
Heparanase as an Additional Tool for Detecting Structural Peculiarities of Heparin Oligosaccharides


Abstract
:1. Introduction
2. Results and Discussion
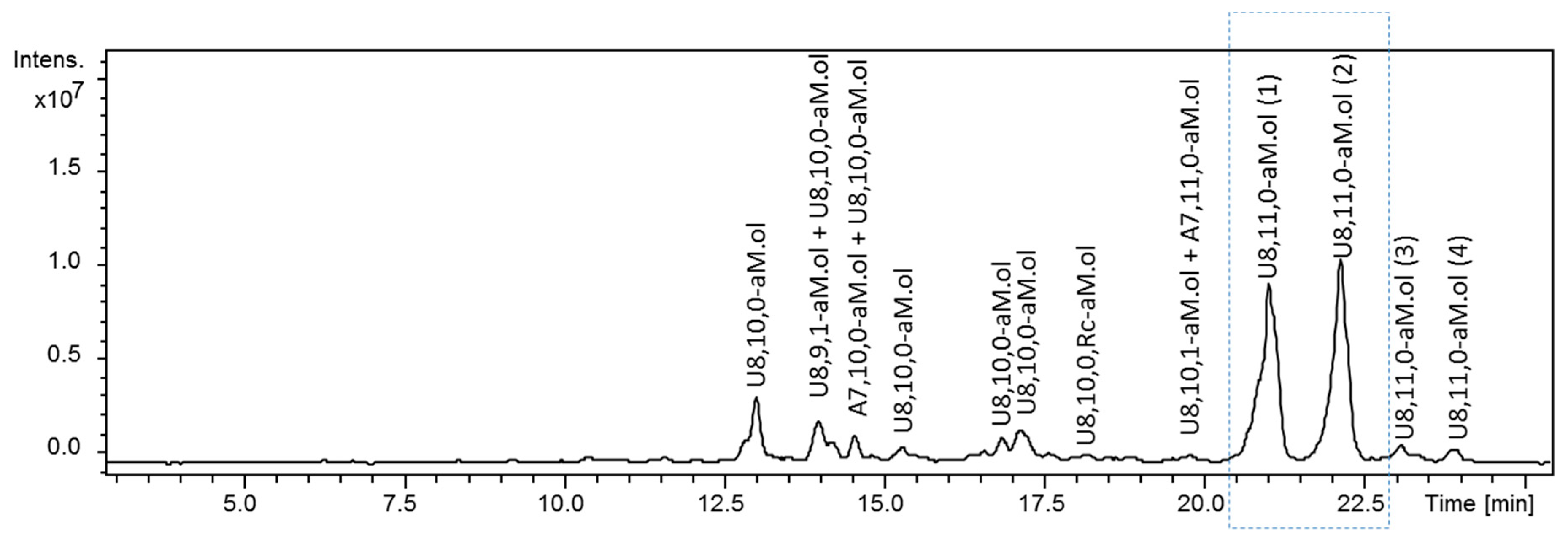
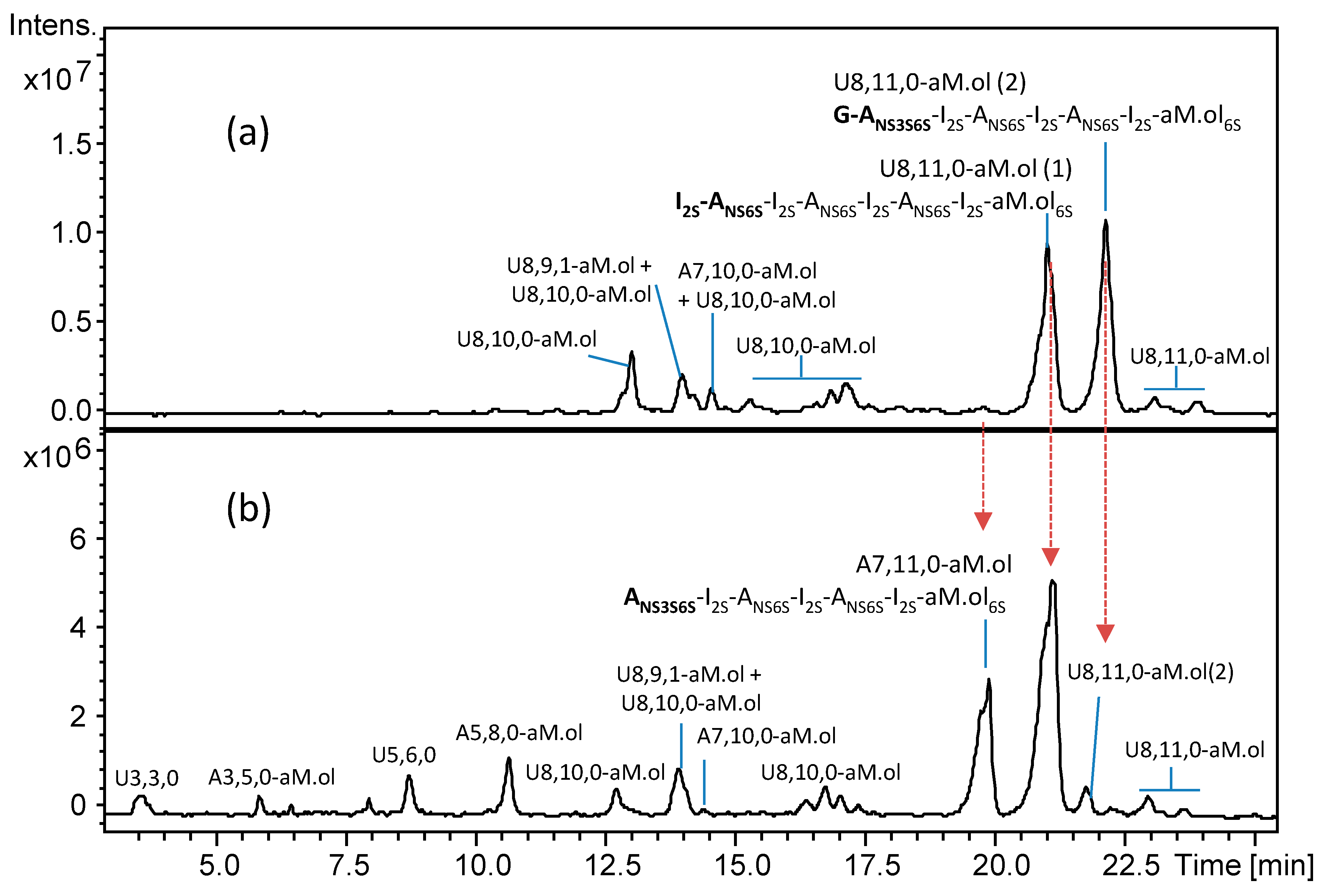
2.1. Identification of G–ANS3S6S Sequence at the NRE of Dalteparin Octasaccharides
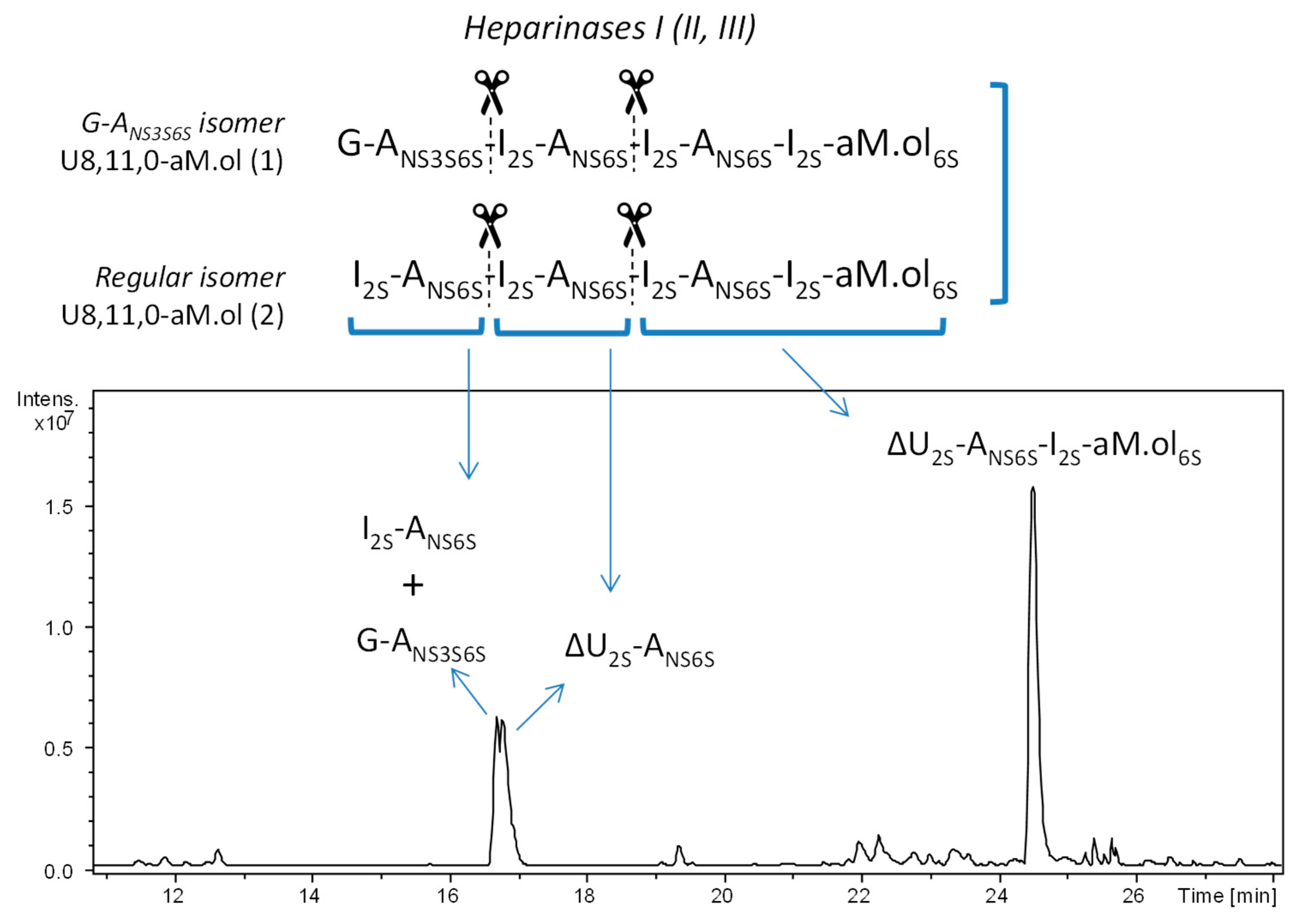
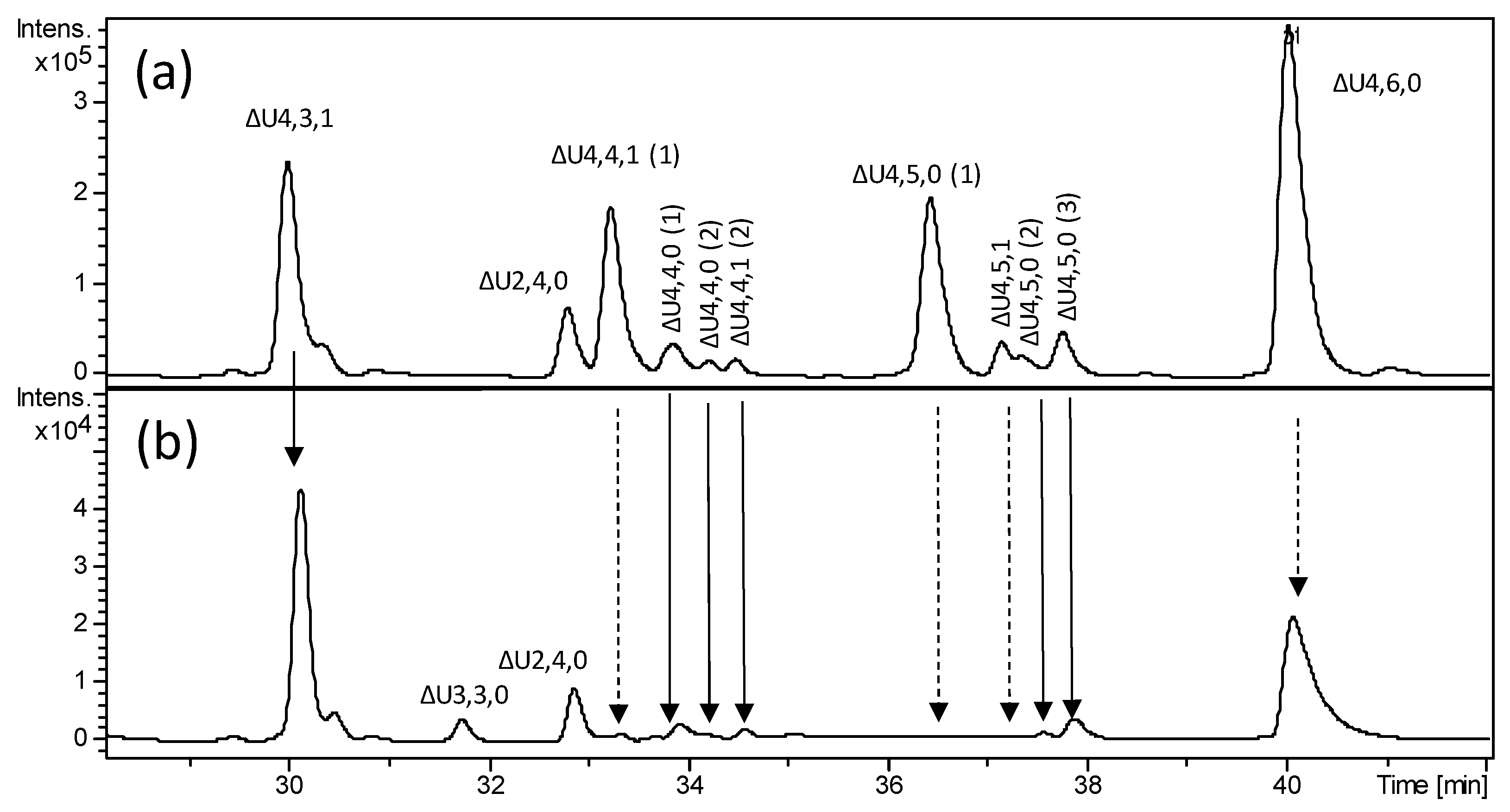
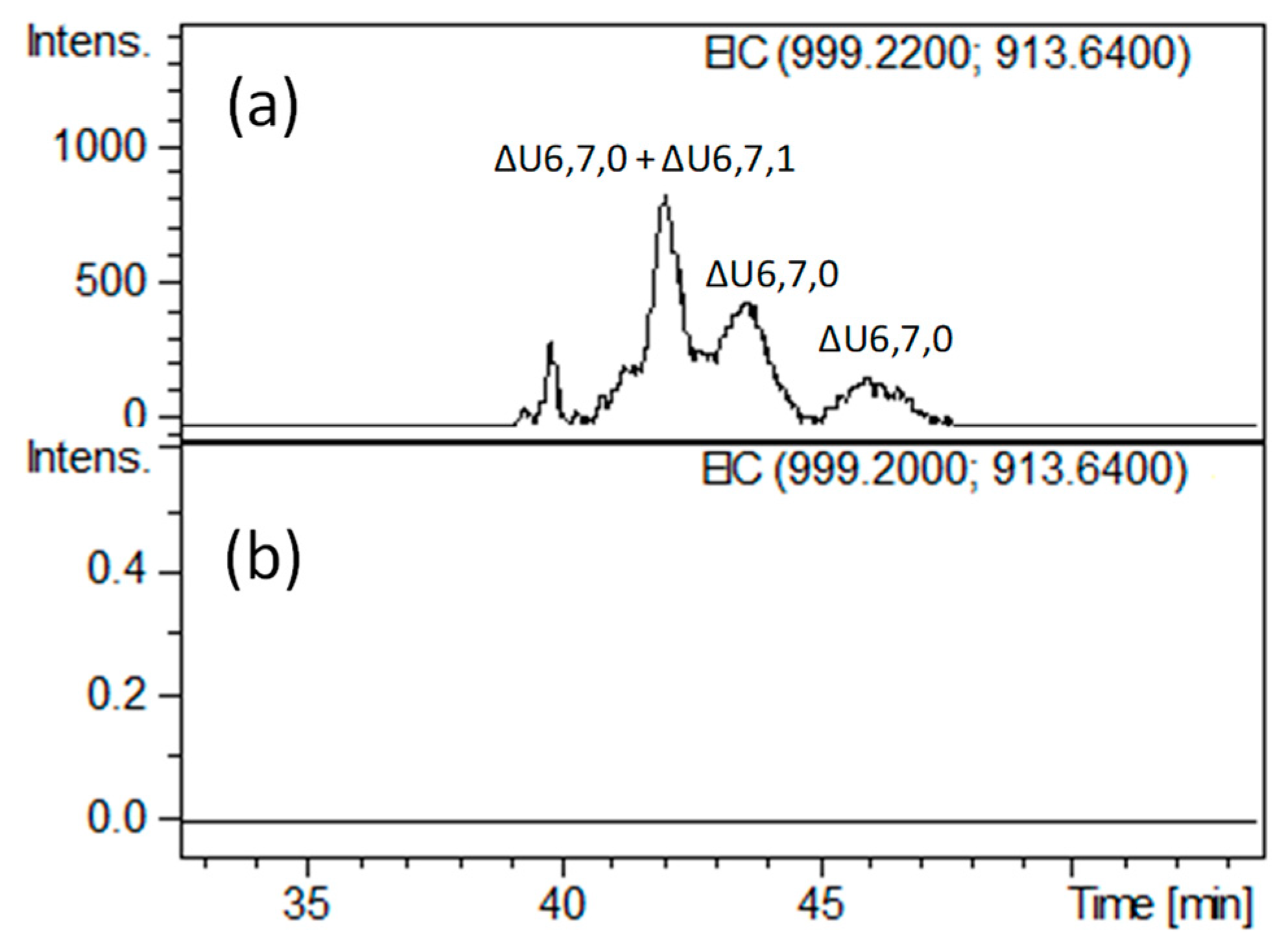
2.2. Structural Elucidation of Heparin G–ANS3S6S Containing Oligomers Resistant to Heparinase Action
3. Materials and Methods
3.1. Materials and Reagents
3.2. Isolation of Dalteparin Fraction by SEC-UV
