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Yang, X.; Yu, S.; Zheng, R. The leptin resistance. Adv. Exp. Med. Biol. 2018, 1090, 145–163. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef]
- Yoon, M.J.; Lee, G.Y.; Chung, J.J.; Ahn, Y.H.; Hong, S.H.; Kim, J.B. Adiponectin increases fatty acid oxidation in skeletal muscle cells by sequential activation of AMP-activated protein kinase, p38 mitogen-activated protein kinase, and peroxisome proliferator-activated receptor alpha. Diabetes 2006, 55, 2562–2570. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin resistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Budavari, A.; Murray, D.; Spiegelman, B.M. Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-alpha. J. Clin. Investig. 1994, 94, 1543–1549. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-α- and obesity-induced insulin resistance. Science 1996, 271, 665–670. [Google Scholar] [CrossRef]
- Uysal, K.T.; Wiesbrock, S.M.; Marino, M.W.; Hotamisligil, G.S. Protection from obesity-induced insulin resistance in mice lacking TNF-α function. Nature 1997, 389, 610–614. [Google Scholar] [CrossRef]
- Bodary, P.F. Links between adipose tissue and thrombosis in the mouse. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2284–2291. [Google Scholar] [CrossRef] [Green Version]
- Nagai, N.; Van Hoef, B.; Lijnen, H.R. Plasminogen activator inhibitor-1 contributes to the deleterious effect of obesity on the outcome of thrombotic ischemic stroke in mice. J. Thromb. Haemost. 2007, 5, 1726–1731. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.J.; Mao, S.L.; Taylor, K.L.; Kanjanabuch, T.; Guan, Y.; Zhang, Y.; Brown, N.J.; Swift, L.L.; McGuinness, O.P.; Wasserman, D.H.; et al. Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes 2004, 53, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Meister, B. Neurotransmitters in key neurons of the hypothalamus that regulate feeding behavior and body weight. Physiol. Behav. 2007, 92, 263–271. [Google Scholar] [CrossRef]
- Arora, S.; Anubhuti. Role of neuropeptides in appetite regulation and obesity- A review. Neuropeptides 2006, 40, 375–401. [Google Scholar] [CrossRef]
- Bray, G.A. Afferent signals regulating food intake. Proc. Nutr. Soc. 2000, 59, 373–384. [Google Scholar] [CrossRef]
- Durnin, J.V.G.A. Basal metabolic rate in man. Joint FAO/WHO/UNU Expert Consultation on Energy and Protein Requirements, Rome, 5 to 17 October 1981. Available online: http://www.fao.org/3/M2845E/M2845E00.htm (accessed on 30 November 2019).
- Poher, A.L.; Altirriba, J.; Veyrat-Durebex, C.; Rohner-Jeanrenaud, F. Brown adipose tissue activity as a target for the treatment of obesity/insulin resistance. Front. Physiol. 2015, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Kajimura, S.; Spiegelman, B.M.; Seale, P. Brown and beige fat: Physiological roles beyond heat generation. Cell Metab. 2015, 22, 546–559. [Google Scholar] [CrossRef] [Green Version]
- Rajan, S.; Gupta, A.; Beg, M.; Shankar, K.; Srivastava, A.; Varshney, S.; Kumar, D.; Gaikwad, A.N. Adipocyte transdifferentiation and its molecular targets. Differentiation 2014, 87, 183–192. [Google Scholar] [CrossRef]
- Nishikawa, S.; Aoyama, H.; Kamiya, M.; Higuchi, J.; Kato, A.; Soga, M.; Kawai, T.; Yoshimura, K.; Kumazawa, S.; Tsuda, T. Artepillin C, a typical Brazilian propolis-derived component, induces brown-like adipocyte formation in C3H10T1/2 cells, primary inguinal white adipose tissue-derived adipocytes, and mice. PLoS ONE 2016, 11, e0162512. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, S.; Kamiya, M.; Aoyama, H.; Yoshimura, K.; Miyata, R.; Kumazawa, S.; Tsuda, T. Co-administration of curcumin and artepillin C induces development of brown-like adipocytes in association with local norepinephrine production by alternatively activated macrophages in mice. J. Nutr. Sci. Vitaminol. 2019, 65, 328–334. [Google Scholar] [CrossRef]
- Bernlohr, D.A.; Angus, C.W.; Lane, M.D.; Bolanowski, M.A.; Kelly, T.J., Jr. Expression of specific mRNAs during adipose differentiation: identification of an mRNA encoding a homologue of myelin P2 protein. Proc. Natl. Acad. Sci. USA 1984, 81, 5468–5472. [Google Scholar] [CrossRef] [Green Version]
- Prusty, D.; Park, B.H.; Davis, K.E.; Farmer, S.R. Activation of MEK/ERK signaling promotes adipogenesis by enhancing peroxisome proliferator-activated receptor gamma (PPARγ) and C/EBPα gene expression during the differentiation of 3T3-L1 preadipocytes. J. Biol. Chem. 2002, 277, 46226–46232. [Google Scholar] [CrossRef] [Green Version]
- Zebisch, K.; Voigt, V.; Wabitsch, M.; Brandsch, M. Protocol for effective differentiation of 3T3-L1 cells to adipocytes. Anal. Biochem. 2012, 425, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.J.; Kim, K.J.; Koh, E.J.; Choi, J.; Lee, B.Y. Anti-adipogenesis mechanism of pterostilbene through the activation of heme oxygenase-1 in 3T3-L1 cells. Phytomedcine 2017, 33, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Gwon, S.Y.; Ahn, J.Y.; Jung, C.H.; Moon, B.K.; Ha, T.Y. Shikonin suppresses ERK 1/2 phosphorylation during the early stages of adipocyte differentiation in 3T3-L1 cells. BMC Complement. Altern. Med. 2013, 13, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Yamamoto, N.; Yamashita, Y.; Ashida, H. The chalcones cardamonin and flavokawain B inhibit the differentiation of preadipocytes to adipocytes by activating ERK. Arch. Biochem. Biophys. 2014, 554, 44–54. [Google Scholar] [CrossRef]
- Kang, Y.H.; Kim, K.K.; Kim, D.J.; Choe, M. Antiobesity effects of the water-soluble fraction of the ethanol extract of Smilax china L. leaf in 3T3-L1 adipocytes. Nutr. Res. Pract. 2015, 9, 606–612. [Google Scholar] [CrossRef] [Green Version]
- Anthonsen, M.W.; Rönnstrand, L.; Wernstedt, C.; Degerman, E.; Holm, C. Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J. Biol. Chem. 1998, 273, 215–221. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.M.; Tucker, D.F.; Gross, D.N.; Easton, R.M.; DiPilato, L.M.; Dean, A.S.; Monks, B.R.; Birnbaum, M.J. Insulin regulates adipocyte lipolysis via an Akt-independent signaling pathway. Mol. Cell. Biol. 2010, 30, 5009–5020. [Google Scholar] [CrossRef] [Green Version]
- Kaestner, K.H.; Christy, R.J.; Lane, M.D. Mouse insulin-responsive glucose transporter gene: Characterization of the gene and trans-activation by the CCAAT/enhancer binding protein. Proc. Natl. Acad. Sci. USA 1990, 81, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Pektaş, M.; Kurt, A.H.; Ün, İ.; Tiftik, R.N.; Büyükafşar, K. Effects of 17β-estradiol and progesterone on the production of adipokines in differentiating 3T3-L1 adipocytes: Role of Rho-kinase. Cytokine 2015, 72, 130–134. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Waki, H.; Murakami, K.; Motojima, K.; Komeda, K.; Ide, T.; Kubota, N.; Terauchi, Y.; Tobe, K.; et al. The mechanisms by which both heterozygous peroxisome proliferator-activated receptor γ (PPARγ) deficiency and PPARγ agonist improve insulin resistance. J. Biol. Chem. 2001, 276, 41245–41254. [Google Scholar] [CrossRef] [Green Version]
- Kliewer, S.A.; Lenhard, J.M.; Willson, T.M.; Patel, I.; Morris, D.C.; Lehmann, J.M. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor γ and promotes adipocyte differentiation. Cell 1995, 83, 813–819. [Google Scholar] [CrossRef] [Green Version]
- Nagy, L.; Tontonoz, P.; Alvarez, J.G.A.; Chen, H.; Evans, R.M. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARγ. Cell 1998, 93, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid. Res. 2014, 53, 124–144. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, M.; Matsuda, M.; Maeda, N.; Funahashi, T.; Matsuzawa, Y.; Makishima, M.; Shimomura, I. Induction of adiponectin, a fat-derived antidiabetic and antiatherogenic factor, by nuclear receptors. Diabetes 2003, 52, 1655–1663. [Google Scholar] [CrossRef] [Green Version]
- Chung, S.W.; Kang, B.Y.; Kim, S.H.; Park, Y.K.; Cho, D.; Trinchieri, G.; Kim, T.S. Oxidized low density lipoprotein inhibits interleukin-12 production in lipopolysaccharide-activated mouse macrophages via direct interactions between peroxisome proliferator-activated receptor-γ and nuclear factor-κB. J. Biol. Chem. 2000, 275, 32681–32687. [Google Scholar] [CrossRef] [Green Version]
- Ichi, I.; Hori, H.; Takashima, Y.; Adachi, N.; Kataoka, R.; Okihara, K.; Hashimoto, K.; Kojo, S. The beneficial effect of propolis on fat accumulation and lipid metabolism in rats fed a high-fat diet. J. Food Sci. 2009, 74, H127–H131. [Google Scholar] [CrossRef]
- Koya-Miyata, S.; Arai, N.; Mizote, A.; Taniguchi, Y.; Ushio, S.; Iwaki, K.; Fukuda, S. Propolis prevents diet-induced hyperlipidemia and mitigates weight gain in diet-induced obesity in mice. Biol. Pharm. Bull. 2009, 32, 2022–2028. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, H.; Naoe, Y.; Kimura, S.; Miyamoto, T.; Okamoto, S.; Toda, C.; Shimamoto, Y.; Iwanaga, T.; Miyoshi, I. Beneficial effects of Brazilian propolis on type 2 diabetes in ob/ob mice. Adipocyte 2013, 2, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Oršolić, N.; Landeka Jurčević, I.; Đikić, D.; Rogić, D.; Odeh, D.; Balta, V.; Perak Junaković, E.; Terzić, S.; Jutrić, D. Effect of propolis on diet-induced hyperlipidemia and atherogenic indices in mice. Antioxidants 2019, 8, 156. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.H.; Seo, S.G.; Min, S.; Yang, H.; Lee, E.; Son, J.E.; Kwon, J.Y.; Yue, S.; Chung, M.Y.; Kim, K.H.; et al. Caffeic acid phenethyl ester, a major component of propolis, suppresses high fat diet-induced obesity through inhibiting adipogenesis at the mitotic clonal expansion stage. Agric. Food Chem. 2014, 62, 4306–4312. [Google Scholar] [CrossRef]
- Roquetto, A.R.; Monteiro, N.E.S.; Moura, C.S.; Toreti, V.C.; de Pace, F.; Santos, A.D.; Park, Y.K.; Amaya-Farfan, J. Green propolis modulates gut microbiota, reduces endotoxemia and expression of TLR4 pathway in mice fed a high-fat diet. Food Res. Int. 2015, 76, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Washio, K.; Shimamoto, Y.; Kitamura, H. Brazilian propolis extract increases leptin expression in mouse adipocytes. Biomed. Res. 2015, 36, 343–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juman, S.; Yasui, N.; Okuda, H.; Ueda, A.; Negishi, H.; Miki, T.; Ikeda, K. Caffeic acid phenethyl ester suppresses the production of adipocytokines, leptin, tumor necrosis factor -alpha and resistin, during differentiation to adipocytes in 3T3-L1 cells. Biol. Pharm. Bull. 2011, 34, 490–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miki, H.; Yamauchi, T.; Suzuki, R.; Komeda, K.; Tsuchida, A.; Kobata, N.; Terauchi, Y.; Kamon, J.; Kaburagi, Y.; Matsui, J.; et al. Essential role of insulin receptor substrate 1(IRS-1) and IRS-2 in adipocyte differentiation. Mol. Cell. Biol. 2001, 21, 2521–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanella, L.; Tibullo, D.; Godos, J.; Pluchinotta, F.R.; Di Giacomo, C.; Sorrenti, V.; Acquaviva, R.; Russo, A.; Li Volti, G.; Barbagallo, I. Caffeic acid phenethyl ester regulates PPAR’s levels in stem cells-derived adipocytes. PPAR Res. 2016, 2016, 7359521. [Google Scholar] [CrossRef]
- Iio, A.; Ohguchi, K.; Inoue, H.; Maruyama, H.; Araki, Y.; Nozawa, Y.; Ito, M. Ethanolic extracts of Brazilian red propolis promote adipocyte differentiation through PPARγ activation. Phytomedicine 2010, 17, 974–979. [Google Scholar] [CrossRef]
- Ikeda, R.; Yanagisawa, M.; Takahashi, N.; Kawada, T.; Kumazawa, S.; Yamaotsu, N.; Nakagome, I.; Hirono, S.; Tsuda, T. Brazilian propolis-derived components inhibit TNF-α-mediated downregulation of adiponectin expression via different mechanisms in 3T3-L1 adipocytes. Biochim. Biophys. Acta. 2011, 10, 695–703. [Google Scholar] [CrossRef]
- Li, F.; Awale, S.; Tezuka, Y.; Esumi, H.; Kadota, S. Study on the constituents of Mexican propolis and their cytotoxic activity against PANC-1 human pancreatic cancer cells. J. Nat. Prod. 2010, 73, 623–627. [Google Scholar] [CrossRef]
