
Radiation Induced One-Electron Oxidation of 2-Thiouracil in Aqueous Solutions



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
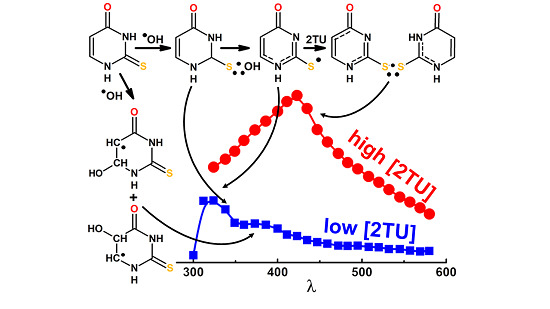
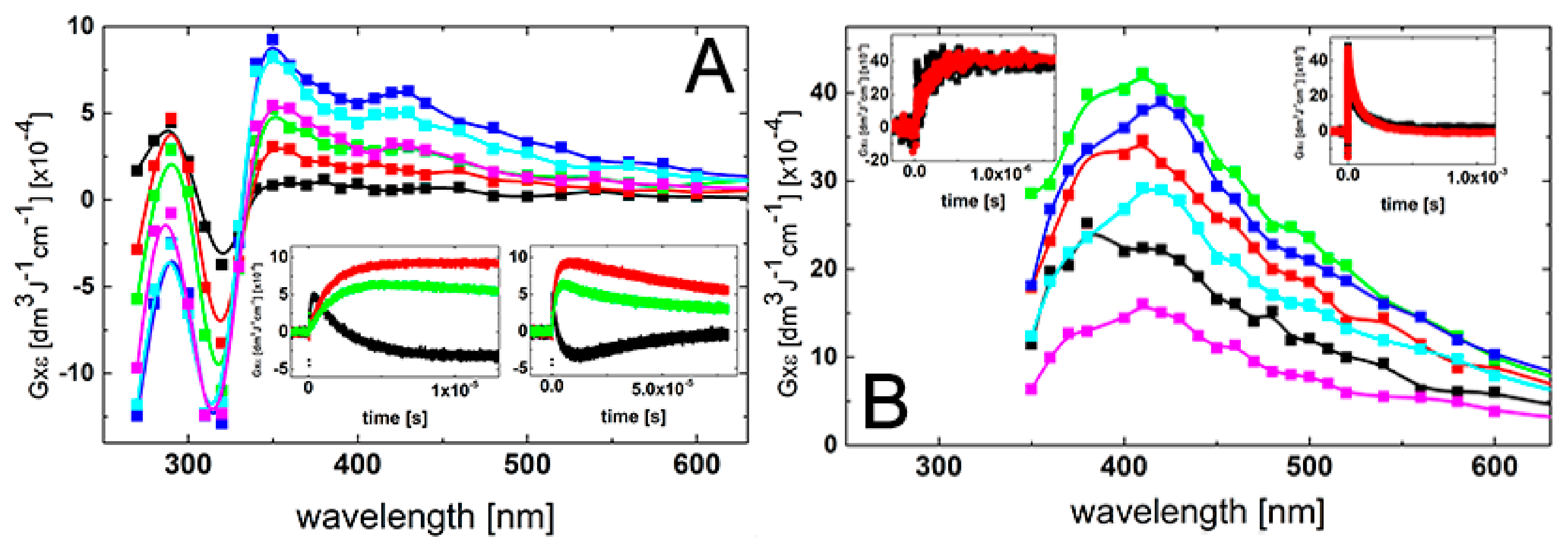
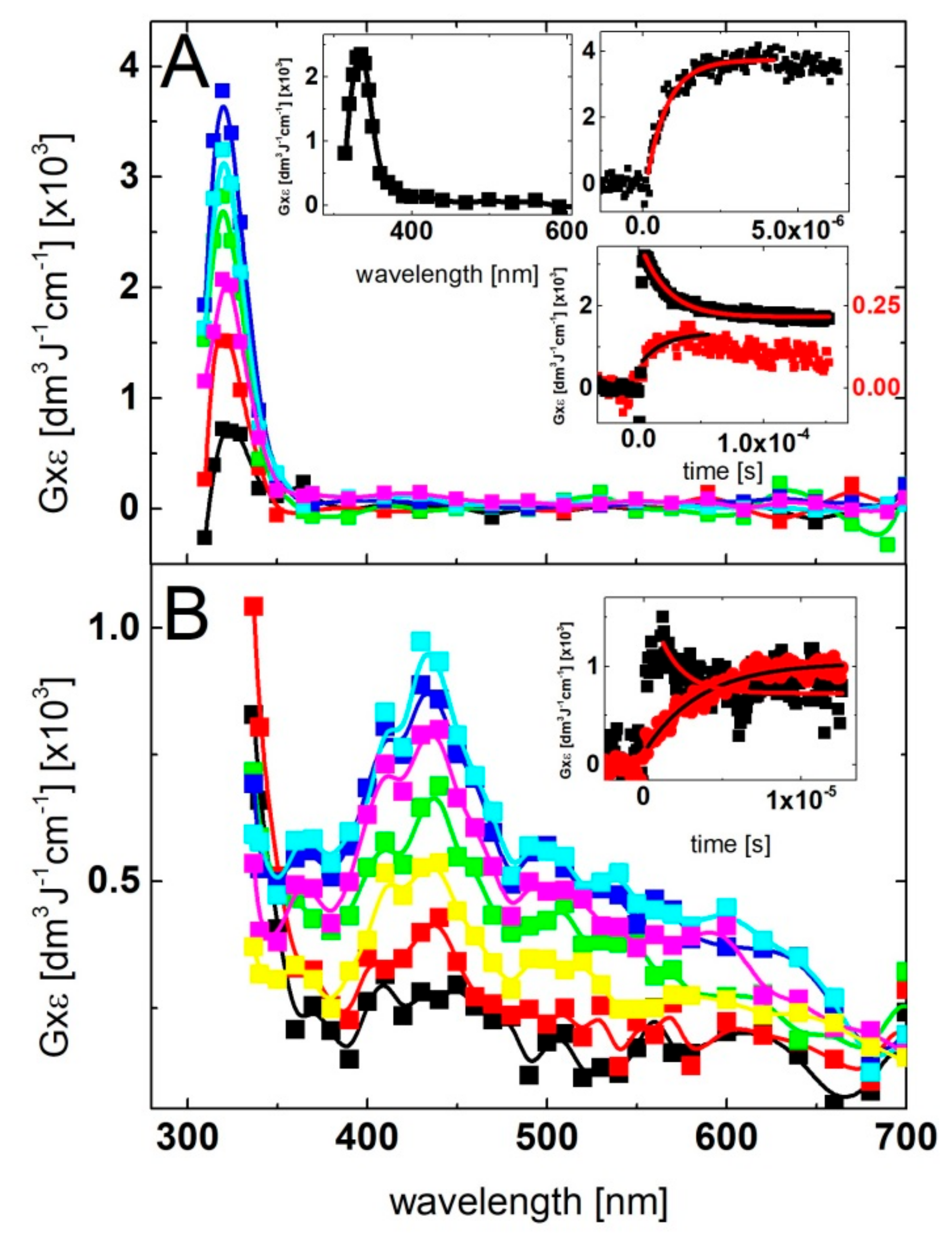
2.1. Oxidation by ●OH Radicals—Influence of 2-TU Concentration on Absorption Spectra
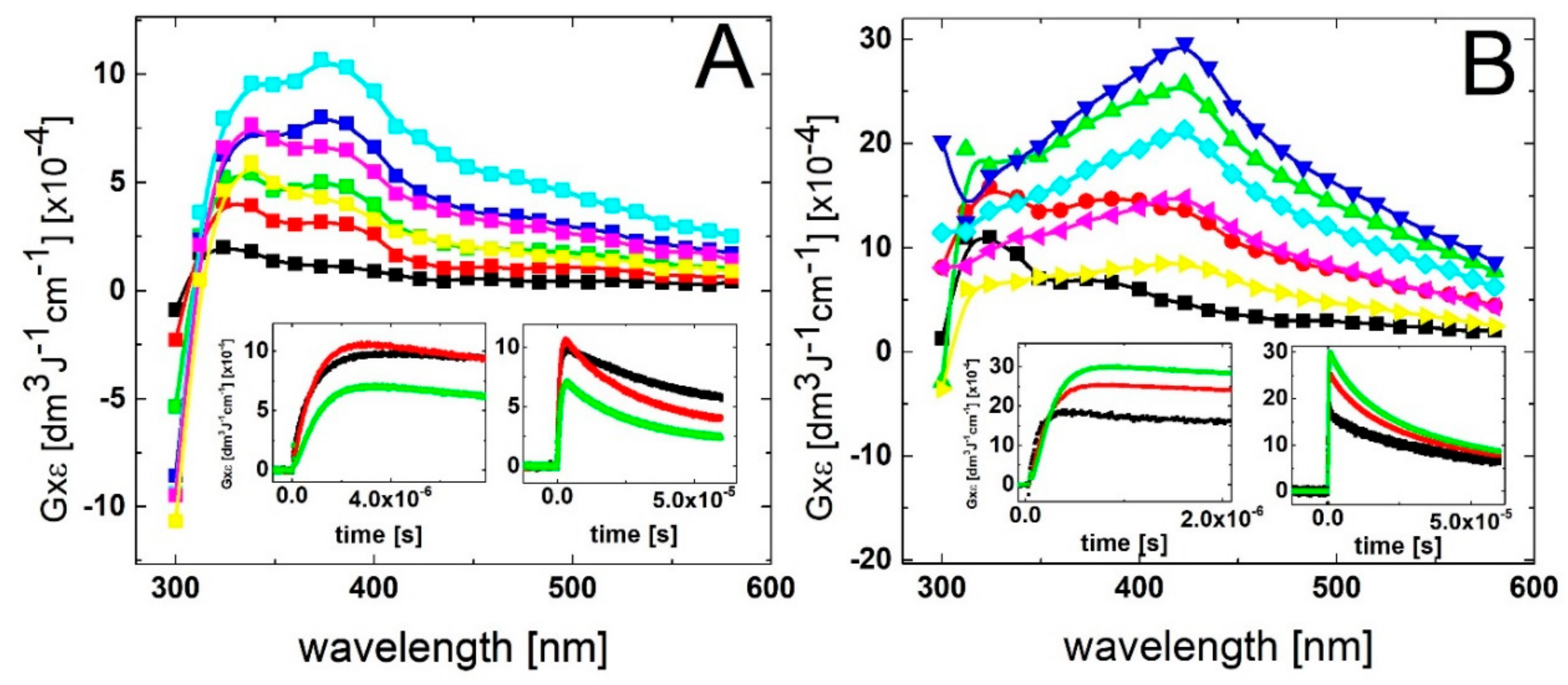
2.1.1. Time Evolution of Absorption Spectra—Low Concentration of 2-TU
2.1.2. Time Evolution of Absorption Spectra—High Concentration of 2-TU
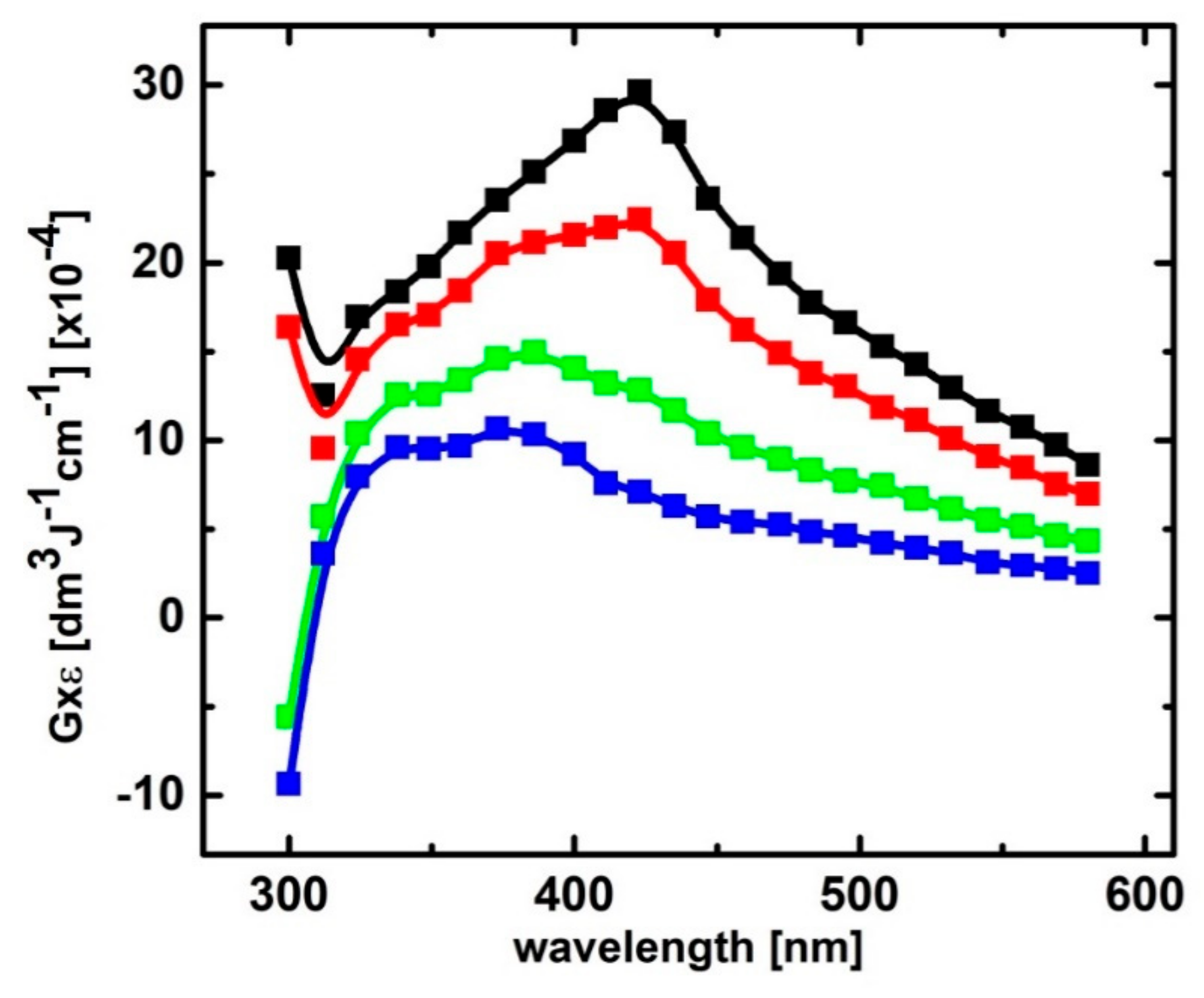
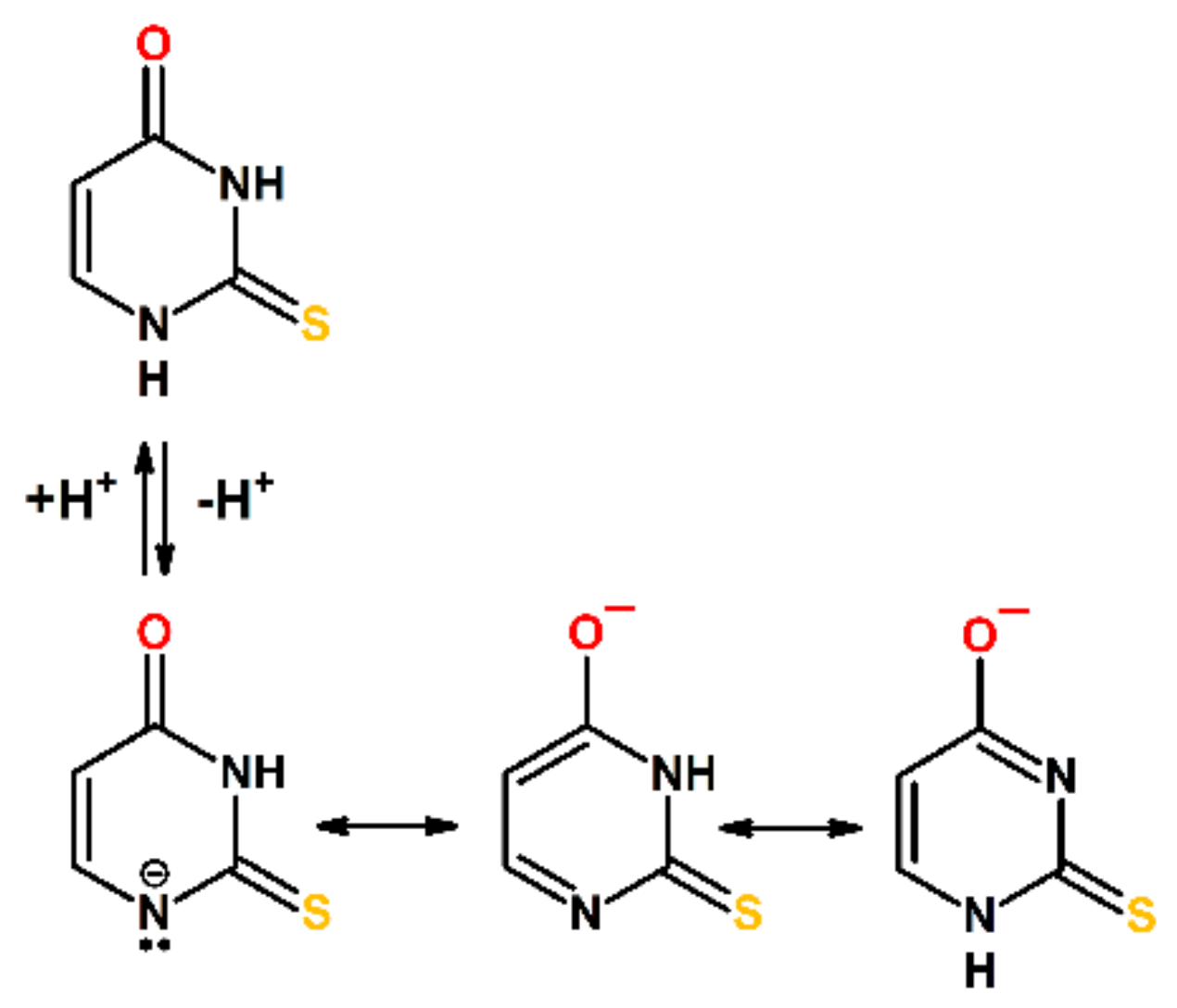
2.1.3. Influence of pH on Time Evolution of Absorption Spectra at Low and High Concentration of 2-TU
2.2. Oxidation by ●OH Radicals—Kinetics
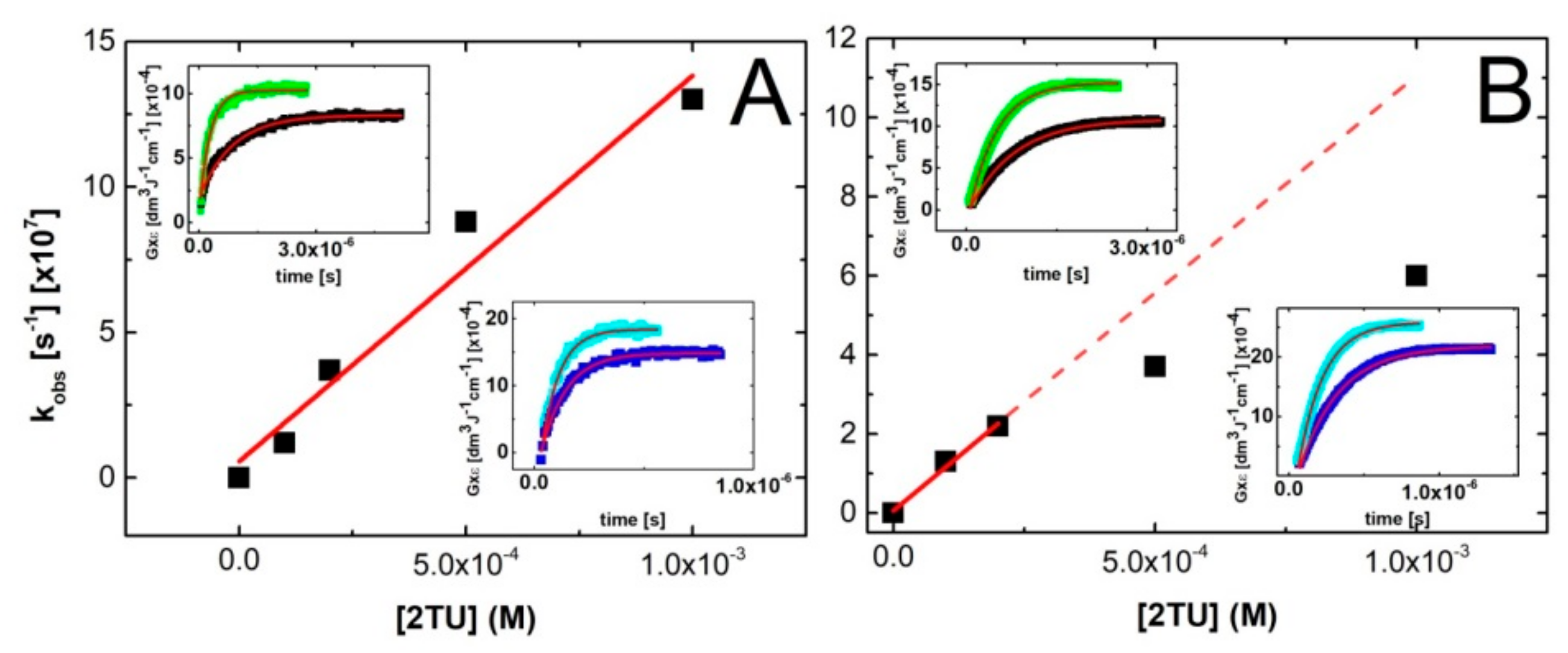
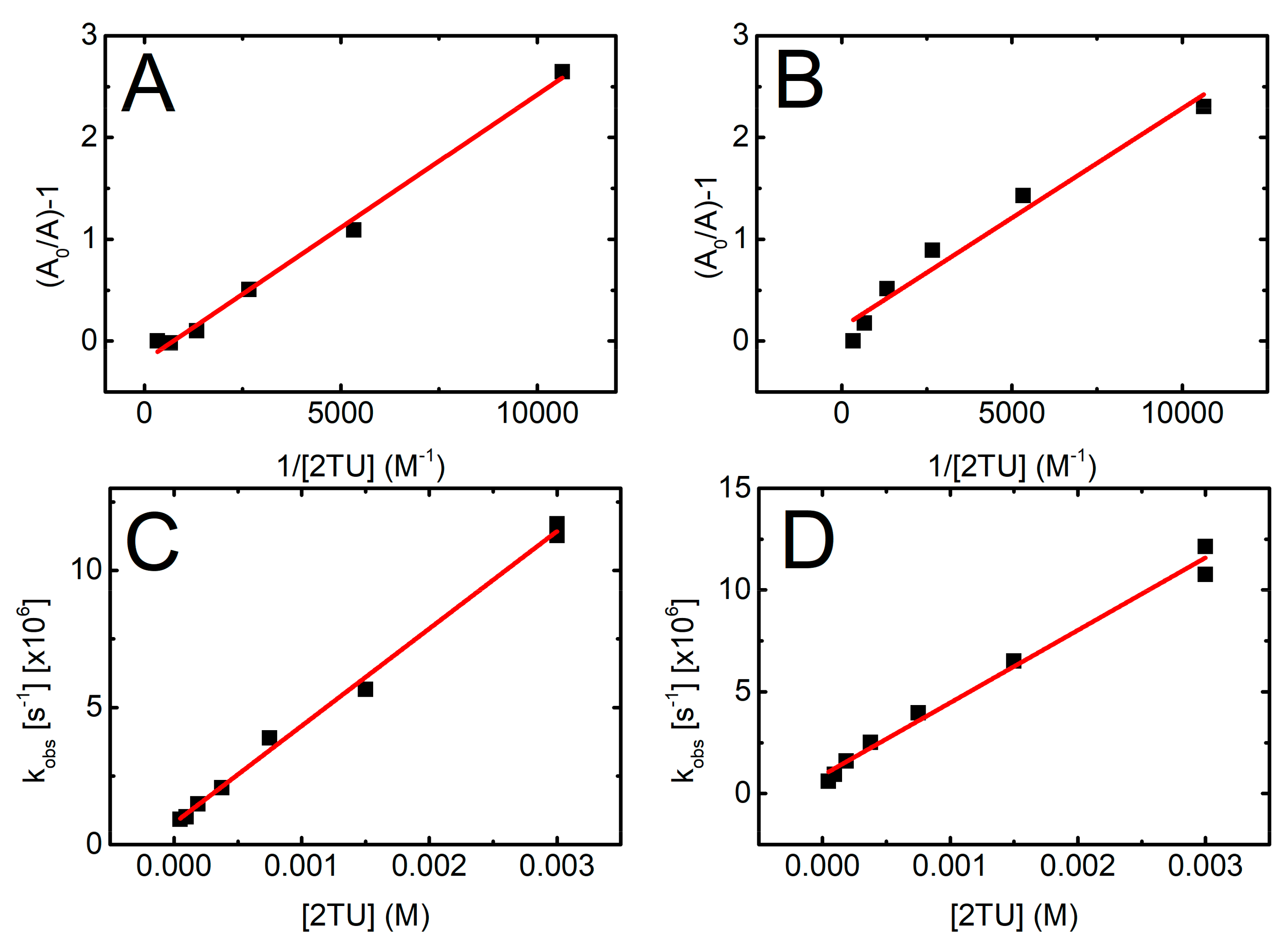
2.2.1. The Rate Constant of the ●OH Reaction with 2-TU
2.2.2. Equilibrium Constant and Rate Constants of Reactions Involved in Equilibrium
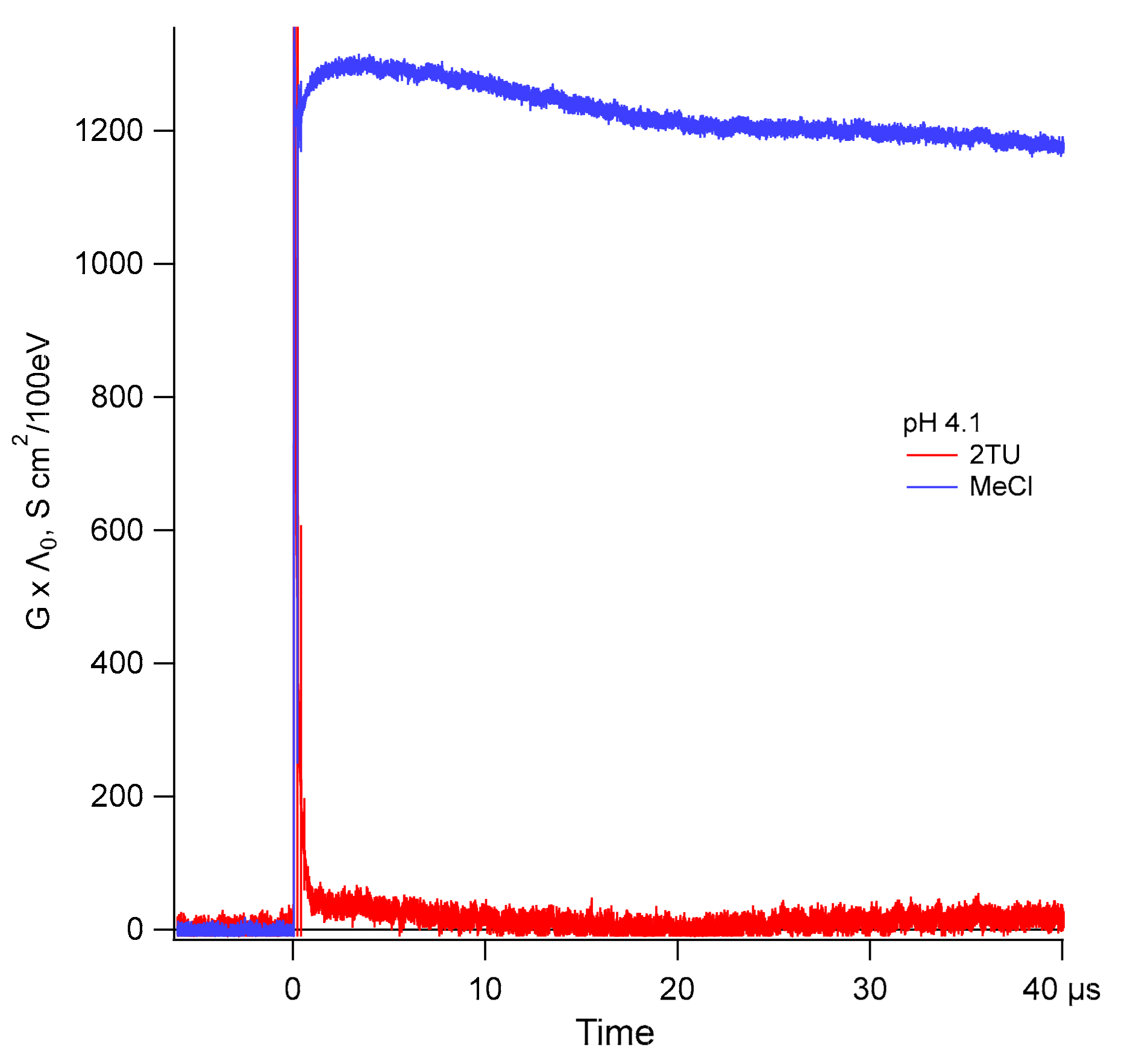
2.3. Oxidation by ●OH Radicals—Time-Resolved Conductivity
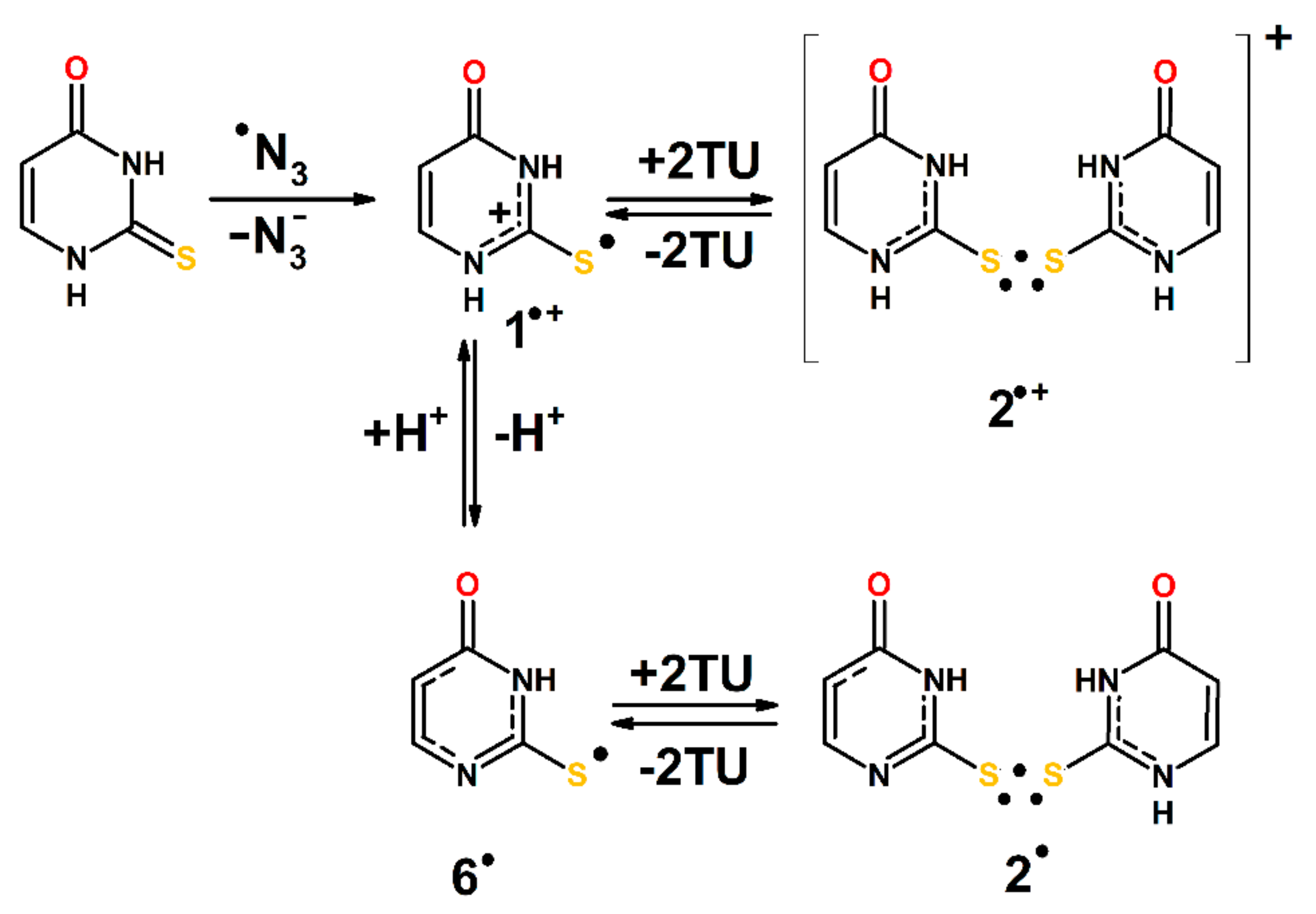
2.4. Oxidation by ●N3 Radicals and CH3CN●+ Radical Cations
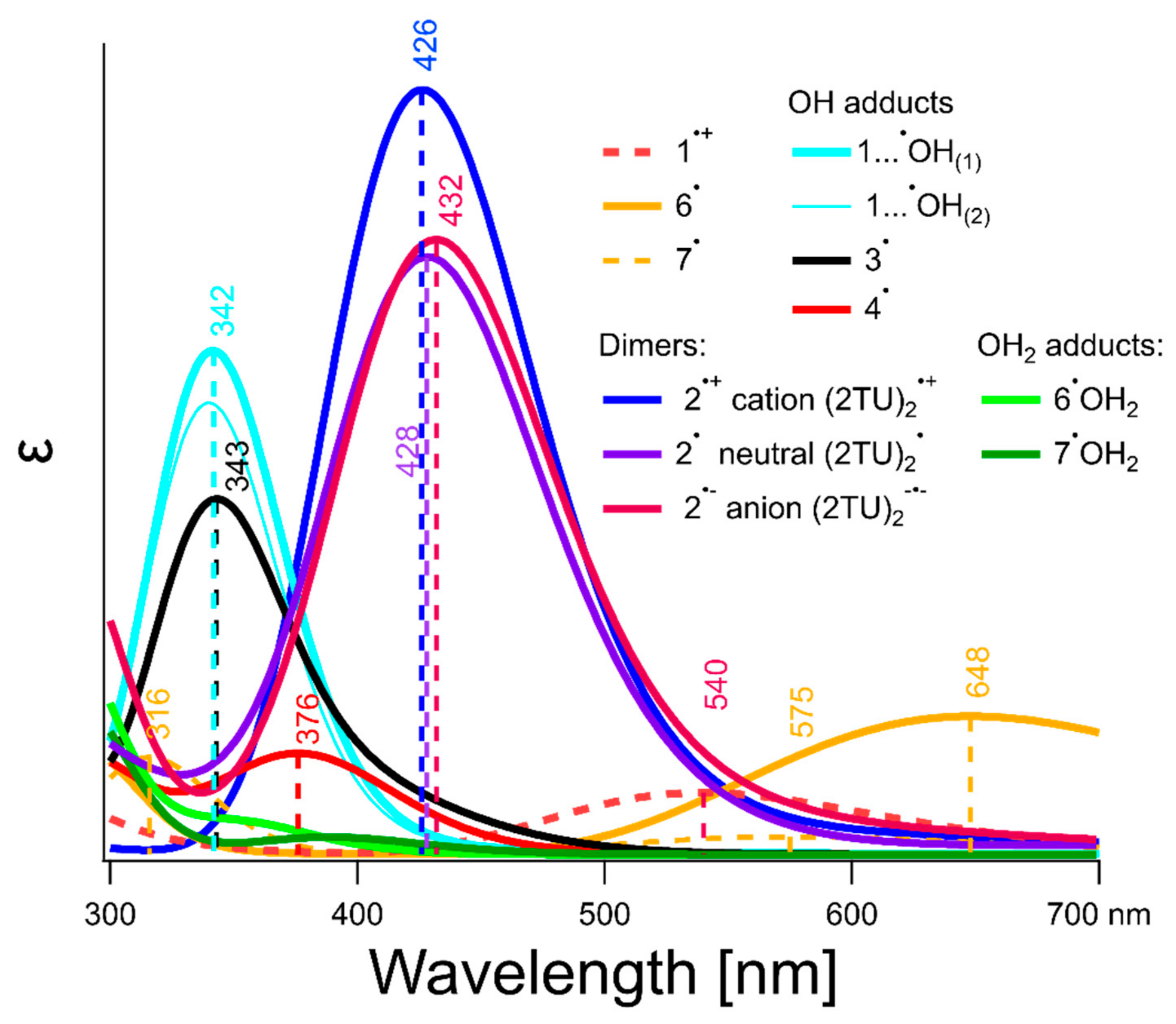
2.5. Theoretical Calculations
3. Discussion
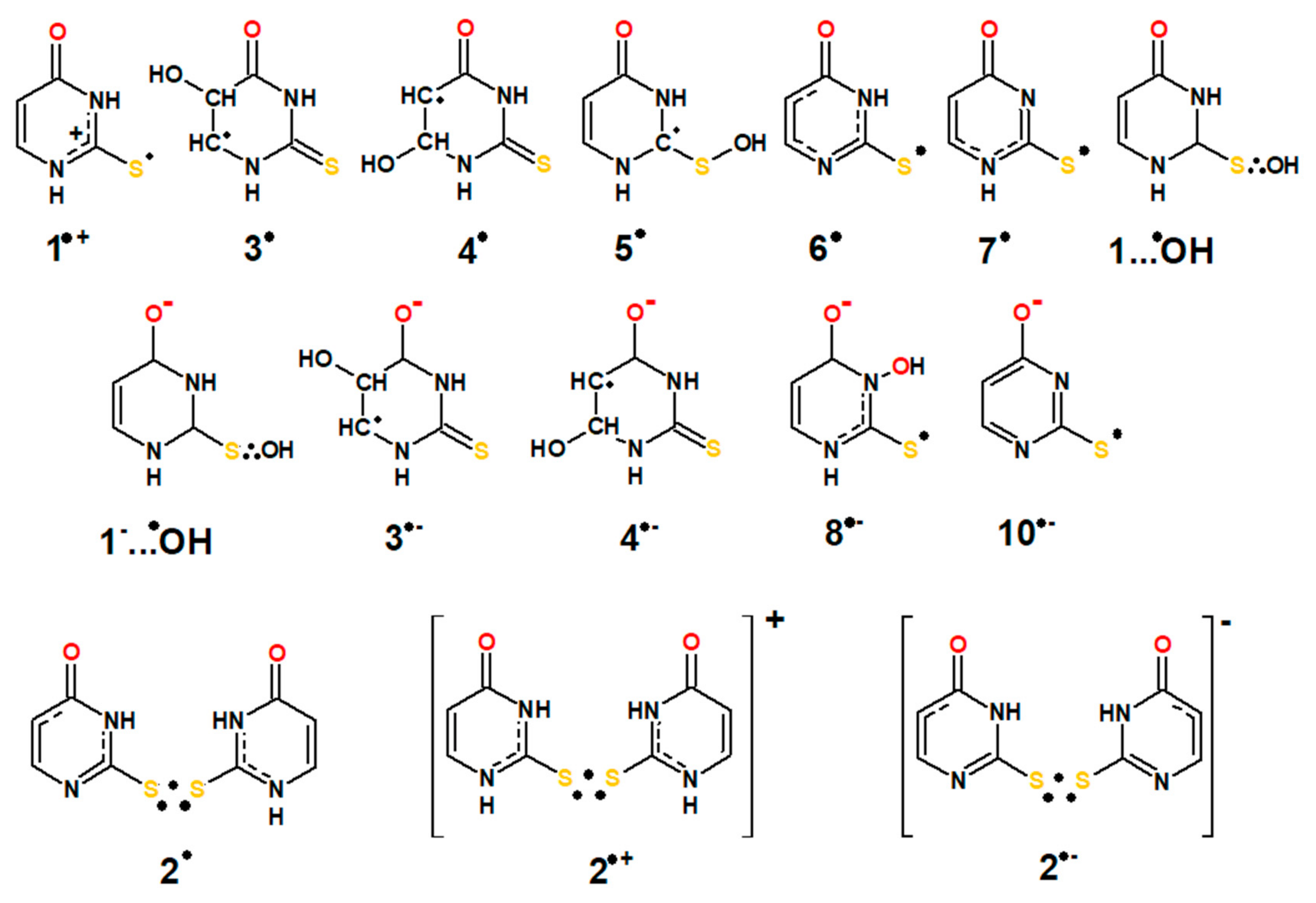
3.1. Assignment of the Absorption Spectra to Respective Intermediates
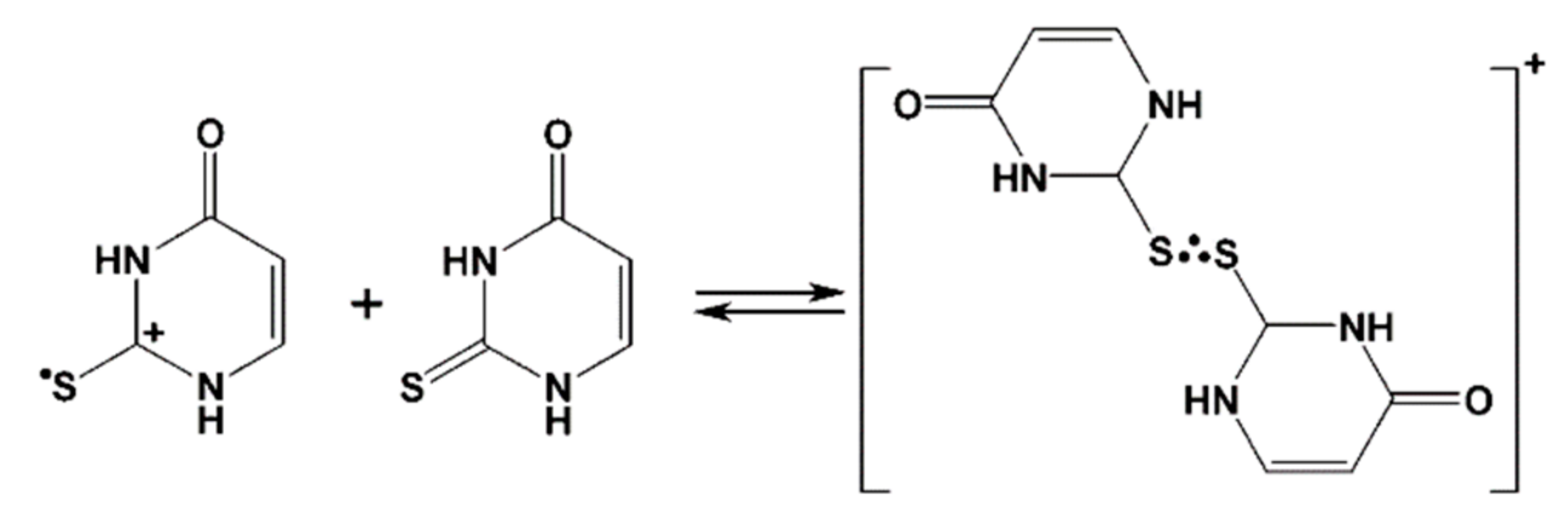
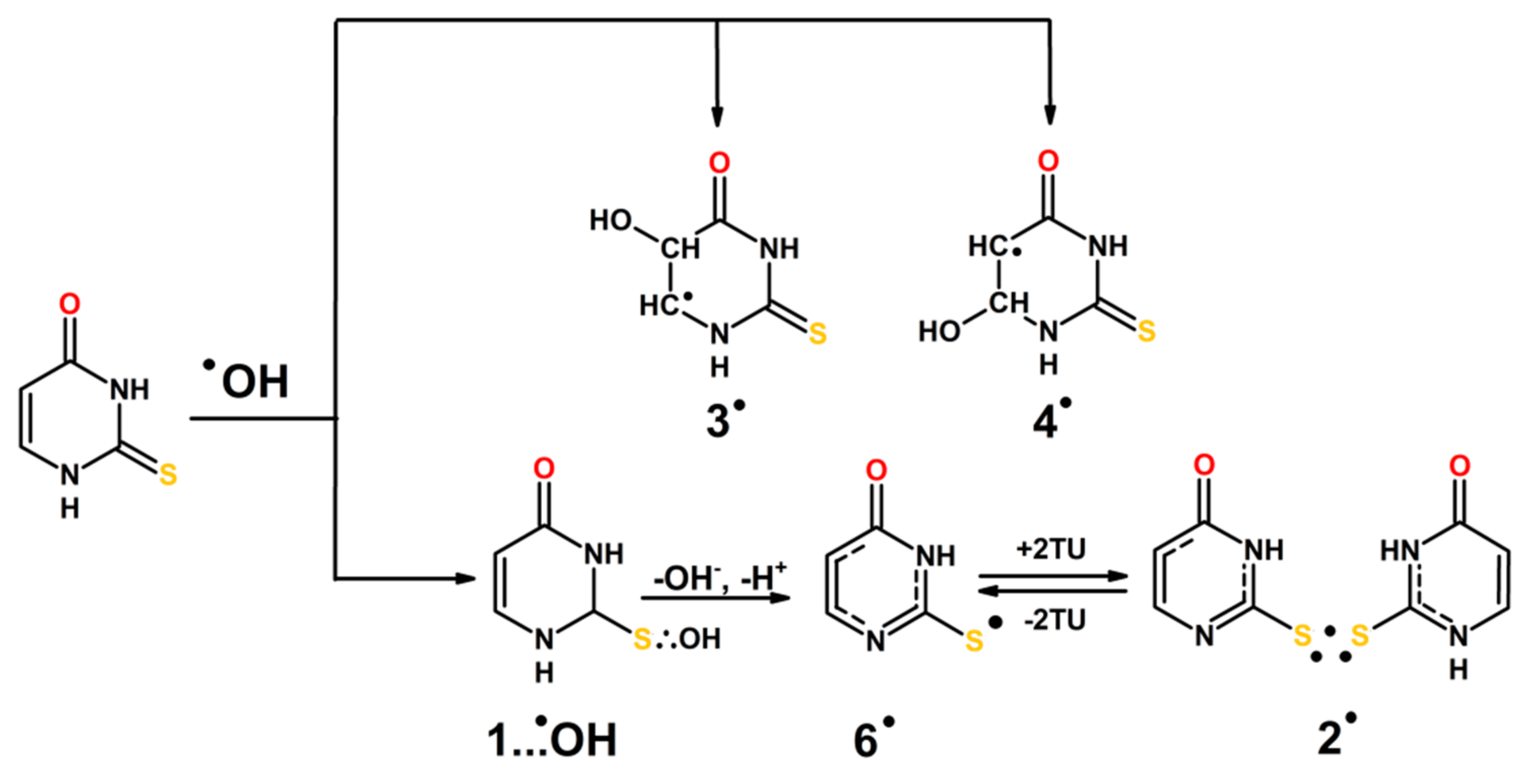
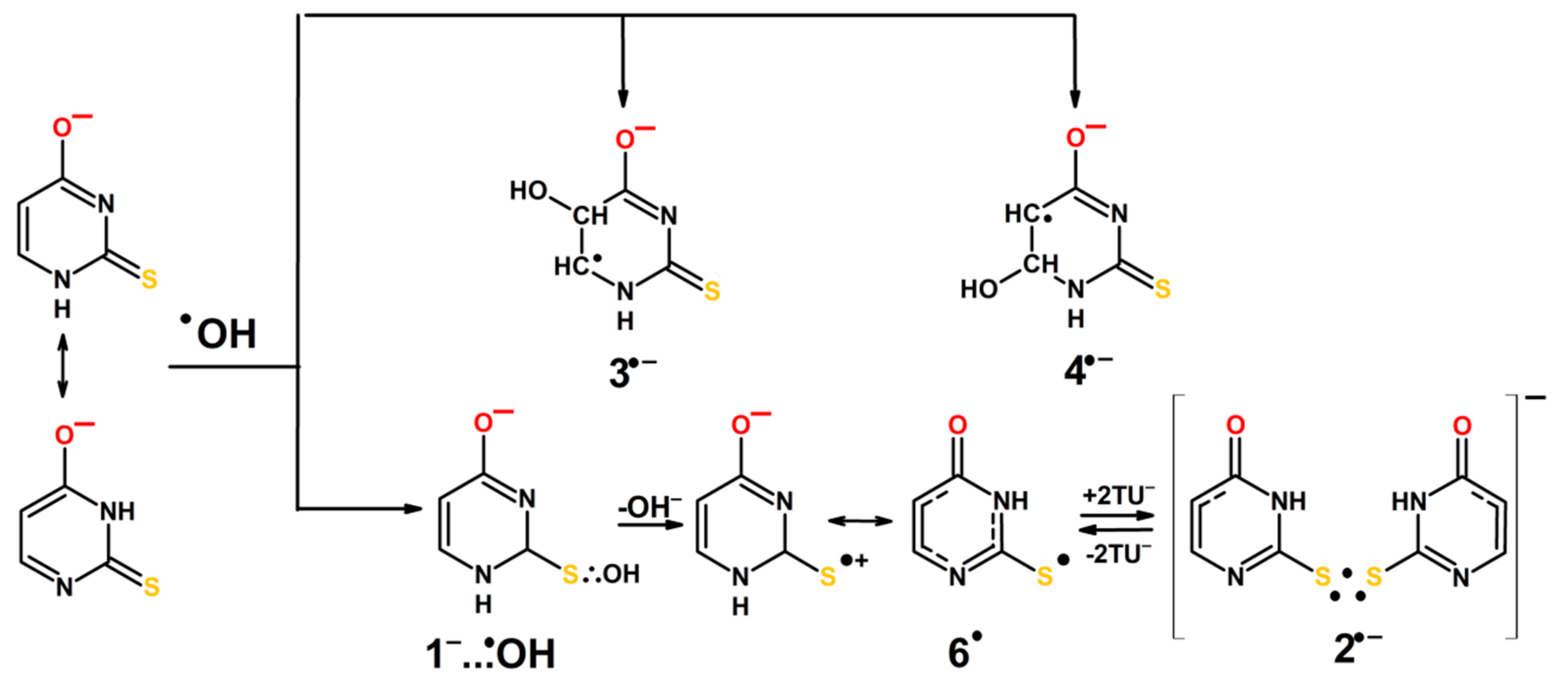
3.2. Justification of the Reaction Pathway Involving Hemibonded ●OH Adducts to Sulfur Atom
3.3. Mechanisms of ●OH and ●N3-Induced Oxidation of 2-Thiouracil
4. Materials and Methods
4.1. Chemicals
4.2. Preparation of Solutions
4.3. Pulse Radiolysis
4.4. Theoretical Procedures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elion, G.B.; Hitchings, G.H. Antagonists of nucleic acid derivatives. 6. Purines. J. Biol. Chem. 1951, 192, 505–518. [Google Scholar] [PubMed]
- Cantara, W.A.; Crain, P.F.; Rozenski, J.; McCloskey, J.A.; Harris, K.A.; Zhang, X.; Vendeix, F.A.P.; Fabris, D.; Agris, P.F. The RNA modification database, RNAMDB:2011 update. Nucleic Acid Res. 2011, 39, D195–D201. [Google Scholar] [CrossRef] [PubMed]
- Ajitkumar, P.; Cherayil, J.D. Thionucleosides in transfer ribonucleic acid -diversity, structure, biosyntheis, and function. Microbiol. Rev. 1988, 52, 103–113. [Google Scholar]
- Wei, F.Y.; Suzuki, T.; Watanabe, S.; Kimura, S.; Kaitsuka, T.; Fujimura, A.; Matsui, H.; Atta, M.; Michiue, H.; Fontecave, M.; et al. Deficit of tRNA(Lys) modification by Cdkal1 causes the development of type 2 diabetes in mice. J. Clin. Investig. 2011, 121, 3598–3608. [Google Scholar] [CrossRef] [PubMed]
- Pollum, M.; Martinez-Fernandez, L.; Crespo-Hernandez, C.E. Photochemistry of Nucleic Acid Bases and Their Thio- and Aza-Analogues in Solution. In Photoinduced Phenomena in Nucleic Acids I: Nucleobases in the Gas Phase and in Solvents; Barbatti, M.B., Borin, A.C., Ullrich, S., Eds.; Springer: Cham, Switzerland, 2015; Volume 355, pp. 245–327. [Google Scholar]
- Kutyavin, L.V.; Rhinehart, R.L.; Lukhtanov, E.A.; Gorn, V.V.; Meyer, R.B.; Gamper, H.B. Oligonucleotides containing 2-aminoadenine and 2-thiothymine act as selectively binding complementary agents. Biochemistry-US 1996, 35, 11170–11176. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Fernandez, L.; Gonzalez, L.; Corral, I. An ab initio mechanism for efficient population of triplet states in cytotoxic sulfur substituted DNA bases: The case of 6-thioguanine. Chem. Commun. 2012, 48, 2134–2136. [Google Scholar] [CrossRef]