3.3. Isolation of a Fraction with High Affinity towards AT(III) from Bovine Mucosal Heparin by Preparative Affinity Chromatography
3.4. Heparinases I, II and III Depolymerization
3.5. Heparanase Depolymerization
3.6. LC-MS Analysis
3.7. Nomenclature
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Casu, B.; Lindahl, U. Structure and biological interactions of heparin and heparan sulfate. Adv. Carbohydr. Chem. Biochem. 2001, 57, 159–206. [Google Scholar] [PubMed]
- Casu, B.; Naggi, A.; Torri, G. Re-visiting the structure of heparin. Carbohydr. Res. 2015, 403, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Murakami, T.; Tsuda, H.; Yoshida, K.; Sugahara, K. Isolation of the porcine heparin tetrasaccharides with glucuronate 2-O-sulfate. Heparinase cleaves glucuronate 2-O-sulfate containing disaccharides in highly sulfated blocks in heparin. J. Biol. Chem. 1995, 270, 8696–8705. [Google Scholar] [PubMed]
- Casu, B.; Oreste, P.; Torri, G.; Zoppetti, G. The structure of heparin oligosaccharide fragments with high anti-(factor Xa) activity containing the minimal antithrombin III-binding sequence Chemical and 13C nuclear-magnetic-resonance studies. Biochem. J. 1981, 197, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Van Boeckel, C.A.A.; Petitou, M. A unique antithrombin binding domain of heparin, a lead to new synthetic antithrombotics. Angew. Chem. Int. Ed. 1993, 32, 1671–1688. [Google Scholar] [CrossRef]