- Juman, S.; Yasui, N.; Okuda, H.; Ueda, A.; Negishi, H.; Miki, T.; Ikeda, K. Caffeic acid phenethyl ester inhibits differentiation to adipocytes in 3T3-L1 mouse fibroblasts. Biol. Pharm. Bull. 2010, 33, 1484–1488. [Google Scholar] [CrossRef] [Green Version]
- Imai, M.; Kumaoka, T.; Hosaka, M.; Sato, Y.; Li, C.; Sudoh, M.; Tamada, Y.; Yokoe, H.; Saito, S.; Tsubuki, M.; et al. Inhibitory effects of hydroxylated cinnamoyl esters on lipid absorption and accumulation. Bioorg. Med. Chem. 2015, 23, 3788–3795. [Google Scholar] [CrossRef]
- Berg, A.H.; Combs, T.P.; Scherer, P.E. ACRP30/adiponectin: An adipokine regulating glucose and lipid metabolism. Trends Endocrinol. Metab. 2002, 13, 84–89. [Google Scholar] [CrossRef]
- Ohkura, N.; Oishi, K.; Kihara-Negishi, F.; Atsumi, G.; Tatefuji, T. Effects of a diet containing Brazilian propolis on lipopolysaccharide-induced increases in plasma plasminogen activator inhibitor-1 levels in mice. J. Intercult. Ethnopharmacol. 2016, 5, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Furman, B.L. Streptozotocin-induced diabetic models in mice and rats. Curr. Protoc. Pharmacol. 2015, 70, 1–20. [Google Scholar] [CrossRef]
- Lenzen, S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia 2008, 51, 216–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicram, A.; Jena, G. S961, an insulin receptor antagonist causes hyperinsulinemia, insulin-resistance and depletion of energy stores in rats. Biochem. Biophys. Res. Commun. 2010, 398, 260–265. [Google Scholar] [CrossRef]
- Rostoker, R.; Bitton-Worms, K.; Caspi, A.; Shen-Orr, Z.; LeRoith, D. Investigating new therapeutic strategies targeting hyperinsulinemia’s mitogenic effects in a female mouse breast cancer model. Endocrinology 2013, 154, 1701–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rifa’I, M.; Widodo, N. Significance of propolis administration for homeostasis of CD4+CD25+ immunoregulatory T cells controlling hyperglycemia. Springerplus 2014, 3, 526. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.Z.; Liu, Y.C.; Zheng, Y.F.; Chen, Y.F.; Si, J.J.; Chen, M.L.; Shou, Q.Y.; Zheng, H.Q.; Hu, F.L. Ethanol extract of chinese propolis attenuates early diabetic retinopathy by protecting the blood-retinal barrier in streptozotocin-induced diabetic rats. J. Food. Sci. 2019, 84, 358–369. [Google Scholar] [CrossRef]
- Zhu, W.; Chen, M.; Shou, Q.; Li, Y.; Hu, F. Biological activities of Chinese propolis and Brazilian propolis on streptozotocin-induced type 1 diabetes mellitus in rats. Evid. Based Complement. Alternat. Med. 2011, 2011, 468529. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Li, Y.H.; Chen, M.L.; Hu, F.L. Protective effects of Chinese and Brazilian propolis treatment against hepatorenal lesion in diabetic rats. Hum. Exp. Toxicol. 2011, 30, 1246–1255. [Google Scholar] [CrossRef]
- Nna, V.U.; Bakar, A.B.A.; Mohamed, M. Malaysian propolis, metformin and their combination, exert hepatoprotective effect in streptozotocin-induced diabetic rats. Life Sci. 2018, 211, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Nna, V.U.; Abu Bakar, A.B.; Md Lazin, M.R.M.L.; Mohamed, M. Antioxidant, anti-inflammatory and synergistic anti-hyperglycemic effects of Malaysian propolis and metformin in streptozotocin-induced diabetic rats. Food Chem. Toxicol. 2018, 120, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Yañez, N.; Rodriguez-Canales, M.; Nieto-Yañez, O.; Jimenez-Estrada, M.; Ibarra-Barajas, M.; Canales-Martinez, M.M.; Rodriguez-Monroy, M.A. Hypoglycaemic and antioxidant effects of propolis of Chihuahua in a model of experimental diabetes. Evid. Based Complement. Alternat. Med. 2018, 2018, 4360356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Rabey, H.A.; Al-Seeni, M.N.; Bakhashwain, A.S. The antidiabetic activity of Nigella sativa and propolis on streptozotocin-induced diabetes and diabetic nephropathy in male rats. Evid. Based Complement. Alternat. Med. 2017, 2017, 5439645. [Google Scholar] [CrossRef] [Green Version]
- Sameni, H.R.; Ramhormozi, P.; Bandegi, A.R.; Taherian, A.A.; Mirmohammadkhani, M.; Safari, M. Effects of ethanol extract of propolis on histopathological changes and anti-oxidant defense of kidney in a rat model for type 1 diabetes mellitus. J. Diabetes Investig. 2016, 7, 506–513. [Google Scholar] [CrossRef]
- El-Sayed, S.M.; Abo-Salem, O.M.; Aly, H.A.; Mansour, A.M. Potential antidiabetic and hypolipidemic effects of propolis extract in streptozotocin-induced diabetic rats. Pak. J. Pharm. Sci. 2009, 22, 168–174. [Google Scholar]
- Abo-Salem, O.M.; El-Edel, R.H.; Harisa, G.E.; El-Halawany, N.; Ghonaim, M.M. Experimental diabetic nephropathy can be prevented by propolis: Effect on metabolic disturbances and renal oxidative parameters. Pak. J. Pharm. Sci. 2009, 22, 205–210. [Google Scholar]
- Okutan, H.; Ozcelik, N.; Yilmaz, H.; Uz, E. Effects of caffeic acid phenethyl ester on lipid peroxidation and antioxidant enzymes in diabetic rat heart. Clin. Biochem. 2005, 38, 191–196. [Google Scholar] [CrossRef]
- Ibrahim, K.A.; Khwanes, S.A.; El-Desouky, M.A.; Elhakim, H.K.A. Propolis relieves the cardiotoxicity of chlorpyrifos in diabetic rats via alleviations of paraoxonase-1 and xanthine oxidase genes expression. Pestic. Biochem. Physiol. 2019, 159, 127–135. [Google Scholar] [CrossRef]
- Usman, U.Z.; Bakar, A.B.A.; Mohamed, M. Propolis improves pregnancy outcomes and placental oxidative stress status in streptozotocin-induced diabetic rats. BMC Complement Altern. Med. 2018, 18, 324. [Google Scholar] [CrossRef]
- Al Ghamdi, A.A.; Badr, G.; Hozzein, W.N.; Allam, A.; Al-Waili, N.S.; Al-Wadaan, M.A.; Garraud, O. Oral supplementation of diabetic mice with propolis restores the proliferation capacity and chemotaxis of B and T lymphocytes towards CCL21 and CXCL12 by modulating the lipid profile, the pro-inflammatory cytokine levels and oxidative stress. BMC Immunol. 2015, 16, 54. [Google Scholar] [CrossRef]
- Aral, C.A.; Kesim, S.; Greenwell, H.; Kara, M.; Çetin, A.; Yakan, B. Alveolar bone protective and hypoglycemic effects of systemic propolis treatment in experimental periodontitis and diabetes mellitus. J. Med. Food 2015, 18, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Matsushige, K.; Basnet, P.; Hase, K.; Kadota, S.; Tanaka, K.; Namba, T. Propolis protects pancreatic β-cells against the toxicity of streptozotocin (STZ). Phytomedicine 1996, 3, 203–209. [Google Scholar] [CrossRef]
- Coskun, O.; Anter, M.; Korkmaz, A.; Oter, S. Quercetin, a flavonoid antioxidant, prevents and protects streptozotocin-induced oxidative stress and β-cell damage in rat pancreas. Pharmacol. Res. 2005, 51, 117–123. [Google Scholar] [CrossRef]
- Granados-Pineda, J.; Uribe-Uribe, N.; García-López, P.; Ramos-Godinez, M.D.P.; Rivero-Cruz, J.F.; Pérez-Rojas, J.M. Effect of pinocembrin isolated from Mexican brown propolis on diabetic nephropathy. Molecules 2018, 23, 852. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Moschen, A.R. Microbiota and diabetes: An evolving relationship. Gut 2014, 1513–1521. [Google Scholar] [CrossRef]
- Ernawati, D.S.; Puspa, A. Expression of vascular endothelial growth factor and matrix metalloproteinase-9 in Apis mellifera Lawang propolis extract gel-treated traumatic ulcers in diabetic rats. Vet. World 2018, 11, 304–309. [Google Scholar] [CrossRef] [Green Version]
- Puspasari, A.; Harijanti, K.; Soebadi, B.; Hendarti, H.T.; Radithia, D.; Ernawati, D.S. Effects of topical application of propolis extract on fibroblast growth factor-2 and fibroblast expression in the traumatic ulcers of diabetic Rattus norvegicus. J. Oral Maxillofac. Pathol. 2018, 22, 54–58. [Google Scholar] [CrossRef]
- Hozzein, W.N.; Badr, G.; Al Ghamdi, A.A.; Sayed, A.; Al-Waili, N.S.; Garraud, O. Topical application of propolis enhances cutaneous wound healing by promoting TGF-beta/Smad-mediated collagen production in a streptozotocin-induced type I diabetic mouse model. Cell. Physiol. Biochem. 2015, 37, 940–954. [Google Scholar] [CrossRef]
- McLennan, S.V.; Bonner, J.; Milne, S.; Lo, L.; Charlton, A.; Kurup, S.; Jia, J.; Yue, D.K.; Twigg, S.M. The anti-inflammatory agent Propolis improves wound healing in a rodent model of experimental diabetes. Wound Repair Regen. 2008, 16, 706–713. [Google Scholar] [CrossRef]
- Picolotto, A.; Pergher, D.; Pereira, G.P.; Machado, K.G.; da Silva Barud, H.; Roesch-Ely, M.; Gonzalez, M.H.; Tasso, L.; Figueiredo, J.G.; Moura, S. Bacterial cellulose membrane associated with red propolis as phytomodulator: Improved healing effects in experimental models of diabetes mellitus. Biomed. Pharmacother. 2019, 112, 108640. [Google Scholar] [CrossRef]
- Voss, G.T.; Gularte, M.S.; Vogt, A.G.; Giongo, J.L.; Vaucher, R.A.; Echenique, J.V.Z.; Soares, M.P.; Luchese, C.; Wilhelm, E.A.; Fajardo, A.R. Polysaccharide-based film loaded with vitamin C and propolis: A promising device to accelerate diabetic wound healing. Int. J. Pharm. 2018, 552, 340–351. [Google Scholar] [CrossRef]
- Oladayo, M.I. Nigerian propolis improves blood glucose, glycated hemoglobin A1c, very low-density lipoprotein, and high-density lipoprotein levels in rat models of diabetes. J. Intercult. Ethnopharmacol. 2016, 5, 233–238. [Google Scholar] [CrossRef]
- Babatunde, I.R.; Abdulbasit, A.; Oladayo, M.I.; Olasile, O.I.; Olamide, F.R.; Gbolahan, B.W. Hepatoprotective and pancreatoprotective properties of the ethanolic extract of Nigerian propolis. J. Intercult. Ethnopharmacol. 2015, 4, 102–108. [Google Scholar] [CrossRef]
- Oršolić, N.; Sirovina, D.; Končić, M.Z.; Lacković, G.; Gregorović, G. Effect of Croatian propolis on diabetic nephropathy and liver toxicity in mice. BMC Complement. Altern. Med. 2012, 12, 117. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Wei, P.; Lu, Q.; Liu, R.; Ding, Y.; Zhang, J. Beneficial effects of poplar buds on hyperglycemia, dyslipidemia, oxidative stress, and inflammation in streptozotocin-induced type-2 diabetes. J. Immunol. Res. 2018, 2018, 7245956. [Google Scholar] [CrossRef] [Green Version]
- Nie, J.; Chang, Y.; Li, Y.; Zhou, Y.; Qin, J.; Sun, Z.; Li, H. Caffeic acid phenethyl ester (propolis extract) ameliorates insulin resistance by inhibiting JNK and NF-κB inflammatory pathways in diabetic mice and HepG2 cell models. J. Agric. Food Chem. 2017, 65, 9041–9053. [Google Scholar] [CrossRef]
- Chen, L.H.; Chien, Y.W.; Chang, M.L.; Hou, C.C.; Chan, C.H.; Tang, H.W.; Huang, H.Y. Taiwanese green propolis ethanol extract delays the progression of type 2 diabetes mellitus in rats treated with streptozotocin/high-fat diet. Nutrients 2018, 10, 503. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, M.; Xuan, H.; Hu, F. Effects of encapsulated propolis on blood glycemic control, lipid metabolism, and insulin resistance in type 2 diabetes mellitus rats. Evid. Based Compliment. Alternat. Med. 2012, 2012, 981896. [Google Scholar] [CrossRef]
- Ahad, A.; Ganai, A.A.; Mujeeb, M.; Siddiqui, W.A. Chrysin, an anti-inflammatory molecule, abrogates renal dysfunction in type 2 diabetic rats. Toxicol. Appl. Pharmacol. 2014, 279, 1–7. [Google Scholar] [CrossRef]
- Gong, P.; Chang, X.; Chen, X.; Bai, X.; Wen, H.; Pi, S.; Yang, W.; Wang, L.; Chen, F. Metabolomics study of cadmium-induced diabetic nephropathy and protective effect of caffeic acid phenethyl ester using UPLC-Q-TOF-MS combined with pattern recognition. Environ. Toxicol. Pharmacol. 2017, 54, 80–92. [Google Scholar] [CrossRef]
- Satyanarayana, K.; Sravanthi, K.; Shaker, I.A.; Ponnulakshmi, R.; Selvaraj, J. Role of chrysin on expression of insulin signaling molecules. J. Ayurveda Integr. Med. 2015, 6, 248–258. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, H.; Saito, N.; Fujimoto, J.; Nakashima, K.; Fujikura, D. Brazilian propolis ethanol extract and its component kaempferol induce myeloid-derived suppressor cells from macrophages of mice in vivo and in vitro. BMC Complement. Altern. Med. 2018, 18, 138. [Google Scholar] [CrossRef] [Green Version]