- Reelfs, O.; Karran, P.; Young, A.R. 4-thiothymidine sensitization of DNA to UVA offers potential for a novel photochemotherapy. Photochem. Photobiol. Sci. 2012, 11, 148–154. [Google Scholar] [CrossRef]
- Brem, R.; Zhang, X.; Xu, Y.-Z.; Karran, P. UVA photoactivation of DNA containing halogenated thiopyrimidines induces DNA lessions. J. Photochem. Photobiol. B Biol. 2015, 145, 1–10. [Google Scholar] [CrossRef]
- Gemenetzidis, E.; Shavorskaya, O.; Xu, Y.-Z.; Trigiante, G. Topical 4-thiothymidine is a viable photosensitiser for the photodynamic therapy of skin malignancies. J. Dermatolog. Treat. 2013, 24, 209–214. [Google Scholar] [CrossRef]
- Reelfs, O.; Macpherson, P.; Ren, X.; Xu, Y.-Z.; Young, A.R. Identification of potentially cytotoxic lesions induced by UVA photoactivation of DNA 4-thithymidine in human cells. Nucleic Acid Res. 2011, 39, 9620–9632. [Google Scholar] [CrossRef]
- Pridgeon, S.W.; Heer, R.; Taylor, G.A.; Newell, D.R.; O’Toole, K.; Robinson, M.; Xu, Y.-Z.; Karran, P.; Boddy, A.V. Thiothymidine combined with UVA as a potential novel therapy for bladder cancer. Br. J. Cancer 2011, 104, 1869–1876. [Google Scholar] [CrossRef] [PubMed]
- Trigiante, G.; Xu, Y.-Z. 4-thiothymidine and its analogues as UVA-activated photosensitizers. In Photodynamic Therapy: Fundamentals, Applications and Health Outcomes; Hugo, A.G., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2015; pp. 193–206. [Google Scholar]
- Ashwood, B.; Pollum, M.; Crespo-Hernandez, C.E. Photochemical and Photodynamical Properties of Sulfur-Substituted Nucleic Acid Bases. Photochem. Photobiol. 2019, 95, 33–58. [Google Scholar] [CrossRef] [PubMed]
- Mai, S.; Pollum, M.; Martinez-Fernandez, L.; Dunn, N.; Marquetand, P.; Corral, I.; Crespo-Hernandez, C.E.; Gonzalez, L. The origin of efficient triplet state population in sulfur-substituted nucleobases. Nat. Commun. 2016, 7, 13077. [Google Scholar] [CrossRef] [PubMed]
- Favre, A.; Saintome, C.; Fourrey, J.L.; Clivio, P.; Laugaa, P. Thionucleobases as intrinsic photoaffinity probes of nucleic acid structure and nucleic acid protein interactions. J. Photochem. Photobiol. B Biol. 1998, 42, 109–124. [Google Scholar] [CrossRef]
- Taras-Goslinska, K.; Wenska, G.; Skalski, B.; Maciejewski, A.; Burdzinski, G.; Karolczak, J. Intra- and intermolecular electronic relaxation of the second excited singlet and the lowest excited triplet states of 1,3-dimethyl-4-thiouracil in solution. Photochem. Photobiol. 2002, 75, 448–456. [Google Scholar] [CrossRef]
- Taras-Goslinska, K.; Wenska, G.; Skalski, B.; Maciejewski, A.; Burdzinski, G.; Karolczak, J. Spectral and photophysical properties of the lowest excited triplet state of 4-thiouridine and its 5-halogeno derivatives. J. Photochem. Photobiol. A Chem. 2004, 168, 227–233. [Google Scholar] [CrossRef]
- Wenska, G.; Taras-Goslinska, K.; Skalski, B.; Maciejewski, A.; Burdzinski, G.; Karolczak, J. Putative phototautomerization of 4-thiouridine in the S2 excited state revealed by fluorescence study using picosecond laser spectroscopy. J. Photochem. Photobiol. A Chem. 2006, 181, 12–18. [Google Scholar] [CrossRef]
- Wenska, G.; Taras-Goslinska, K.; Filipiak, P.; Hug, G.L.; Marciniak, B. Photochemical reactions of 4-thiouridine disulfide and 4-benzylthiouridine the involvement of the 4-pyrimidinylthiyl radical. Photochem. Photobiol. Sci. 2008, 7, 250–256. [Google Scholar] [CrossRef]