- Aoi, W.; Hosogi, S.; Niisato, N.; Yokoyama, N.; Hayata, H.; Miyazaki, H.; Kusuzaki, K.; Fukuda, T.; Fukui, M.; Nakamura, N.; et al. Improvement of insulin resistance, blood pressure and interstitial pH in early developmental stage of insulin resistance in OLETF rats by intake of propolis extracts. Biochem. Biophys. Res. Commun. 2013, 432, 650–653. [Google Scholar] [CrossRef]
- Zamami, Y.; Fujiwara, H.; Hosoda, M.; Hino, H.; Hirai, K.; Okamoto, K.; Jin, X.; Takatori, S.; Doi-Takaki, S.; Kawasaki, H. Ameliorative effect of propolis on insulin resistance in Otsuka Long-Evans Tokushima Fatty (OLETF) rat. Yakugaku Zasshi 2010, 130, 833–840. [Google Scholar] [CrossRef] [Green Version]
- Winzell, M.S.; Ahrén, B. The high-fat diet-fed mouse. Diabetes 2004, 53, S215–S219. [Google Scholar] [CrossRef] [Green Version]
- Olefsky, J.M.; Glass, C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef]
- Strissel, K.J.; Stancheva, Z.; Miyoshi, H.; Perfield, J.W., II; DeFuria, J.; Jick, Z.; Greenberg, A.S.; Obin, M.S. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 2007, 56, 2910–2918. [Google Scholar] [CrossRef] [Green Version]
- Kahle, M.; Horsch, M.; Fridrich, B.; Seelig, A.; Schultheiß, J.; Leonhardt, J.; Irmler, M.; Beckers, J.; Rathkolb, B.; Wolf, E.; et al. Phenotypic comparison of common mouse strains developing high-fat diet-induced hepatosteatosis. Mol. Metab. 2013, 2, 435–446. [Google Scholar] [CrossRef]
- Magalhães, D.A.; Kume, W.T.; Correia, F.S.; Queiroz, T.S.; Allebrandt Neto, E.W.; Santos, M.P.D.; Kawashita, N.H.; França, S.A. High-fat diet and streptozotocin in the induction of type 2 diabetes mellitus: New proposal. An. Acad. Bras. Cienc. 2019, 91, e20180314. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Chandrasekera, P.C.; Pippin, J.J. Leptin- and leptin receptor-deficient rodent models: Relevance for human type 2 diabetes. Curr. Diabetes Rev. 2014, 10, 131–145. [Google Scholar] [CrossRef] [Green Version]
- Coleman, D.L.; Hummel, K.P. Hyperinsulinemia in pre-weaning diabetes (db) mice. Diabetologia 1974, 10, 607–610. [Google Scholar] [CrossRef] [Green Version]
- Zucker, L.M.; Antoniades, H.N. Insulin and obesity in the Zucker genetically obese rat “fatty”. Endocrinology 1972, 90, 1320–1330. [Google Scholar] [CrossRef]
- Clark, J.B.; Palmer, C.J.; Shaw, W.N. The diabetic Zucker Fatty rats. Proc. Soc. Exp. Biol. Med. 1983, 173, 68–75. [Google Scholar] [CrossRef]
- Takiguchi, S.; Takata, Y.; Funakoshi, A.; Miyasaka, K.; Kataoka, K.; Fujimura, Y.; Goto, T.; Kono, A. Disrupted cholecystokinin type-A receptor (CCKAR) gene in OLETF rats. Gene 1997, 197, 169–175. [Google Scholar] [CrossRef]
- Moran, T.H.; Katz, L.F.; Plata-Salaman, C.R.; Schwartz, G.J. Disordered food intake and obesity in rats lacking cholecystokinin A receptors. Am. J. Physiol. 1998, 274, R618–R625. [Google Scholar] [CrossRef]
- Kawano, K.; Hirashima, T.; Mori, S.; Saitoh, Y.; Kurosumi, M.; Natori, T. Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka Long-Evans Tokushima Fatty (OLETF) strain. Diabetes 1992, 41, 1422–1448. [Google Scholar] [CrossRef]
- Goto, Y.; Kakizaki, M.; Masaki, N. Production of spontaneous diabetic rats by repetition of selective breeding. Tohoku J. Exp. Med. 1976, 119, 85–90. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.W.; Sun, G.D.; Sun, J.; Liu, S.J.; Wang, J.; Xu, X.H.; Miao, L.N. Spontaneous type 2 diabetic rodent models. J. Diabetes Res. 2013, 2013, 401723. [Google Scholar] [CrossRef]
- Agardh, C.D.; Agardh, E.; Zhang, H.; Ostenson, C.G. Altered endothelial/pericyte ratio in Goto-Kakizaki rat retina. J. Diabetes Complications 1997, 11, 158–162. [Google Scholar] [CrossRef]
- Ohneda, M.; Johnson, J.H.; Inman, L.R.; Chen, L.; Suzuki, K.; Goto, Y.; Alam, T.; Ravazzola, M.; Orci, L.; Unger, R.H. GLUT2 expression and function in beta-cells of GK rats with NIDDM. Dissociation between reductions in glucose transport and glucose-stimulated insulin secretion. Diabetes 1993, 42, 1065–1072. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, W.M.T.; Panveloski-Costa, A.C.; Yokota, C.N.F.; Pereira, J.N.B.; Filho, J.M.; Torres, R.P.; Hirabara, S.M.; Curi, R.; Alba-Loureiro, T.C. Comparison of Goto-Kakizaki rats and high fat diet-induced obese rats: Are they reliable models to study type 2 diabetes mellitus? PLoS ONE 2017, 12, e0189622. [Google Scholar] [CrossRef] [PubMed]
- Turer, E.E.; San Miguel, M.; Wang, K.W.; McAlpine, W.; Ou, F.; Li, X.; Tang, M.; Zang, Z.; Wang, J.; Hayse, B.; et al. A viable hypomorphic Arnt2 mutation causes hyperphagic obesity, diabetes and hepatic steatosis. Dis. Model Mech. 2018, 11, dmm035451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Z.; Liu, M.; Chen, Z.; Shao, Y.; Pan, H.; Wei, G.; Yu, C.; Zhang, L.; Li, X.; Wang, P.; et al. High-efficiency and heritable gene targeting in mouse by transcription activator-like effector nucleases. Nucleic Acids Res. 2013, 41, e120. [Google Scholar] [CrossRef] [PubMed]
- Seth, A.; Stemple, D.L.; Barroso, I. The emerging use of zebrafish to model metabolic disease. Dis. Model Mech. 2013, 6, 1080–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Li, X.; Liu, L.; Canet, M.J.; Guan, Y.; Fan, Y.; Zhou, Y. Effect of fasting time on measuring mouse glucose level. Int. J. Clin. Exp. Med. 2016, 9, 4186–4189. [Google Scholar]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Antunes, L.C.; Elkfury, J.L.; Jornada, M.N.; Foletto, K.C.; Bertoluci, M.C. Validation of HOMA-IR in a model of insulin-resistance induced by a high-fat diet in Wistar rats. Arch. Endocrinol. Metab. 2016, 60, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Baribault, H. Mouse models of type 2 diabetes mellitus in drug discovery. Methods Mol. Biol. 2016, 1438, 153–175. [Google Scholar] [CrossRef]
- Andrikopoulos, S.; Blair, A.R.; Deluca, N.; Fam, B.C.; Proietto, J. Evaluating the glucose tolerance test in mice. Am J. Physiol. 2008, 295, E1323–E1332. [Google Scholar] [CrossRef] [Green Version]
- Sadowska-Bartosz, I.; Bartosz, G. Prevention of protein glycation by natural compounds. Molecules 2015, 20, 3309–3334. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.K.; Jeong, J.S.; Kwon, H.S.; Baek, K.H.; Song, K.H. Concordance the hemoglobin glycation index with glycation gap using glycated albumin in patients with type 2 diabetes. J. Diabetes Complicat. 2017, 31, 1127–1131. [Google Scholar] [CrossRef] [PubMed]
- Yazdanpanah, S.; Rabiee, M.; Tahriri, M.; Abdolrahim, M.; Rajab, A.; Jazayeri, H.E.; Tayebi, L. Evaluation of glycated albumin (GA) and GA/HbA1c ratio for diagnosis of diabetes and glycemic control: A comprehensive review. Crit. Rev. Clin. Lab. Sci. 2017, 54, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Beltran Del Rio, M.; Tiwari, M.; Amodu, L.I.; Cagliani, J.; Rodriguez Rilo, H.L. Glycated hemoglobin, plasma glucose, and erythrocyte aging. J. Diabetes Sci. Technol. 2016, 10, 1303–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saudek, C.D.; Kalyani, R.R.; Derr, R.L. Assessment of glycemia in diabetes mellitus: Hemoglobin A1c. J. Assoc. Physicians India 2005, 53, 299–305. [Google Scholar]
- Tahara, Y.; Shima, K. Kinetics of HbA1c, glycated albumin, and fructosamine and analysis of their weight functions against preceding plasma glucose level. Diabetes Care 1995, 18, 440–447. [Google Scholar] [CrossRef]
- Kaneto, H.; Katakami, N.; Matsushita, M.; Matsuoka, T. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediators Inflamm. 2010, 2010, 453892. [Google Scholar] [CrossRef] [Green Version]
- Patche, J.; Girard, D.; Catan, A.; Boyer, F.; Dobi, A.; Planesse, C.; Diotel, N.; Guerin-Dubourg, A.; Baret, P.; Bravo, S.B.; et al. Diabetes-induced hepatic oxidative stress: A new pathogenic role for glycated albumin. Free Radic. Biol. Med. 2017, 102, 133–148. [Google Scholar] [CrossRef]
- Jha, J.C.; Banal, C.; Chow, B.S.; Cooper, M.E.; Jandeleit-Dahm, K. Diabetes and kidney disease: Role of oxidative stress. Antioxid. Redox Signal. 2016, 25, 657–684. [Google Scholar] [CrossRef] [Green Version]
- Muriach, M.; Flores-Bellver, M.; Romero, F.J.; Barcia, J.M. Diabetes and the brain: Oxidative stress, inflammation, and autophagy. Oxid. Med. Cell Longev. 2014, 2014, 102158. [Google Scholar] [CrossRef] [Green Version]
- Fuliang, H.U.; Hepburn, H.R.; Xuan, H.; Chen, M.; Daya, S.; Radloff, S.E. Effects of propolis on blood glucose, blood lipid and free radicals in rats with diabetes mellitus. Pharmacol. Res. 2005, 51, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Tsikas, D. Assessment of lipid peroxidation by measuring malondialdehyde (MDA) and relatives in biological samples: Analytical and biological challenges. Anal. Biochem. 2017, 524, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanwar, M.; Kowluru, R.A. Role of glyceraldehyde 3-phosphate dehydrogenese in the development and progression of diabetic retinopathy. Diabetes 2009, 58, 227–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.Y.; Zhang, F.; Hong, C.Q.; Giuliano, A.E.; Cui, X.J.; Zhou, G.J.; Zhang, G.J.; Cui, Y.K. Critical protein GAPDH and its regulatory mechanisms in cancer cells. Cancer Biol. Med. 2015, 12, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Thornalley, P.J.; Giardino, I.; Beisswenger, P.; Thorpe, S.R.; Onorato, J.; Brownlee, M. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J. Clin. Investig. 1998, 101, 1142–1147. [Google Scholar] [CrossRef] [PubMed]
- Khalifah, R.G.; Baynes, J.W.; Hudson, B.G. Amadorins: Novel post-Amadori inhibitors of advanced glycation reactions. Biochem. Biophys. Res. Commun. 1999, 257, 251–258. [Google Scholar] [CrossRef]
- Morita, M.; Yano, S.; Yamaguchi, T.; Sugimoto, T. Advanced glycation end products-induced reactive oxygen species generation is partly through NF-kappa B activation in human aortic endothelial cells. J. Diabetes Complicat. 2013, 27, 11–15. [Google Scholar] [CrossRef]
- Yamomoto, Y.; Gaynor, R.B. Therapeutic potential of inhibition of the NF-κB pathway in the treatment of inflammation and cancer. J. Clin. Investig. 2001, 107, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.F.; Yan, S.D.; Ramasamy, R.; Schmidt, A.M. Tempering the wrath of RAGE: An emerging therapeutic strategy against diabetic complications, neurodegeneration, and inflammation. Ann. Med. 2009, 41, 408–422. [Google Scholar] [CrossRef] [Green Version]
- Brett, J.; Schmidt, A.M.; Yan, S.D.; Zou, Y.S.; Weidman, E.; Pinsky, D.; Nowygrod, R.; Neeper, M.; Przysiecki, C.; Shaw, A.; et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am. J. Pathol. 1993, 143, 1699–1712. [Google Scholar] [PubMed]
- Bettiga, A.; Fiorio, F.; Di Marco, F.; Trevisani, F.; Romani, A.; Porrini, E.; Salonia, A.; Montorsi, F.; Vago, R. The modern western diet rich in advanced glycation end-products (AGEs): An overview of its impact on obesity and early progression of renal pathology. Nutrients 2019, 11, 1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, I.; Tupe, R.S.; Wallner, A.K.; Kanwar, Y.S. Contribution of myo-inositol oxygenase in AGE:RAGE-mediated renal tubulointerstitial injury in the context of diabetic nephropathy. Am. J. Physiol. 2018, 314, F107–F121. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Joo, J.; Jeon, Y.; Lee, J.; In, J.; Kim, D.; Kang, E.; Kim, Y.; Lim, Y.; Kang, J.; et al. Advanced glycation end products downregulate glucokinase in mice. Diabetes Metab. Res. Rev. 2011, 27, 557–563. [Google Scholar] [CrossRef]
- Vegiopoulos, A.; Rohm, M.; Herzig, S. Adipose tissue: Between the extremes. EMBO J. 2017, 36, 1999–2017. [Google Scholar] [CrossRef]
- Zimmet, P.; Boyko, E.J.; Collier, G.R.; de Courten, M. Etiology of the metabolic syndrome: Potential role of insulin resistance, leptin resistance, and other players. Ann. N. Y. Acad. Sci. 1999, 892, 25–44. [Google Scholar] [CrossRef]
- Kraakman, M.J.; Murphy, A.J.; Jandeleit-Dahm, K.; Kammoun, H.L. Macrophage polarization in obesity and type 2 diabetes: Weighing down our understanding of macrophage function? Front. Immunol. 2014, 5, 470. [Google Scholar] [CrossRef]
- Engin, A.B. Adipocyte-macrophage cross-talk in obesity. Adv. Exp. Med. Biol. 2017, 960, 327–343. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The metabolic signature of macrophage responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [Green Version]