- Wenska, G.; Koput, J.; Burdzinski, G.; Taras-Goslinska, K.; Maciejewski, A. Photophysical and photochemical properties of the T1 excited state of thioinosine. J. Photochem. Photobiol. A Chem. 2009, 206, 93–101. [Google Scholar] [CrossRef]
- Wenska, G.; Filipiak, P.; Taras-Goslinska, K.; Sobierajska, A.; Gdaniec, Z. Orientation-dependent quenching of the triplet excited 6-thiopurine by nucleobases. J. Photochem. Photobiol. A Chem. 2011, 217, 55–61. [Google Scholar] [CrossRef]
- Wenska, G.; Taras-Goslinska, K.; Lukaszewicz, A.; Burdzinski, G.; Koput, J.; Maciejewski, A. Mechanism and dynamics of intramolecular triplet state decay of 1-propyl-4-thiouracil and its alpha-methyl-substituted derivatives studied in perfluoro-1,3-dimethylcyclohexane. Photochem. Photobiol. Sci. 2011, 10, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Taras-Goslinska, K.; Burdzinski, G.; Wenska, G. Relaxation of the T1 excited state of 2-thiothymine, its riboside and deoxyriboside-enhanced nonradiative decay rate induced by sugar substituent. J. Photochem. Photobiol. A Chem. 2014, 275, 89–95. [Google Scholar] [CrossRef]
- Pollum, M.; Crespo-Hernandez, C.E. Communication: The dark singlet state as a doorway state in the ultrafast and efficient intersystem crossing dynamics in 2-thiothymine and 2-thiouracil. J. Chem. Phys. 2014, 140, 071101. [Google Scholar] [CrossRef] [PubMed]
- Pollum, M.; Jockusch, S.; Crespo-Hernandez, C.E. 2,4-Dithiothymine as a Potent UVA Chemotherapeutic Agent. J. Am. Chem. Soc. 2014, 136, 17930–17933. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Hernandez, C.E.; Martinez-Fernandez, L.; Rauer, C.; Reichardt, C.; Mai, S.; Pollum, M.; Marquetand, P.; Gonzalez, L.; Corral, I. Electronic and Structural Elements That Regulate the Excited-State Dynamics in Purine Nucleobase Derivatives. J. Am. Chem. Soc. 2015, 137, 4368–4381. [Google Scholar] [CrossRef] [PubMed]
- Pollum, M.; Jockusch, S.; Crespo-Hernandez, C.E. Increase in the photoreactivity of uracil derivatives by doubling thionation. Phys. Chem. Chem. Phys. 2015, 17, 27851–27861. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Rodriguez, J.A.; Mohamadzade, A.; Mai, S.; Ashwood, B.; Pollum, M.; Marquetand, P.; Gonzalez, L.; Crespo-Hernandez, C.E.; Ullrich, S. 2-Thiouracil intersystem crossing photodynamics studied by wavelength-dependent photoelectron and transient absorption spectroscopies. Phys. Chem. Chem. Phys. 2017, 19, 19756–19766. [Google Scholar] [CrossRef]
- Karran, P.; Attard, N. Thiopurines in current medical practice: Molecular mechanisms and contributions to therapy-related cancer. Nat. Rev. Cancer 2008, 8, 24–36. [Google Scholar] [CrossRef]
- Herak, J.N.; Sankovic, K.; Hutterman, J. Thiocytosine as a radiation energy trap in a single crystal of cytosine hydrochloride. Int. J. Radiat. Biol. 1994, 66, 3–9. [Google Scholar] [CrossRef]
- Ling, L.; Ward, J.F. Radiosensitization of Chinese Hamster V79 Cells by Bromodeoxyuridine Substitution of Thymidine: Enhancement of Radiation-Induced Toxicity and DNA Strand Break Production by Monofilar and Bifilar Substitution. Radiat. Res. 1990, 121, 76–83. [Google Scholar] [CrossRef]
- Abdoul-Carime, H.; Dugal, P.-C.; Sanche, L. Damage Induced by 1-30 eV Electrons on Thymine- and Bromouracil-Substituted Oligonucleotides. Radiat. Res. 2000, 153, 23–28. [Google Scholar] [CrossRef]