- Saito, N.; Kimura, S.; Miyamoto, T.; Fukushima, S.; Amagasa, M.; Shimamoto, Y.; Nishioka, C.; Okamoto, S.; Toda, C.; Washio, K.; et al. Macrophage ubiquitin-specific protease 2 modifies insulin sensitivity in obese mice. Biochem. Biophys. Rep. 2017, 9, 322–329. [Google Scholar] [CrossRef]
- Taguchi, K.; Okada, A.; Kitamura, H.; Yasui, T.; Naiki, T.; Hamamoto, S.; Ando, R.; Mizuno, K.; Kawai, N.; Tozawa, K.; et al. Colony-stimulating factor-1 suppresses renal crystal formation. J. Am. Soc. Nephrol. 2014, 25, 1680–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, T.J.; Skiba, D.S.; Touyz, R.M.; Harrison, D.G. The role of infiltrating immune cells in dysfunctional adipose tissue. Cardiovasc. Res. 2017, 113, 1009–1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, H.; Kimura, S.; Shimamoto, Y.; Okabe, J.; Ito, M.; Miyamoto, T.; Naoe, Y.; Kikuguchi, C.; Meek, B.; Toda, C.; et al. Ubiquitin-specific protease 2-69 in macrophages potentially modulates metainflammation. FASEB J. 2013, 27, 4940–4953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, L.J.; Lee, H.B.; Bae, H.J.; Lee, S.G. Antidiabetic effect of propolis: Reduction of expression of glucose-6-phosphatase through inhibition of Y279 and Y216 autophosphorylation of GSK-3α/β in HepG2 cells. Phytother. Res. 2010, 24, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, X.; Zhang, G.; Kong, L.; Peng, W.; Zhang, H. Galangin and pinocembrin from propolis ameliorate insulin resistance in HepG2 cells via regulating Akt/mTOR signaling. Evid. Based Complement. Alternat. Med. 2018, 2018, 7971842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, K.J.; Han, H.S.; Kim, M.J.; Koo, S.H. CREB and FoxO1: Two transcription factors for regulation of hepatic gluconeogenesis. BMB Rep. 2013, 46, 567–574. [Google Scholar] [CrossRef] [Green Version]
- Liberman, Z.; Eldar-Finkelman, H. Serine 332 phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 attenuates insulin signaling. J. Biol. Chem. 2005, 280, 4422–4428. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S.; Takamura, T.; Matsuzawa-Nagata, N.; Takayama, H.; Misu, H.; Noda, H.; Nabemoto, S.; Kurita, S.; Ota, T.; Ando, H.; et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J. Biol. Chem. 2009, 284, 14809–14818. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Nong, S.; Huang, X.; Lu, Y.; Zhao, H.; Lin, Y.; Man, Y.; Wang, S.; Yang, J.; Li, J. The effects of palmitate on hepatic insulin resistance are mediated by NADPH Oxidase 3-derived reactive oxygen species through JNK and p38MAPK pathways. J. Biol. Chem. 2010, 285, 29965–29973. [Google Scholar] [CrossRef] [Green Version]
- Sinacore, D.R.; Gulve, E.A. The role of skeletal muscle in glucose transport, glucose homeostasis, and insulin resistance: Implications for physical therapy. Phys. Ther. 1993, 73, 878–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snell, K. Muscle alanine synthesis and hepatic gluconeogenesis. Biochem. Soc. Trans. 1980, 8, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, A.F.; Fazakerley, D.J.; James, D.E. Mapping insulin/GLUT4 circuitry. Traffic 2011, 12, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Habegger, K.M.; Hoffman, N.J.; Ridenour, C.M.; Brozinick, J.T.; Elmendorf, J.S. AMPK enhances insulin-stimulated GLUT4 regulation via lowering membrane cholesterol. Endocrinology 2012, 153, 2130–2141. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M.; Hayashibara, K.; Ashida, H. Propolis extract promotes translocation of glucose transporter 4 and glucose uptake through both PI3K- and AMPK-dependent pathways in skeletal muscle. Biofactors 2013, 39, 457–466. [Google Scholar] [CrossRef]
- Washio, K.; Kobayashi, M.; Saito, N.; Amagasa, M.; Kitamura, H. Propolis ethanol extract stimulates cytokine and chemokine production through NF-κB cctivation in C2C12 myoblasts. Evid. Based Complement. Alternat. Med. 2015, 2015, 349751. [Google Scholar] [CrossRef] [Green Version]
- Hamaguchi, K.; Gaskins, H.R.; Leiter, E.H. NIT-1, a pancreatic β-cell line established from a transgenic NOD/Lt mouse. Diabetes 1991, 40, 842–849. [Google Scholar] [CrossRef]
- Yang, S.Y.; Lee, J.J.; Lee, J.H.; Lee, K.; Oh, S.H.; Lim, Y.M.; Lee, M.S.; Lee, K.J. Secretagogin affects insulin secretion in pancreatic β-cells by regulating actin dynamics and focal adhesion. Biochem. J. 2016, 473, 1791–1803. [Google Scholar] [CrossRef] [Green Version]
- QiNan, W.; XiaGuang, G.; XiaoTian, L.; WuQuan, D.; Ling, Z.; Bing, C. Par-4/NF-κB mediates the apoptosis of islet β cells induced by glucolipotoxicity. J. Diabetes Res. 2016, 2016, 4692478. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, Q.; Huan, Y.; Li, R.; Li, C.; Sun, S.; Guo, N.; Yang, M.; Liu, S.; Shen, Z. Sirtuin 5 overexpression attenuates glucolipotoxicity-induced pancreatic β cells apoptosis and dysfunction. Exp. Cell Res. 2018, 371, 205–213. [Google Scholar] [CrossRef]
- Yuan, H.; Lu, Y.; Huang, X.; He, Q.; Man, Y.; Zhou, Y.; Wang, S.; Li, J. Suppression of NADPH oxidase 2 substantially restores glucose-induced dysfunction of pancreatic NIT-1 cells. FEBS J. 2010, 277, 5061–5071. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.K.; Tesch, G.H. Inflammation in diabetic nephropathy. Mediators Inflamm. 2012, 2012, 146154. [Google Scholar] [CrossRef] [PubMed]
- Zubair, M.; Ahmad, J. Role of growth factors and cytokines in diabetic foot ulcer healing: A detailed review. Rev. Endocr. Metab. Disord. 2019, 20, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Martinelli, G.; Soldano, S.; Paolino, S.; Pacini, G.; Patane, M.; Alessandri, E.; Smith, V.; Cutolo, M. Macrophage M1/M2 polarization and rheumatoid arthritis: A systemic review. Autoimmun. Rev. 2019, 18, 102397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemonnot, A.L.; Hua, L.; Ulmann, L.; Hirbec, H. Microglia in Alzheimer disease: Well-known targets and new opportunities. Front. Aging Neurosci. 2019, 11, 233. [Google Scholar] [CrossRef] [Green Version]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, mechanisms, and significance of macrophage plasticity. Ann. Rev. Pathol. 2019, in press. [Google Scholar] [CrossRef]
- Bachiega, T.F.; Orsatti, C.L.; Pagliarone, A.C.; Sforcin, J.M. The effects of propolis and its isolated compounds on cytokine production by murine macrophages. Phytother. Res. 2012, 26, 1308–1313. [Google Scholar] [CrossRef]
- Jabir, M.S.; Sulaiman, G.M.; Taqi, Z.J.; Li, D. Iraqi propolis increases degradation of IL-1β and NLRC4 by autophagy following Pseudomonas aeruginosa infection. Microbes Infect. 2018, 20, 89–100. [Google Scholar] [CrossRef]
- Zhang, J.; Cao, X.; Ping, S.; Wang, K.; Shi, J.; Zhang, C.; Zheng, H.; Hu, F. Comparisons of ethanol extracts of chinese propolis (poplar type) and poplar gums based on the antioxidant activities and molecular mechanism. Evid. Based Complement. Alternat. Med. 2015, 2015, 307594. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wu, Z.; Zhang, X.; Ni, J.; Yu, W.; Zhou, Y.; Nakanishi, H. Leptomeningeal cells transduce peripheral macrophages inflammatory signal to microglia in reponse to Porphyromonas gingivalis LPS. Mediators Inflamm. 2013, 2013, 407562. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, P.A.; Coghlan, M.; Rice, S.Q.; Sutherland, C. Inhibition of GSK-3 selectively reduces glucose-6-phosphatase and phosphatase and phosphoenolypyruvate carboxykinase gene expression. Diabetes 2001, 50, 937–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkhondeh, T.; Abedi, F.; Samarghandian, S. Chrysin attenuates inflammatory and metabolic disorder indices in aged male rat. Biomed. Pharmaother. 2019, 109, 1120–1125. [Google Scholar] [CrossRef] [PubMed]
- El-Bassossy, H.M.; Abo-Warda, S.M.; Fahmy, A. Chrysin and luteolin attenuate diabetes-induced impairment in endothelial-dependent relaxation: Effect on lipid profile, AGEs and NO generation. Phytother. Res. 2013, 27, 1678–1684. [Google Scholar] [CrossRef]
- Kim, J.S.; Kwon, C.S.; Son, K.H. Inhibition of alpha-glucosidase and amylase by luteolin, a flavonoid. Biosci. Biotechnol. Biochem. 2000, 64, 2458–2461. [Google Scholar] [CrossRef] [PubMed]
- Li, J.M.; Che, C.T.; Lau, C.B.; Leung, P.S.; Cheng, C.H. Inhibition of intestinal and renal Na+-glucose cotransporter by naringenin. Int. J. Biochem. Cell Biol. 2006, 38, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Umeda, M.; Hiramoto, M.; Watanabe, A.; Tsunoda, N.; Imai, T. Arginine-induced insulin secretion in endoplasmic reticulum. Biochem. Biophys. Res. Commun. 2015, 466, 717–722. [Google Scholar] [CrossRef] [Green Version]
- Oršolić, N.; Sirovina, D.; Gajski, G.; Garaj-Vrhovac, V.; Jazvinšćak Jembrek, M.; Kosalec, I. Assessment of DNA damage and lipid peroxidation in diabetic mice: Effects of propolis and epigallocatechin gallate (EGCG). Mutat. Res. 2013, 757, 36–44. [Google Scholar] [CrossRef]
- El-Awady, M.S.; El-Agamy, D.S.; Suddek, G.M.; Nader, M.A. Propolis protects against high glucose-induced vascular endothelial dysfunction in isolated rat aorta. J. Physiol. Biochem. 2014, 70, 247–254. [Google Scholar] [CrossRef]
- Sorrenti, V.; Raffaele, M.; Vanella, L.; Acquaviva, R.; Salerno, L.; Pittalà, V.; Intagliata, S.; Di Giacomo, C. Protective effects of caffeic acid phenethyl ester (CAPE) and novel Cape analogue as inducers of heme oxygenase-1 in streptozotocin-induced type 1 diabetic rats. Int. J. Mol. Sci. 2019, 20, 2441. [Google Scholar] [CrossRef] [Green Version]
- Fuke, N.; Nagata, N.; Suganuma, H.; Ota, T. Regulation of gut microbiota and metabolic endotoxemia with dietary factors. Nutrients 2019, 10, 2277. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, M.; Arimatsu, K.; Minagawa, T.; Matsuda, Y.; Sato, K.; Takahashi, N.; Nakajima, T.; Yamazaki, K. Brazilian propolis mitigates impaired glucose and lipid metabolism in experimental periodontitis in mice. BMC Complement. Altern. Med. 2016, 16, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Molofsky, A.B.; Liang, H.E.; Ricardo-Gonzalez, R.R.; Jouihan, H.A.; Bando, J.K.; Chawla, A.; Locksley, R.M. Eosinophils sustain adipose alternatively activated macrophages associated with glucose homeostasis. Science 2011, 332, 243–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izcue, A.; Coombes, J.L.; Powrie, F. Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol. Rev. 2006, 212, 256–271. [Google Scholar] [CrossRef] [PubMed]
- Borriello, M.; Iannuzzi, C.; Sirangelo, I. Pinocembrin protects from AGE-induced cytotoxicity and inhibits non-enzymatic glycation in human insulin. Cells 2019, 8, 385. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Kanazashi, M.; Maeshige, N.; Kondo, H.; Ishihara, A.; Fujino, H. Protective effects of Brazilian propolis supplementation on capillary regression in the soleus muscle of hindlimb-unloaded rats. J. Physiol. Sci. 2019, 69, 223–233. [Google Scholar] [CrossRef]
- Izuta, H.; Shimazawa, M.; Tsuruma, K.; Araki, Y.; Mishima, S.; Hara, H. Bee products prevent VEGF-induced angiogenesis in human umbilical vein endothelial cells. BMC Complement. Altern. Med. 2009, 9, 45. [Google Scholar] [CrossRef] [Green Version]
- Sena, C.M.; Pereira, A.M.; Seiça, R. Endothelial dysfunction - a major mediator of diabetic vascular disease. Biochim. Biophys. Acta 2013, 132, 2216–2231. [Google Scholar] [CrossRef] [Green Version]
- Soulis, T.; Thallas, V.; Youssef, S.; Gilbert, R.E.; McWilliam, B.G.; Murray-McIntosh, R.P.; Cooper, M.E. Advanced glycation end products and their receptors co-localise in rat organs susceptible to diabetic microvascular injury. Diabetologia 1997, 40, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Czech, M.P. Insulin action and resistance in obesity and type 2 diabetes. Nat. Med. 2017, 23, 804–814. [Google Scholar] [CrossRef]
- Raz, I.; Eldor, R.; Cernea, S.; Shafrir, E. Diabetes: Insulin resistance and derangements in lipid metabolism. Cure through intervention in fat transport and storage. Diabetes Metab. Res. Rev. 2005, 21, 3–14. [Google Scholar] [CrossRef]