- Abdoul-Carime, H.; Huels, M.A.; Illenberger, E.; Sanche, L. Sensitizing DNA to secondary electron damage: Resonant oxidative radicals from 5-halouracils. J. Am. Chem. Soc. 2001, 123, 5354–5355. [Google Scholar] [CrossRef] [PubMed]
- Kopyra, J.; Freza, S.; Abdoul-Carime, H.; Marchaj, M.; Skurski, P. Dissociative electron attachment to gas phase thiothymine: Experimental and theoretical approaches. Phys. Chem. Chem. Phys. 2014, 16, 5342–5348. [Google Scholar] [CrossRef] [PubMed]
- Kopyra, J.; Abdoul-Carime, H.; Kossoski, F.; Varella, M.T.d.N. Electron driven reactions in sulphur containing analogues of uracil: The case of 2-thiouracil. Phys. Chem. Chem. Phys. 2014, 16, 25054–25061. [Google Scholar] [CrossRef] [PubMed]
- Kopyra, J.; Abdoul-Carime, H. Dissociative electron attachment to gas phase nucleobases: Comparision of thymine and thiothymine. J. Phys. Conf. Ser. 2015, 635, 072066. [Google Scholar] [CrossRef] [Green Version]
- Kopyra, J.; Abdoul-Carime, H.; Skurski, P. Temperature Dependence of the Dissociative Electron Attachment to 2-Thiothymine. J. Phys. Chem. A 2016, 120, 7130–7136. [Google Scholar] [CrossRef]
- Kopyra, J.; Abdoul-Carime, H. Unusual temperature dependence of the dissociative electron attachment cross section of 2-thiouracil. J. Chem. Phys. 2016, 144, 034306. [Google Scholar] [CrossRef]
- Kopyra, J.; Kopyra, K.K.; Abdoul-Carime, H.; Branowska, D. Insights into the dehydrogenation of 2-thiouracil induced by slow electrons: Comparison of 2-thiouracil and 1-methyl-2-thiouracil. J. Chem. Phys. 2018, 148, 234301. [Google Scholar] [CrossRef]
- Besic, E. EPR study of the free radicals in the single crystals of 2-thithymine g-irradiated at 300 K. J. Mol. Struct. 2009, 917, 71–75. [Google Scholar] [CrossRef]
- Besic, E.; Gomzi, V. EPR study of sulfur-centered p-radical in g-irradiated single crystal of 2-thiothymine. J. Mol. Struct. 2008, 876, 234–239. [Google Scholar] [CrossRef]
- Besic, E.; Sankovic, K.; Gomzi, V.; Herak, J.N. Sigma radicals in gamma-irradiated single crystals of 2-thiothymine. Phys. Chem. Chem. Phys. 2001, 2723–2725. [Google Scholar] [CrossRef]
- Herak, J.N.; Sankovic, K.; Hole, E.O.; Sagstuen, E. ENDOR study of a thiocytosine oxidation product in cytosine monohydrate crystals doped with 2-thiocytosine, X-irradiated at 15 K. Phys. Chem. Chem. Phys. 2000, 2, 4971–4975. [Google Scholar] [CrossRef]
- Herak, J.N.; Sankovic, K.; Krilov, D.; Hutterman, J. An EPR Study of the Transfer and Trapping of Holes Produced by Radiation in Guanine(Thioguanine) Hydrochloride Single Crystals. Radiat. Res. 1999, 151, 319–324. [Google Scholar] [CrossRef]
- Herak, J.N.; Hutterman, J. Long-range hole migration in irradiated crystals of nucleic acid bases. An EPR study. IntJ. Radiat. Biol. 1991, 60, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Sankovic, K.; Herak, J.N.; Krilov, D. EPR spectroscopy of the sulphur-centered radicals derived from thiocytosine. J. Mol. Struct. 1988, 190, 277–286. [Google Scholar] [CrossRef]
- Jorgensen, J.-P.; Sagstuen, E. ESR of Irradiated 2-Thiouracil Single Crystals. A 3a-Hydrogen Radical. Radiat. Res. 1981, 88, 29–36. [Google Scholar] [CrossRef]
- Claesson, O.; Lund, A.; Jorgensen, J.-P.; Sagstuen, E. Electron spin resonance of irradiated crystals of 2-thiouracil; hyperfine coupling tensor for nuclei with I = 1/2. J. Magn. Reson. 1980, 41, 229–239. [Google Scholar] [CrossRef]
- Kumar, A.; Sevilla, M.D. SOMO-HOMO Level Inversion in Biologically Important Radicals. J. Phys. Chem. B 2018, 122, 98–105. [Google Scholar] [CrossRef]
- Kumar, A.; Sevilla, M.D. p vs. s-Radical States of One-Electron-Oxidized DNA/RNA Bases: A Density Functional Theory Study. J. Phys. Chem. B 2013, 117, 11623–11632. [Google Scholar] [CrossRef] [Green Version]
- Gomzi, V. DFT study of radicals formed in 2-thithymine single crystals at 77 K: 1- and 2-molecule models revised. Comput. Theor. Chem. 2011, 963, 497–502. [Google Scholar] [CrossRef]
- Wenska, G.; Filipiak, P.; Asmus, K.-D.; Bobrowski, K.; Koput, J.; Marciniak, B. Formation of a Sandwich-Structure Assisted, Relatively Long-Lived Sulfur-Centered Three-electron Bonded Radical Anion in the Reduction of a Bis(1-substituted-uracilyl) Disulfide in Aqueous Solution. J. Phys. Chem. B 2008, 112, 100045–100053. [Google Scholar] [CrossRef] [PubMed]
- Prasanthkumar, K.P.; Alvarez-Idaboy, J.R.; Kumar, P.V.; Singh, B.G.; Priyadarsini, K.I. Contrasting reactions of hydrated electron and formate radical with 2-thioanalogues of cytosine and uracil. Phys. Chem. Chem. Phys. 2016, 18, 28781–28790. [Google Scholar] [CrossRef] [PubMed]
- Taras-Goslinska, K.; Vetica, F.; Barata-Vallejo, S.; Triantakostanti, V.; Marciniak, B.; Chatgilialoglu, C. Converging Fate of the Oxidation and Reduction of 8-Thioguanosine. Molecules 2019, 24, 3143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makurat, S.; Spisz, P.; Kozak, W.; Rak, J.; Zdrowowicz, M. 5-iodo-4-thio-2’-deoxyuridine as a sensitizer of X-ray induced cancer cell killing. Int. J. Mol. Sci. 2019, 20, 1308. [Google Scholar] [CrossRef] [Green Version]
- Spisz, P.; Zdrowowicz, M.; Makurat, S.; Kozak, W.; Skotnicki, K.; Bobrowski, K.; Rak, J. Why Does the Type of Halogen Atom Matter for the Radiosensitizing Properties of 5-Halogen Substituted 4-Thio-2’-Deoxyuridines? Molecules 2019, 24, 2819. [Google Scholar] [CrossRef] [Green Version]
- Prasanthkumar, K.P.; Suresh, C.H.; Aravindakumar, C.T. Oxidation Reactions of 2-Thiouracil: A Theoretical and Pulse Radiolysis Study. J. Phys. Chem. A 2012, 116, 10712–10720. [Google Scholar] [CrossRef]
- Prasanthkumar, K.P.; Suresh, C.H.; Aravindakumar, C.T. Dimer radical cation of 4-thiouracil: A pulse radiolysis and theoretical study. J. Phys. Org. Chem. 2013, 26, 510–516. [Google Scholar] [CrossRef]
- Giuliano, B.M.; Feyer, V.; Prince, K.C.; Coreno, M.; Evangelisti, L.; Melandri, S.; Caminati, W. Tautomerism in 4-Hydroxypyrimidine, S-Methyl-2-thiouracil, and 2-Thiouracil. J. Phys. Chem. A 2010, 114, 12725–12730. [Google Scholar] [CrossRef]
- Katrizky, A.R.; Szafran, M.; Stevens, J. The Tautomeric Equilibria of Thio Analogues of Nucleic Acid Bases. Part 2. AM1 and ab initio Calculations of 2-Thiouracil and its Methyl Derivatives. J. Chem. Soc. Perkin Trans. II 1989, 1507–1511. [Google Scholar] [CrossRef]
- Yekeler, H. Ab initio study on tautomerism of 2-thiouracil in the gas phase and in solution. J. Comput. -Aided Mol. Des. 2000, 14, 243–250. [Google Scholar] [CrossRef]
- Babu, N.S. Theoretical Study of Stability, Tautomerism, Equilibrium Constants (pKT) of 2-Thiouracil in Gas Phase and Different Solvents (Water and Acetonitrile) by the Density Functional Theory Method. Amer. Chem. Sci. J. 2013, 3, 137–150. [Google Scholar] [CrossRef]
- Marino, T.; Russo, N.; Sicilia, E.; Toscano, M. Tautomeric Equilibria of 2- and 4-Thiouracil in Gas Phase and in Solvent: A Density Functional Study. Int. J. Quantum Chem. 2001, 82, 44–52. [Google Scholar] [CrossRef]