- Kandimalla, R.; Thirumala, V.; Reddy, P.H. Is Alzheimer’s disease a type 3 diabetes? A critical appraisal. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998, 352, 837–853. [Google Scholar] [CrossRef]
- Makita, Z.; Radoff, S.; Rayfield, E.J.; Yang, Z.; Skolnik, E.; Delaney, V.; Friedman, E.A.; Cerami, A.; Vlassara, H. Advanced glycosylation end products in patients with diabetic nephropathy. New Engl. J. Med. 1991, 325, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Tesch, G.; Sourris, K.C.; Summers, S.A.; McCarthy, D.; Ward, M.S.; Borg, D.J.; Gallo, L.A.; Fotheringham, A.K.; Pettit, A.R.; Yap, F.Y.; et al. Deletion of bone-marrow-derived receptor for AGEs (RAGE) improves renal function in an experimental mouse model of diabetes. Diabetologia 2014, 57, 1977–1985. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Higashimoto, Y.; Nishino, Y.; Nakamura, N.; Fukami, K.; Yamagishi, S.I. RAGE-aptamer blocks the development and progression of experimental diabetic nephropathy. Diabetes 2017, 66, 1683–1695. [Google Scholar] [CrossRef] [Green Version]
- Brouwers, O.; Niessen, P.M.; Miyata, T.; Østergaard, J.A.; Flyvbjerg, A.; Peutz-Kootstra, C.J.; Sieber, J.; Mundel, P.H.; Brownlee, M.; Janssen, B.J.; et al. Glyoxalase-1 overexpression reduces endothelial dysfunction and attenuates early renal impairment in a rat model of diabetes. Diabetologia 2014, 57, 224–235. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Chen, Y.; Tan, X.; Zhang, L.; Zhang, H.; Li, Z.; Liu, S.; Li, R.; Lin, T.; Liao, R.; et al. Advanced glycation end-products suppress autophagic flux in podocytes by activating mammalian target of rapamycin and inhibiting nuclear translocation of transcription factor EB. J. Pathol. 2018, 245, 235–248. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Li, C.; Cai, L. Advanced glycation end-products induce connective tissue growth factor-mediated renal fibrosis predominantly through transforming growth factor beta-independent pathway. Am. J. Pathol. 2004, 165, 2033–2043. [Google Scholar] [CrossRef]
- Tuttle, K.R.; Johnson, E.C.; Cooney, S.K.; Anderberg, R.J.; Johnson, E.K.; Clifton, G.D.; Meek, R.L. Amino acids injure mesangial cells by advanced glycation end products, oxidative stress, and protein kinase C. Kidney Int. 2005, 67, 953–968. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Huang, J.; Xie, X.; Wang, S.; Chen, C.; Shen, X.; Liu, P.; Huang, H. Sirt1 resists advanced glycation end products-induced expressions of fibronectin and TGF-β1 by activating the Nrf2/ARE pathway in glomerular mesangial cells. Free Radic. Biol. Med. 2013, 65, 528–540. [Google Scholar] [CrossRef]
- Chen, C.; Gong, W.; Li, C.; Xiong, F.; Wang, S.; Huang, J.; Wang, Y.; Chen, Z.; Chen, Q.; Liu, P.; et al. Sphingosine kinase 1 mediates AGEs-induced fibronectin upregulation in diabetic nephropathy. Oncotarget 2017, 8, 78660–78676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chuang, P.Y.; Yu, Q.; Fang, W.; Uribarri, J.; He, J.C. Advanced glycation endproducts induce podocyte apoptosis by activation of the FOXO4 transcription factor. Kidney Int. 2007, 72, 965–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondeva, T.; Wojciech, S.; Wolf, G. Advanced glycation end products inhibit adhesion ability of differentiated podocytes in a neuropilin-1-dependent manner. Am. J. Physiol. 2011, 301, F852–F870. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.A.; Welsh, G.I.; Raghu, G.; Menon, R.K.; Saleem, M.A.; Reddy, G.B. Carboxymethyl lysine induces EMT in podocytes through transcription factor ZEB2: Implications for podocyte depletion and proteinuria in diabetes mellitus. Arch. Biochem. Biophys. 2016, 590, 10–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hills, C.E.; Squires, P.E. The role of TGF-β and epithelial-to mesenchymal transition in diabetic nephropathy. Cytokine Growth Factor Rev. 2011, 22, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Fong, D.S.; Aiello, L.; Gardner, T.W.; King, G.L.; Blankenship, G.; Cavallerano, J.D.; Ferris, F.L., 3rd; Klein, R.; American Diabetes Association. Retinopathy in diabetes. Diabetes Care 2004, 27 (Suppl. 1), S84–S87. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Miller, C.M.; Kern, T.S. Hyperglycemia increases mitochondrial superoxide in retina and retinal cells. Free Radic. Biol. Med. 2003, 35, 1491–1499. [Google Scholar] [CrossRef]
- Price, T.O.; Sheibani, N.; Shah, G.N. Regulation of high glucose-induced apoptosis of brain pericytes by mitochondrial CA VA: A specific target for prevention of diabetic cerebrovascular pathology. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 929–935. [Google Scholar] [CrossRef]
- Yamagishi, S.; Okamoto, T.; Amano, S.; Inagaki, Y.; Koga, K.; Koga, M.; Choei, H.; Sasaki, N.; Kikuchi, S.; Takeuchi, M.; et al. Palmitate-induced apoptosis of microvascular endothelial cells and pericytes. Mol. Med. 2002, 8, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Mizutani, M.; Kern, T.S.; Lorenzi, M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J. Clin. Investig. 1996, 97, 2883–2890. [Google Scholar] [CrossRef]
- Barot, M.; Gokulgandhi, M.R.; Patel, S.; Mitra, A.K. Microvascular complications and diabetic retinopathy: Recent advances and future implications. Future Med. Chem. 2013, 5, 301–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.K.; Lee, E.J.; Kim, Y.H.; Kim, D.Y.; Oh, H.; Kim, S.I.; Kang, Y.H. Chrysin ameliorates malfunction of retinoid visual cycle through blocking activation of AGE-RAGE-ER stress in glucose-stimulated retinal pigment epithelial cells and diabetic eyes. Nutrients 2018, 10, 1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, W.; An, Y.; He, X.; Zhang, D.; He, W. Protection of kaempferol on oxidative stress-induced retinal pigment epithelial cell damage. Oxid. Med. Cell. Longev. 2018, 2018, 1610751. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.; Nissen, E.; Geiger, A. Migratory, metabolic and functional alterations of fibrocytes in type 2 diabetes. IUBMB Life 2018, 70, 1122–1132. [Google Scholar] [CrossRef]
- Yoshida, S.; Matsumoto, K.; Tomioka, D.; Bessho, K.; Itami, S.; Yoshikawa, K.; Nakamura, T. Recombinant hepatocyte growth factor accelerates cutaneous wound healing in a diabetic mouse model. Growth Factors 2004, 22, 111–119. [Google Scholar] [CrossRef]
- Qi, W.; Poronnik, P.; Young, B.; Jackson, C.J.; Field, M.J.; Pollock, C.A. Human cortical fibroblast responses to high glucose and hypoxia. Nephron Physiol. 2004, 96, 121–129. [Google Scholar] [CrossRef]
- Okonkwo, U.; DiPietro, L.A. Diabetes and wound angiogenesis. Int. J. Mol. Sci. 2017, 18, 1419. [Google Scholar] [CrossRef] [Green Version]
- Garraud, O.; Hozzein, W.H.; Badr, G. Wound healing: Time to look for intelligent, “natural” immunological approaches? BMC Immunol. 2017, 18, 23. [Google Scholar] [CrossRef] [Green Version]
- Portillo-Sanchez, P.; Bril, F.; Maximos, M.; Lomonaco, R.; Biernacki, D.; Orsak, B.; Subbarayan, S.; Webb, A.; Hecht, J.; Cusi, K. High prevalence of nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus and normal plasma aminotransferase levels. J. Clin. Endocrinol. Metab. 2015, 100, 2231–2238. [Google Scholar] [CrossRef]
- Hansen, H.H.; Feigh, M.; Veidal, S.S.; Rigbolt, K.T.; Vrang, N.; Fosgerau, K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov. Today 2017, 22, 1701–1718. [Google Scholar] [CrossRef]
- Pai, S.A.; Munshi, R.P.; Panchal, F.H.; Gaur, I.S.; Juvekar, A.R. Chrysin ameliorates nonalcoholic fatty liver disease in rats. Naunyn Schmiedebergs Arch. Pharmacol. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Kismet, K.; Ozcan, C.; Kuru, S.; Gencay Celemli, O.; Celepli, P.; Senes, M.; Guclu, T.; Sorkun, K.; Hucumenoglu, S.; Besler, T. Does propolis have any effect on non-alcoholic fatty liver disease? Biomed. Pharmacother. 2017, 90, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Zhang, T.; Lou, G.; Xu, W.; Dong, F.; Chen, G.; Liu, Y. Plasma miR-17, miR-20a, miR-20b and miR-122 as potential biomarkers for diagnosis of NAFLD in type 2 diabetes mellitus patients. Life Sci. 2018, 208, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Zakerkish, M.; Jenabi, M.; Zaeemzadeh, N.; Hemmati, A.A.; Neisi, N. The effect of Iranian propolis on glucose metabolism, lipid profile, insulin resistance, renal function and inflammatory biomarkers in patients with type 2 diabetes mellitus: A randomized double-blind clinical trial. Sci. Rep. 2019, 9, 7289. [Google Scholar] [CrossRef]
- Samadi, N.; Mozaffari-Khosravi, H.; Rahmanian, M.; Askarishahi, M. Effects of bee propolis supplementation on glycemic control, lipid profile and insulin resistance indices in patients with type 2 diabetes: A randomized, double-blind clinical trial. J. Integr. Med. 2017, 15, 124–134. [Google Scholar] [CrossRef]
- Afsharpour, F.; Javadi, M.; Hashemipour, S.; Koushan, Y.; Haghighian, H.K. Propolis supplementation improves glycemic and antioxidant status in patients with type 2 diabetes: A randomized, double-blind, placebo-controlled study. Complement. Ther. Med. 2019, 43, 283–288. [Google Scholar] [CrossRef]
- Hesami, S.; Hashemipour, S.; Shiri-Shahsavar, M.R.; Koushan, Y.; Khadem Haghighian, H. Administration of Iranian Propolis attenuates oxidative stress and blood glucose in type II diabetic patients: A randomized, double-blind, placebo-controlled, clinical trial. Caspian J. Intern. Med. 2019, 10, 48–54. [Google Scholar] [CrossRef]
- Gao, W.; Pu, L.; Wei, J.; Yao, Z.; Wang, Y.; Shi, T.; Zhao, L.; Jiao, C.; Guo, C. Serum antioxidant parameters are significantly increased in patients with type 2 diabetes mellitus after consumption of Chinese propolis: A randomized controlled trial based on basting serum glucose level. Diabetes Ther. 2018, 9, 101–111. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, T.; Fukui, M.; Tanaka, M.; Senmaru, T.; Iwase, H.; Yamazaki, M.; Aoi, W.; Inui, T.; Nakamura, N.; Marunaka, Y. Effects of Brazilian green propolis in patients with type 2 diabetes: A double-blind randomized placebo-controlled study. Biomed. Rep. 2015, 3, 355–360. [Google Scholar] [CrossRef]
- Zhao, L.; Pu, L.; Wei, J.; Li, J.; Wu, J.; Xin, Z.; Gao, W.; Guo, C. Brazilian green propolis improves antioxidant function in patients with type 2 diabetes mellitus. Int. J. Environ. Res. Public Health 2016, 13, 498. [Google Scholar] [CrossRef] [Green Version]
- El-Sharkawy, H.M.; Anees, M.M.; Van Dyke, T.E. Propolis improves periodontal status and glycemic control in patients with type 2 diabetes mellitus and chronic periodontitis: A randomized clinical trial. J. Periodontal. 2016, 87, 1418–1426. [Google Scholar] [CrossRef] [PubMed]
- Silveira, M.A.D.; Teles, F.; Berretta, A.A.; Sanches, T.R.; Rodrigues, C.E.; Seguro, A.C.; Andrade, L. Effects of Brazilian green propolis on proteinuria and renal function in patients with chronic kidney disease: A randomized, double-blind, placebo-controlled trial. BMC Nephrol. 2019, 20, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henshaw, F.R.; Bolton, T.; Nube, V.; Hood, A.; Veldhoen, D.; Pfrunder, L.; McKew, G.L.; Macleod, C.; McLennan, S.V.; Twigg, S.M. Topical application of the bee hive protectant propolis is well tolerated and improves human diabetic foot ulcer healing in a prospective feasibility study. J. Diabetes Complicat. 2014, 28, 850–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afkhamizadeh, M.; Aboutorabi, R.; Ravari, H.; Fathi Najafi, M.; Ataei Azimi, S.; Javadian Langaroodi, A.; Yaghoubi, M.A.; Sahebkar, A. Topical propolis improves wound healing in patients with diabetic foot ulcer: A randomized controlled trial. Nat. Prod. Res. 2018, 32, 2096–2099. [Google Scholar] [CrossRef] [PubMed]
- Mujica, V.; Orrego, R.; Fuentealba, R.; Leiva, E.; Zúñiga-Hernández, J. Propolis as an adjuvant in the healing of human diabetic foot wounds receiving care in the diagnostic and treatment centre from the regional hospital of Talca. J. Diabetes Res. 2019, 2019, 2507578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ (accessed on 24 November 2019).
- Bankova, V. Chemical diversity of propolis and the problem of standardization. J. Ethnopharmacol. 2005, 100, 114–117. [Google Scholar] [CrossRef]
- Popova, M.; Bankova, V.; Butovska, D.; Petkov, V.; Nikolova-Damyanova, B.; Sabatini, A.G.; Marcazzan, G.L.; Bogdanov, S. Validated methods for the quantification of biologically active constituents of poplar-type propolis. Phytochem. Anal. 2004, 15, 235–240. [Google Scholar] [CrossRef]
- Popova, M.P.; Graikou, K.; Chinou, I.; Bankova, V.S. GC-MS profiling of diterpene compounds in Mediterranean propolis from Greece. J. Agric. Food Chem. 2010, 58, 3167–3176. [Google Scholar] [CrossRef]
- Georgieva, K.; Popova, M.; Dimitrova, L.; Trusheva, B.; Thanh, L.N.; Phuong, D.T.L.; Lien, N.T.P.; Najdenski, H.; Bankova, V. Phytochemical analysis of Vietnamese propolis produced by the stingless bee Lisotrigona cacciae. PLoS ONE 2019, 14, e0216074. [Google Scholar] [CrossRef] [Green Version]
- Solorzano, E.R.; Di Gangi, I.M.; Roverso, M.; Favaro, G.; Bogialli, S. Low level of allergens in the Argentinean plant Zuccagnia punctata Cav: Screening and quality control of north-western propolis using an LC-DAD-QTOF system. Appl. Sci. 2019, 9, 3546. [Google Scholar] [CrossRef] [Green Version]
- Corazza, M.; Borghi, A.; Gallo, R.; Schena, D.; Pigatto, P.; Lauriola, M.M.; Guarneri, F.; Stingeni, L.; Vincenzi, C.; Foti, C.; et al. Topical botanically derived products: Use, skin reactions, and usefulness of patch tests. A multicentre Italian study. Contact Dermat. 2014, 70, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Hata, T.; Tazawa, S.; Ohta, S.; Rhyu, M.R.; Misaka, T.; Ichihara, K. Artepillin C, a major ingredient of Brazilisn propolis, induces a pungent taste by activating TRPA1 channels. PLoS ONE 2012, 7, e48072. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Jia, J.; Xie, C.; Wu, Y.; Tu, W. Transient receptor potential ankyrin 1 and substance P mediate the development of gastric mucosal lesions in a water immersion restraint stress rat model. Digestion 2018, 97, 228–239. [Google Scholar] [CrossRef] [PubMed]







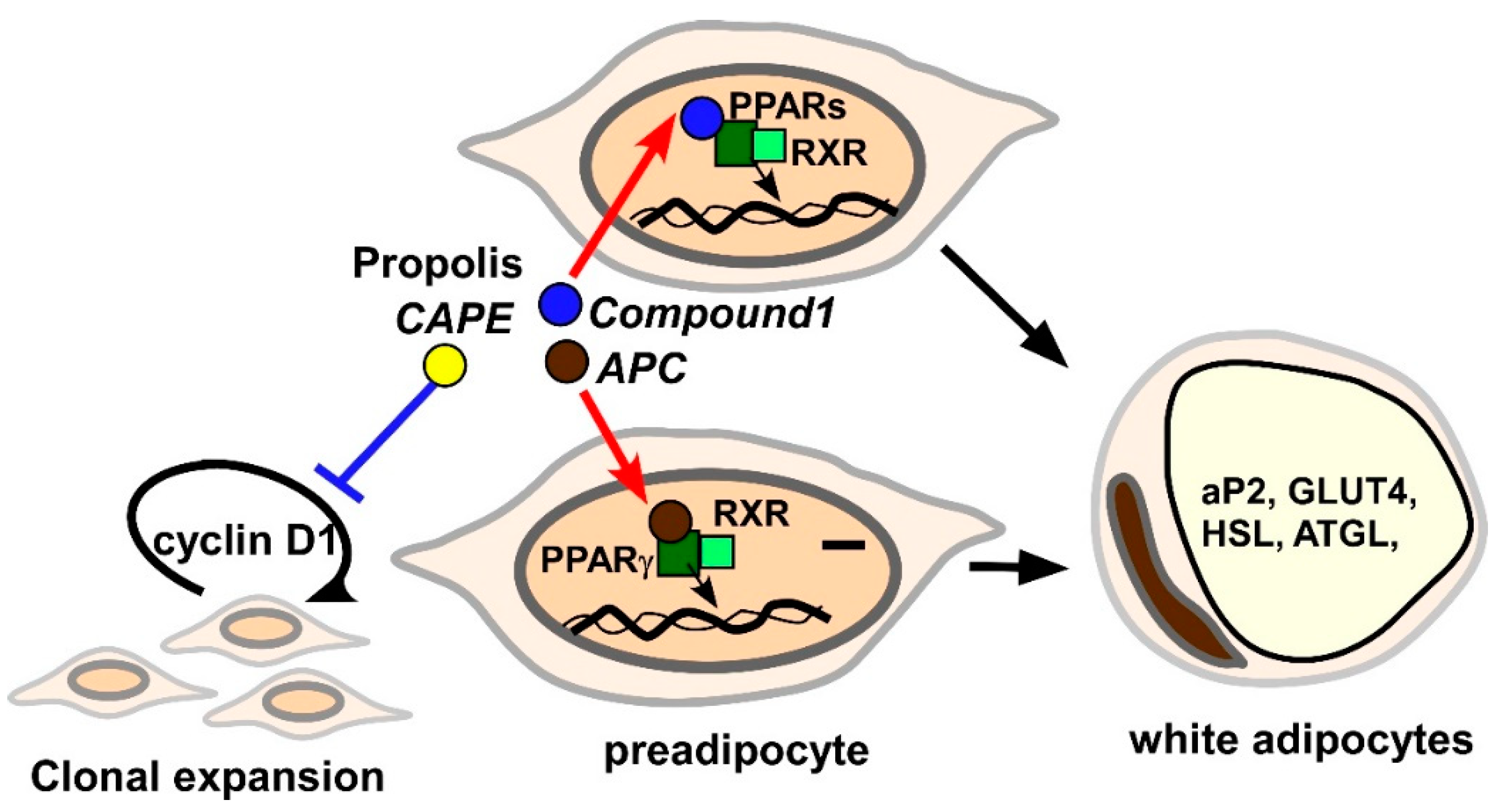{kind=link}
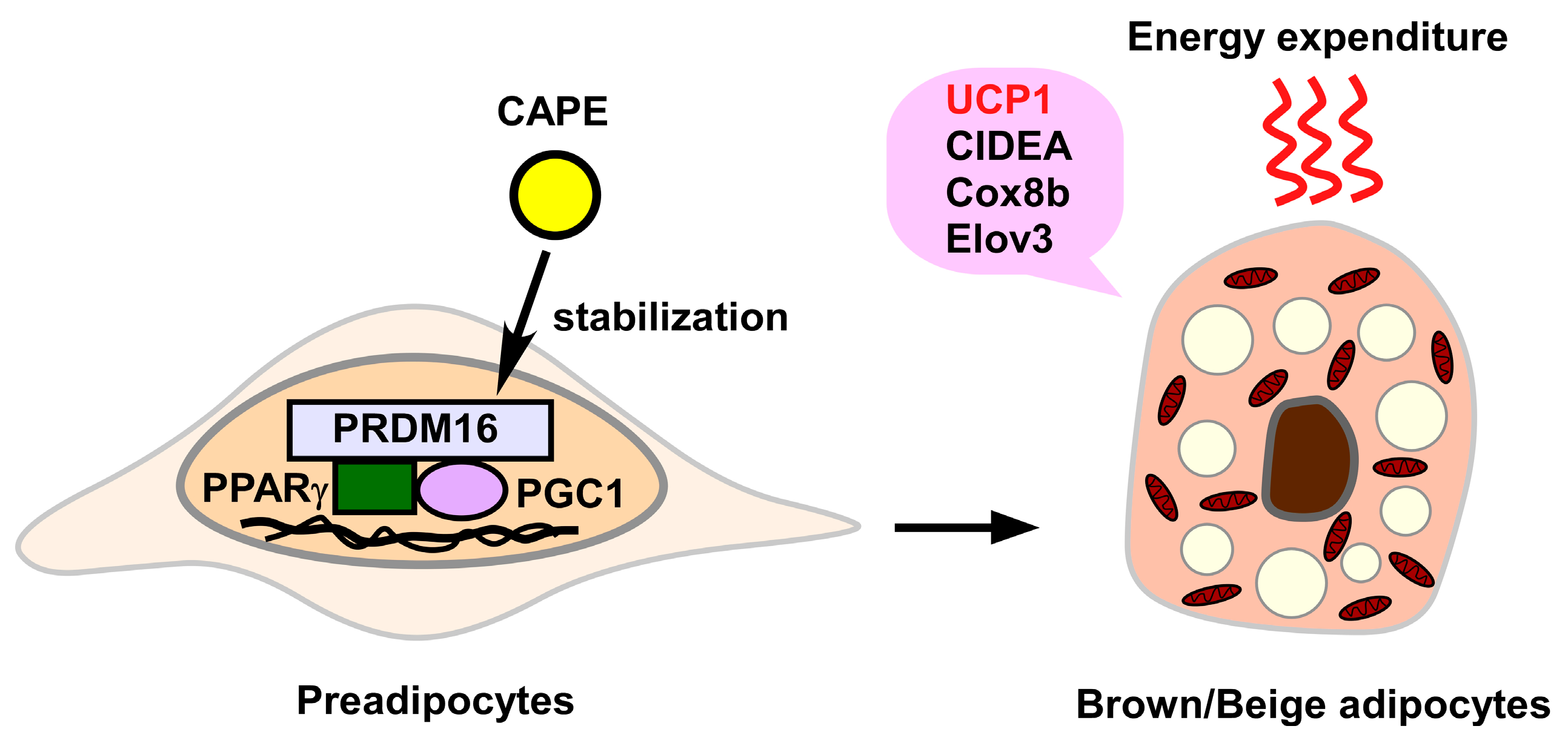{kind=link}
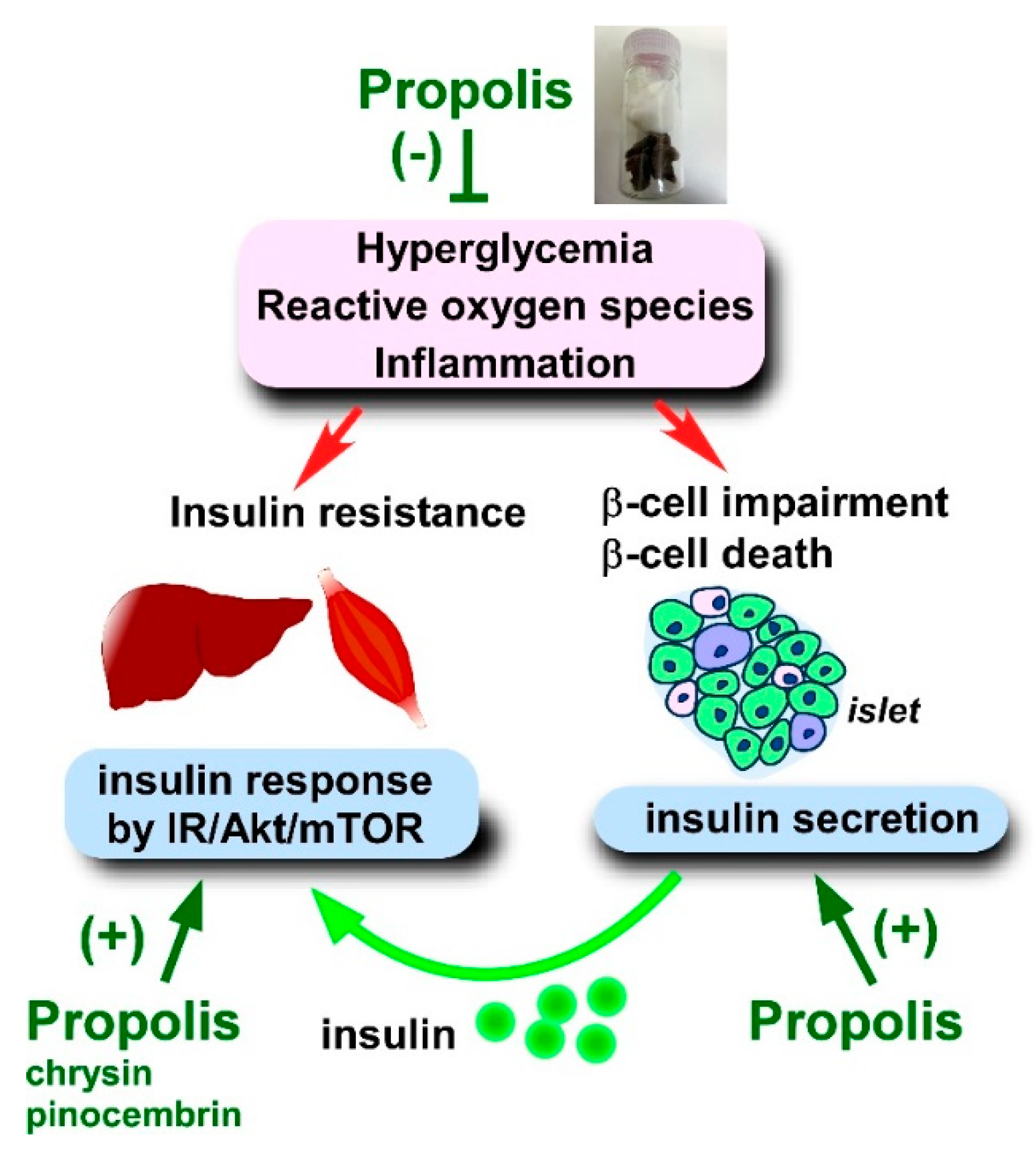{kind=link}
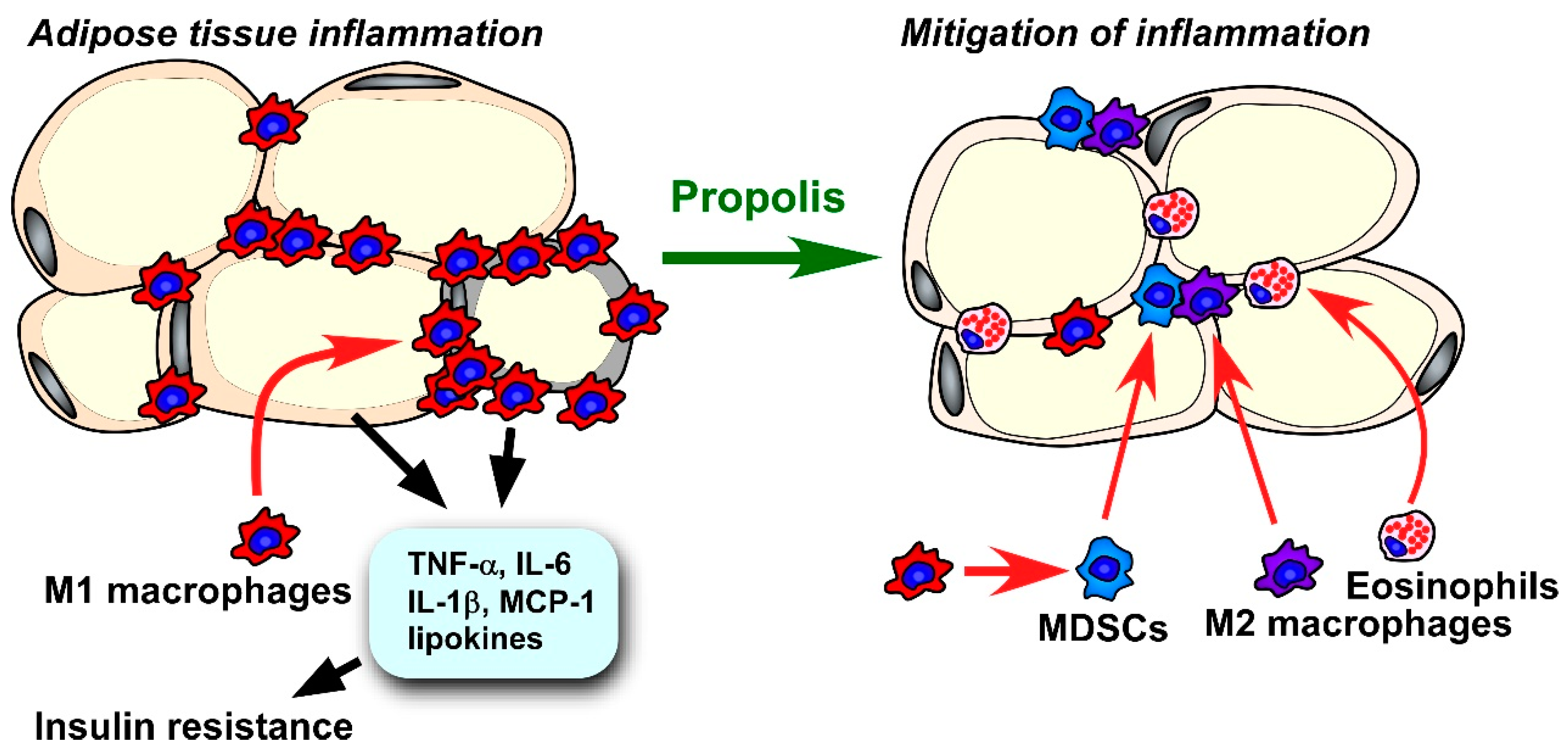{kind=link}
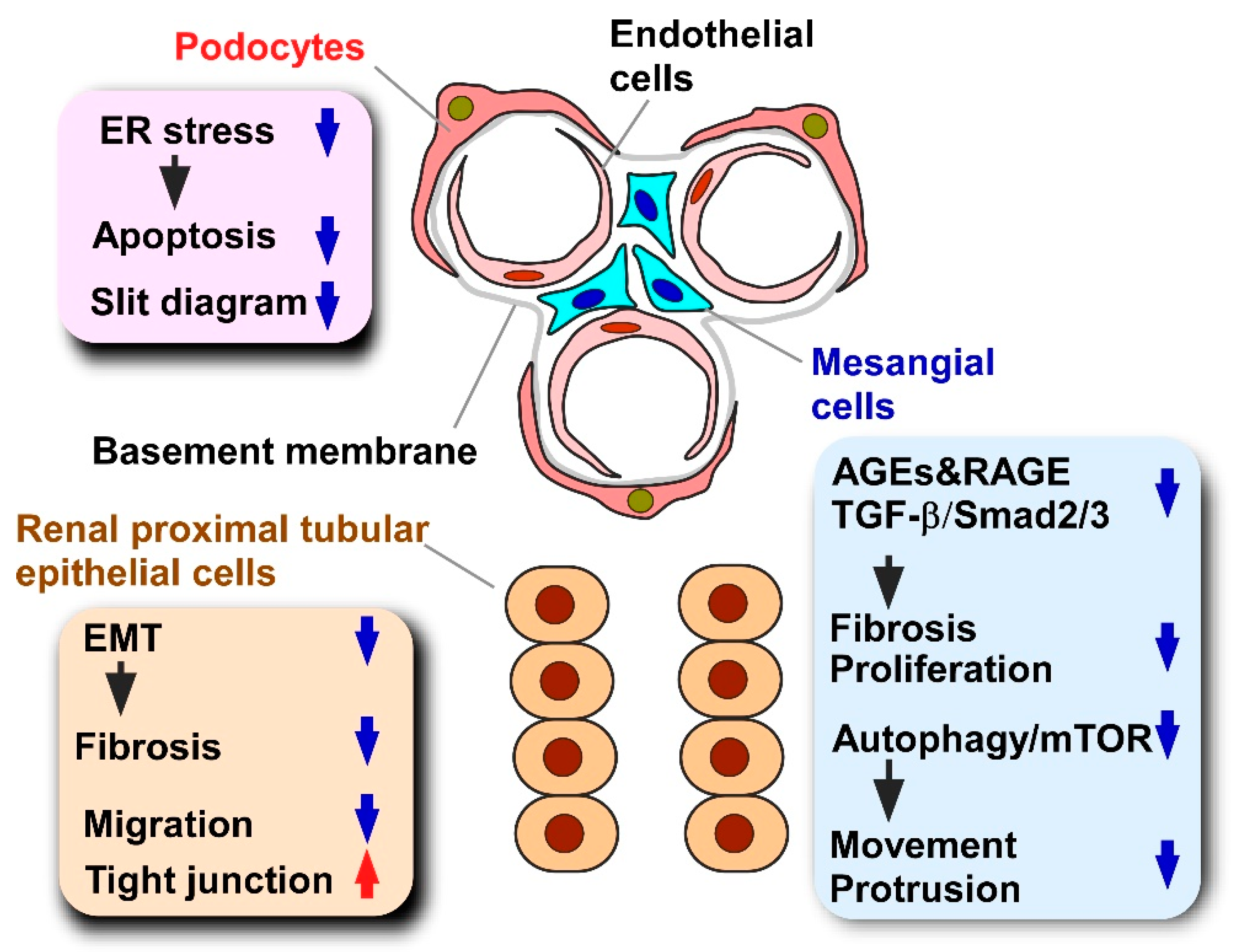{kind=link}
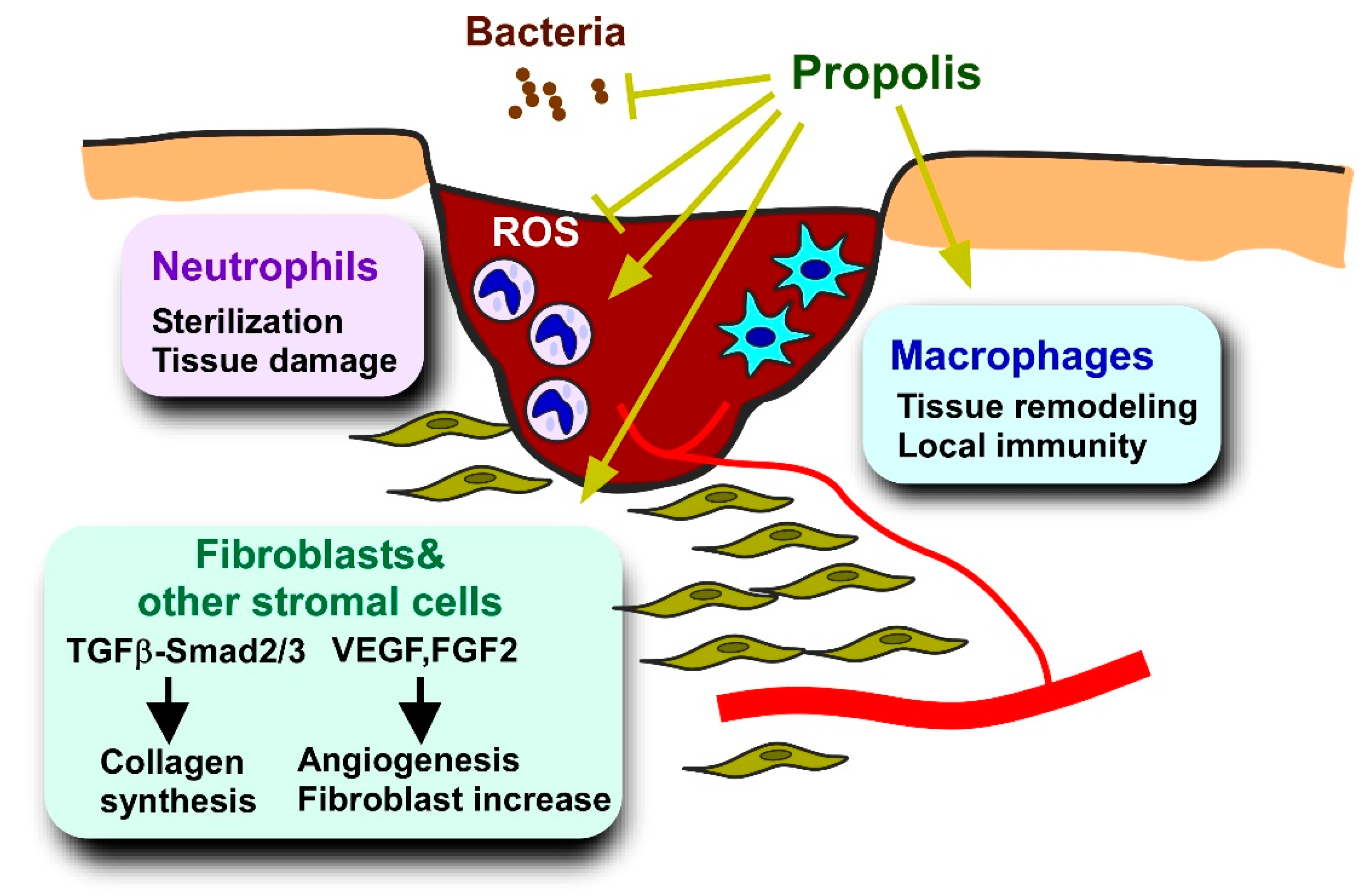{kind=link}
| Administrated Materials | Mouse/Rat Models | Attenuation |
|---|---|---|
| Brazilian propolis | HFD | Body weight gain [108] Visceral adipose tissue weight [107,108] Serum triglyceride [107] Serum cholesterol [107] |
| Brazilian propolis | Leptin deficient | Visceral adipose tissue weight [109] Total cholesterol [109] |
| Croatian propolis | HFD | Body weight gain [110] Serum triglyceride [110] Serum LDL-C [110] |
| CAPE | HFD | Body weight gain [111] Visceral adipose tissue weight [111] |
| Administrated Material | Model | Adipocytokine | Expressional Change |
|---|---|---|---|
| Brazilian propolis | 3T3-L1 HFD mouse | Leptin | Increase [113] |
| CAPE | 3T3-L1, ASCs-derived adipocytes | Leptin | Decrease [114,116] |
| Brazilian propolis | 3T3-L1 | Adiponectin | Increase [117] |
| Artepillin C | 3T3-L1 | Adiponectin | Increase [49,118] |
| Compound 1 | BM-derived adipocytes | Adiponectin | Increase [51] |
| CAPE | ASCs-derived adipocytes | Adiponectin | Increase [116] |
| CAPE | 3T3-L1, ASCs-derived adipocytes | TNF-α | Decrease [114,116] |
| CAPE | 3T3-L1 | Resistin | Decrease [114] |