- Psoda, A.; Shugar, D. Structure and Tautomerism of The Neutral and Monoanionic Forms of 2-Thiouracil, 2,4-Dithiouracil, Their Nucleosides, and Some Related Derivatives. Acta Biochim. Pol. 1979, 26, 55–72. [Google Scholar]
- Ghomi, M.; Letellier, R.; Taillandier, E.; Chinsky, L.; Laigle, A.; Turpin, P. Interpretation of the vibrational modes of uracil and its 18O-substituted and thio derivatives studied by resonance Raman spectroscopy. J. Raman Spectrosc. 1986, 17, 249–255. [Google Scholar] [CrossRef]
- Igarashi-Yamamoto, N.; Tajiri, A.; Hatano, M.; Shibuya, S.; Ueda, T. Ultraviolet absorption, circular dichroism studies of sulfur-containing nucleic acid bases and their nucleosides. Biochim. Biophys. Acta 1981, 656, 1–15. [Google Scholar] [CrossRef]
- Christensen, H.N. Ultrafiltrability of Thiouracil in Human Serum; Determination of thiouracil. J. Biol. Chem. 1945, 160, 425–433. [Google Scholar]
- Wang, W.; Schuchmann, M.N.; Schuchmann, H.-P.; Knolle, W.; Von Sonntag, J.; Von Sonntag, C. Radical Cations in the OH-Radical-Induced Oxidation of Thiourea and Tetramethylthiourea in Aqueous Solution. J. Am. Chem. Soc. 1999, 121, 238–245. [Google Scholar] [CrossRef]
- Schuler, R.H.; Hartzell, A.L.; Behar, B. Track effects in radiation chemistry. Concentration dependence for the scavenging of OH by ferrocyanide in N2O-saturated aqueous solutions. J. Phys. Chem. 1981, 85, 192–199. [Google Scholar] [CrossRef]
- Schöneich, C.; Pogocki, D.; Hug, G.L.; Bobrowski, K. Free radical reactions of methionine in peptides: Mechanisms relevant to b-amyloid oxidation and Alzheimer’s disease. J. Am. Chem. Soc. 2003, 125, 13700–13713. [Google Scholar] [CrossRef]
- Bobrowski, K.; Hug, G.L.; Pogocki, D.; Marciniak, B.; Schoneich, C. Stabilization of sulfide radical cations through complexation with the peptide bond: Mechanisms relevant to oxidation of proteins containing multiple methionine residues. J. Phys. Chem. B. 2007, 111, 9608–9620. [Google Scholar] [CrossRef]
- Asmus, K.-D. Some conductivity experiments in pulse radiolysis. Int. J. Radiat. Phys. Chem. 1972, 4, 417–437. [Google Scholar] [CrossRef]
- Eyring, E.M. Fast Reactions in Solutions. Surv. Prog. Chem. 1964, 2, 57–89. [Google Scholar]
- Goyal, R.N.; Singh, U.P.; Abdullah, A.A. Electrochemical oxidation of 2-thiouracil at pyrolitic graphite electrode. Bioelectrochemistry 2005, 67, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Wardman, P. The reduction potentials of one-electron couples involving free radicals in aqueous solution. J. Phys. Chem. Ref. Data 1989, 18, 1637–1753. [Google Scholar] [CrossRef] [Green Version]
- Skotnicki, K.; De la Fuente, J.R.; Cañete, A.; Berrios, E.; Bobrowski, K. Radical Ions of 3-Styryl-quinoxalin-2-one Derivatives Studied by Pulse Radiolysis in Organic Solvents. J. Phys. Chem. B 2018, 122, 4051–4066. [Google Scholar] [CrossRef] [PubMed]
- Janik, I.; Carmichael, I.; Tripathi, G.N.R. Transient Raman spectra, structure, and thermochemistry of the thiocyanate dimer radical anion in water. J. Chem. Phys. 2017, 146, 214305. [Google Scholar] [CrossRef] [Green Version]
- Janik, I.; Tripathi, G.N.R. The selenocyanate dimer radical anion in water: Transient Raman spectra, structure, and reaction dynamics. J. Chem. Phys. 2019, 150, 094304. [Google Scholar] [CrossRef]