| CAPE | ASCs-derived adipocytes | IL-1β, IL-6, IL-8 | Decrease [116] |
| Model | Primary Defect | Diabetes Type | References |
|---|---|---|---|
| Streptozotocin injection | β-cell destruction | Type 1 | [129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153] |
| Alloxan injection | β-cell destruction | Type 1 | [137,147,154,155,156] |
| S961 injection | β-cell destruction | Type 1 | [128] |
| High fat diet with low dose streptozotocin injection | β-cell impairment Adipocity | Type 2 | [157,158,159,160,161] |
| High fat diet | Adipocity | Type 2 | [107,162,163] |
| ob/ob mouse | Aberrant leptin signal | Type 2 | [109,164] |
| db/db mouse | Aberrant leptin signal | Type 2 | [45,46,47,48] |
| OLETF rat | Aberrant cholecystokinin signal | Type 2 | [165,166] |
| Propolis Production Area | Diabetes Type | Tissues | Index | Change |
|---|---|---|---|---|
| Croatia | T1DM | Liver, | MDA | Decrease [156,258] |
| Kidney | ||||
| Spleen | ||||
| China | T1DM | Blood | MDA | Decrease [129,130,202] |
| SOD | Increase [131,202] | |||
| ROS, NOS, RNS | Decrease [129,202] | |||
| Kidney | MDA | Decrease [131] | ||
| CAT | Increase [131] | |||
| GPx | Decrease [130] | |||
| Liver | GPx | Increase [131] | ||
| Brazil | T1DM | Blood | MDA | Decrease [131,138] |
| SOD, CAT, GSH | Increase [130,131,137] | |||
| NOS | Decrease [137] | |||
| Kidney | MDA | Decrease [130,131,138] | ||
| SOD, CAT, GSH | Increase [138,195] | |||
| GPx | Decrease [131] | |||
| Liver | MDA | Decrease [131] | ||
| SOD, GSH, GPx | Increase [130,131] | |||
| Pancreas | MDA | Decrease [137] | ||
| SOD | Increase [137] | |||
| Mexico | T1DM | Pancreas | SOD, CAT, GPx | Increase [134] |
| Malaysia | T1DM | Liver | MDA | Decrease [132] |
| SOD, CAT, GSH, GPx, GST, GR | Increase [132] | |||
| Total antioxidant activity | Increase [132] | |||
| Pancreas | MDA | Decrease [133] | ||
| SOD, CAT, GSH, GPx, GST, GR | Increase [133] | |||
| Total antioxidant activity | Increase [133] | |||
| Iran | T1DM | Kidney | MDA | Decrease [136] |
| SOD, GPx | Increase [136] | |||
| FRAP | Decrease [136] | |||
| Nigeria | T1DM | Blood | MDA | Decrease [155] |
| SOD | Increase [155] | |||
| Saudi Arabia | T1DM | Blood | MDA | Decrease [142] |
| Blood | ROS | Decrease [142] | ||
| Liver | ||||
| Spleen | ||||
| Bone marrow | ||||
| Saudi Arabia(?) 1 | T1DM | Blood | MDA | Decrease [135] |
| SOD, CAT, GST | Increase [135] | |||
| Kidney | MDA | Decrease [135] | ||
| SOD, CAT, GST | Increase [135] | |||
| Taiwan | T2DM | Blood | MDA | Decrease [159] |
| SOD, GPx | Increase [159] |
| Administrated Material | Diabetes Type | Tissues/Samples | Index | Change |
|---|---|---|---|---|
| Chrysin | T2DM | Skeletal muscle | MDA | Decrease [241] |
| ROS | Decrease [241] | |||
| Kidney | MDA | Decrease [242] | ||
| SOD, CAT, GSH, GPx, GR | Increase [242] | |||
| Pinocembrin | T1DM | Blood | MDA | Decrease [246] |
| Kidney | ||||
| Quercetin | T1DM | Blood | MDA | Decrease [245] |
| RNS | Decrease [245] | |||
| Pancreas | MDA | Decrease [245] | ||
| SOD, CAT, GPx | Increase [245] | |||
| CAPE | T2DM and cadmium | Kidney | MDA, protein carbonyl | Decrease [171] |
| SOD, CAT, GSH | Increase [171] | |||
| RNS | Decrease [171] | |||
| T1DM | Heart | MDA | Decrease [178] | |
| SOD, CAT | Decrease [178] | |||
| GPx | Increase [178] | |||
| Blood | LOOH | Decrease [252] | ||
| RSH | Increase [252] | |||
| ADMA | Decrease [252] | |||
| RNS | Decrease [252] | |||
| Pancreas | LOOH | Decrease [252] | ||
| RSH | Increase [252] | |||
| ADMA | Decrease [252] | |||
| RNS | Decrease [252] | |||
| NOS | Decrease [252] | |||
| DDAH, HO-1, GGCL | Increase [252] |
| Administrated Materials | Dose (Duration) | Number of Patients | Index | vs. before Treatment (Propolis Group) | vs. Placebo (after Treatment) | References |
|---|---|---|---|---|---|---|
| Iranian propolis | 1 g/day (90 days) | Propolis: n = 50 Placebo: n = 44 | HbA1c Insulin, HOMA-IR, HOMA-β, TNF-α | Decrease | Decrease | [305] |
| BUN, ALT, AST, IL-1β | Decrease | Unchanged | [305] | |||
| 2hp, ALP, CRP | Unchanged | Decrease | [305] | |||
| HDL-C | Increase | Increase | [305] | |||
| Body weight, BMI, FBG, Triglyceride, Total-C, LDL-C, VLDL-C, Creatinine, Uric acid, IL-6 | Unchanged | Unchanged | [305] | |||
| 900 mg/day (12 weeks) | Propolis: n = 30 Placebo: n = 27 | FBG, HbA1c | Decrease | Decrease | [306] | |
| BMI | Decrease | Unchanged | [306] | |||
| Total-C, LDL-C | Unchanged | Decrease | [306] | |||
| Body weight, Triglyceride, HDL-C, VLDL-C | Unchanged | Unchanged | [306] | |||
| Insulin | Increase | Unchanged | [306] | |||
| 1.5 g/day (8 weeks) | Propolis: n = 30 Placebo: n = 30 | FBG, 2hp, Insulin, HOMA-IR, HbA1c | Decrease | Decrease | [307] | |
| TAC, SOD, GPx | Increase | Increase | [307] | |||
| 1.5 g/day (8 weeks) | Propolis: n = 30 Placebo: n = 30 | Fructosamine Ox-LDL | Decrease | Decrease | [308] | |
| CAT | Increase | Increase | [308] | |||
| Body weight, BMI | Unchanged | Unchanged | [308] | |||
| Chinese propolis | 900 mg/day | Propolis: n = 25 Placebo: n = 30 | GSH, Flavonoids, IL-6 | N.S. | Increase | [309] |
| LDH | N.S. | Decrease | [309] | |||
| Blood glucose, Glycosylated hemoglobin, Insulin | Unchanged | Unchanged | [309] | |||
| FRAP, Polyphenols, SOD, GPx, MDA, Carbonyls, IL-1β, TNF-α | N.S. | Unchanged | [309] | |||
| Brazilian green propolis | 226.8 mg/day (8 weeks) | Propolis: n = 41 Placebo: n = 39 | HOMA-IR, FBG, HbA1c, Insulin, Uric acid, Total-C, LDL-C, RLP-C, Triglyceride, TNF-α, IL-6, CRP | Unchanged | Unchanged | [310] |
| 900 mg/day (18 weeks) | Propolis: n = 33 Placebo: n = 32 | GSH, Polyphenols, IL-1β, IL-6 | N.S. | Increase | [311] | |
| Carbonyls, LDH, TNF-α | N.S. | Decrease | [311] | |||
| Blood glucose, Glycosylated hemoglobin, Insulin, FRAP, SOD, GPx, MDA, Aldose reductase, Adiponectin, Ox-LDL | N.S. | Unchanged | [311] | |||
| Commercial propolis (BioPropolis) | 400 mg/day (3 and 6 months) | Propolis: n = 24 Placebo: n = 26 | HbA1c, Plasma glucose, CML, | Decrease | Decrease | [312] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kitamura, H. Effects of Propolis Extract and Propolis-Derived Compounds on Obesity and Diabetes: Knowledge from Cellular and Animal Models. Molecules 2019, 24, 4394. https://doi.org/10.3390/molecules24234394
Kitamura H. Effects of Propolis Extract and Propolis-Derived Compounds on Obesity and Diabetes: Knowledge from Cellular and Animal Models. Molecules. 2019; 24(23):4394. https://doi.org/10.3390/molecules24234394
Chicago/Turabian StyleKitamura, Hiroshi. 2019. "Effects of Propolis Extract and Propolis-Derived Compounds on Obesity and Diabetes: Knowledge from Cellular and Animal Models" Molecules 24, no. 23: 4394. https://doi.org/10.3390/molecules24234394
APA StyleKitamura, H. (2019). Effects of Propolis Extract and Propolis-Derived Compounds on Obesity and Diabetes: Knowledge from Cellular and Animal Models. Molecules, 24(23), 4394. https://doi.org/10.3390/molecules24234394