- Janik, I.; Tripathi, G.N.R. The early events in the OH radical oxidation of dimethyl sulfide in water. J. Chem. Phys. 2013, 138, 044506. [Google Scholar] [CrossRef]
- Schöneich, C.; Bobrowski, K. Intramolecular hydrogen transfer as the key step in the dissociation of hydroxyl radical adducts of (alkylthio)ethanol derivatives. J. Am. Chem. Soc. 1993, 115, 6538–6547. [Google Scholar] [CrossRef]
- Wang, F.; Pernot, P.; Marignier, J.-L.; Archirel, P.; Mostafavi, M. Mechanism of (SCN)2·− Formation and Decay in Neutral and Basic KSCN Solution under Irradiation from a Pico- to Microsecond Range. J. Phys. Chem. B 2019, 123, 6599–6608. [Google Scholar] [CrossRef]
- Bobrowski, K.; Schöneich, C. Hydroxyl radical adduct at sulfur in substituted organic sulfides stabilized by internal hydrogen bond. J. Chem. Soc., Chem. Commun. 1993, 795–797. [Google Scholar] [CrossRef]
- Chipman, D.M. Hemibonding between Hydroxyl Radical and Water. J. Phys. Chem. A 2011, 115, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Treinin, A.; Hayon, E. Charge transfer spectra of halogen atoms in water. Correlation of the electronic transition energies of iodine, bromine, chlorine, hydroxyl, and hydrogen radicals with their electron affinities. J. Am. Chem. Soc. 1975, 97, 1716–1721. [Google Scholar] [CrossRef]
- Prasanthkumar, K.P.; Suresh, C.H.; Aravindakumar, C.T. Theoretical study of the addition and abstraction reactions of hydroxyl radical with uracil. Radiat. Phys. Chem. 2012, 81, 267–272. [Google Scholar] [CrossRef]
- Francés-Monerris, A.; Merchán, M.; Roca-Sanjuán, D. Theoretical Study of the Hydroxyl Radical Addition to Uracil and Photochemistry of the Formed U6OH• Adduct. J. Phys. Chem. B 2014, 118, 2932–2939. [Google Scholar] [CrossRef]
- Fujita, S.; Steenken, S. Pattern of OH radical addition to uracil and methyl-, and carboxyl-substituted uracils. Electron transfer of OH adduct with N,N,N’,N’-tetramethyl-p-phenylene diamine and tetranitromethane. J. Am. Chem. Soc. 1981, 103, 2540–2545. [Google Scholar] [CrossRef]
- Domin, D.; Braida, B.; Berges, J. Influence of Water on the Oxidation of Dimethyl Sulfide by the •OH Radical. J. Phys. Chem. B 2017, 121, 9321–9330. [Google Scholar] [CrossRef] [Green Version]
- Fourré, I.; Bergés, J. Structural and topological characterization of three-electron bond: The S\O radicals. J. Phys. Chem. A 2004, 108, 898–906. [Google Scholar] [CrossRef]
- Bobrowski, K.; Pogocki, D.; Schöneich, C. Mechanism of the OH radical-induced decarboxylation of 2-(alkylthio)ethanoic acid derivatives. J. Phys. Chem. 1993, 97, 13677–13684. [Google Scholar] [CrossRef]
- Schöneich, C.; Pogocki, D.; Wisniowski, P.; Hug, G.L.; Bobrowski, K. Intramolecular Sulfur-Oxygen Bond Formation in Radical Cations of N-Acetylmethionine Amide. J. Am. Chem. Soc. 2000, 122, 10224–10225. [Google Scholar] [CrossRef]
- Bobrowski, K. Free radicals in chemistry, biology and medicine: Contribution of radiation chemistry. Nukleonika 2005, 50 (Supplement 3), S67–S76. [Google Scholar]
- Mirkowski, J.; Wisniowski, P.; Bobrowski, K. INCT Annual Report 2000; INCT: Warsaw, Poland, 2001; pp. 31–33. [Google Scholar]
- Hug, G.L.; Wang, Y.; Schöneich, C.; Jiang, P.-Y.; Fessenden, R.W. Multiple time scales in pulse radiolysis. Application to bromide solutions and dipeptides. Radiat. Phys. Chem. 1999, 54, 559–566. [Google Scholar] [CrossRef]
- Schuler, R.H.; Patterson, L.K.; Janata, E. Yield for the scavenging of hydroxyl radicals in the radiolysis of nitrous oxide-saturated aqueous solutions. J. Phys. Chem. 1980, 84, 2088–2090. [Google Scholar] [CrossRef]
- Janata, E. Pulse radiolysis conductivity measurements in aqueous solutions with nanosecond time resolution. Radiat. Phys. Chem. 1982, 19, 17–21. [Google Scholar] [CrossRef]
- Veltwisch, D.; Janata, E.; Amus, K.-D. Primary processes in the reaction of OH-radicals with sulphoxides. J.C.S. Perkin Trans. II 1980, 146–153. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Vydrov, A.; Scuseria, G.E. Assessment of a long-range corrected hybrid functional. J. Chem. Phys. 2006, 125, 234109. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef] [Green Version]
- Tomasi, J.; Mennuci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Laurent, A.D.; Jacquemin, D. TD-DFT benchmarks: A review. Int. J. Quantum Chem. 2013, 113, 2019–2039. [Google Scholar] [CrossRef]
- Dupont, C.; Dumont, É.; Jacquemin, D. Superior Performance of Range-Separated Hybrid Functionals for Describing σ* ← σ UV–Vis Signatures of Three-Electron Two-Center Anions. J. Phys. Chem. A 2012, 116, 3237–3246. [Google Scholar] [CrossRef] [PubMed]
- Milhøj, B.O.; Sauer, S.P.A. Kinetics and Thermodynamics of the Reaction between the •OH Radical and Adenine: A Theoretical Investigation. J. Phys. Chem. A 2015, 119, 6516–6527. [Google Scholar] [CrossRef] [PubMed]
- Francés-Monerris, A.; Merchán, M.; Roca-Sanjuán, D. Mechanism of the OH Radical Addition to Adenine from Quantum-Chemistry Determinations of Reaction Paths and Spectroscopic Tracking of the Intermediates. J. Org. Chem. 2017, 82, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Frances-Monerris, A.; Merchan, M.; Roca-Sanjuan, D. Communication: Electronic UV-Vis transient spectra of the center dot OH reaction products of uracil, thymine, cytosine, and 5,6-dihydrouracil by using the complete active space self-consistent field second-order perturbation (CASPT2//CASSCF) theory. J. Chem. Phys. 2013, 139. [Google Scholar] [CrossRef] [Green Version]
- Ipatov, A.; Cordova, F.; Doriol, L.C.; Casid, M.E. Excited-state spin-contamination in time-dependent density-functional theory for molecules with open-shell ground states. J. Mol. Struc.-Theochem. 2009, 60–73. [Google Scholar] [CrossRef]
- Creating UV/Visible Plots from the Results of Excited States Calculations. Available online: http://gaussian.com/uvvisplot/ (accessed on 29 November 2019).
Sample Availability: Samples of the 2-thiouracil are available from the authors. |














© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skotnicki, K.; Taras-Goslinska, K.; Janik, I.; Bobrowski, K. Radiation Induced One-Electron Oxidation of 2-Thiouracil in Aqueous Solutions. Molecules 2019, 24, 4402. https://doi.org/10.3390/molecules24234402
Skotnicki K, Taras-Goslinska K, Janik I, Bobrowski K. Radiation Induced One-Electron Oxidation of 2-Thiouracil in Aqueous Solutions. Molecules. 2019; 24(23):4402. https://doi.org/10.3390/molecules24234402
Chicago/Turabian StyleSkotnicki, Konrad, Katarzyna Taras-Goslinska, Ireneusz Janik, and Krzysztof Bobrowski. 2019. "Radiation Induced One-Electron Oxidation of 2-Thiouracil in Aqueous Solutions" Molecules 24, no. 23: 4402. https://doi.org/10.3390/molecules24234402
APA StyleSkotnicki, K., Taras-Goslinska, K., Janik, I., & Bobrowski, K. (2019). Radiation Induced One-Electron Oxidation of 2-Thiouracil in Aqueous Solutions. Molecules, 24(23), 4402. https://doi.org/10.3390/molecules24234402