- Petitou, M.; Van Boeckel, C.A.A. Synthetic antithrombin III binding pentasaccharide is now a drug! What comes next? Angew. Chem. Int. Ed. 2004, 42, 3118–3133. [Google Scholar] [CrossRef]
- Naggi, A.; Gardini, C.; Pedrinola, G.; Mauri, L.; Urso, E.; Alekseeva, A.; Casu., B.; Cassinelli, G.; Guerrini, M.; Iacomini, M.; et al. Structural peculiarity and antithrombin binding region profile of mucosal bovine and porcine heparins. J. Pharm. Biomed. Anal. 2016, 118, 52–64. [Google Scholar] [CrossRef]
- Guerrini, M.; Guglieri, S.; Naggi, A.; Sasisekharan, R.; Torri, G. Low molecular weight heparins: structural differentiation by bidimentional nuclear magnetic resonance spectroscopy. Semin. Thromb. Hemost. 2007, 33, 478–487. [Google Scholar] [CrossRef]
- Bisio, A.; Vecchietti, D.; Citterio, L.; Guerrini, M.; Raman, R.; Bertini, S.; Eisele, G.; Naggi, A.; Sasisekharan, R.; Torri, G. Structural features of low-molecular weight heparins affecting their affinity to antithrombin. Thromb. Haemost. 2009, 102, 865–873. [Google Scholar] [CrossRef]
- Mourier, P.; Viskov, C. Chromatographic analysis and sequencing approach of heparin oligosaccharides using cetyltrimethylammonium dynamically coated stationary phases. Anal. Biochem. 2004, 332, 299–313. [Google Scholar] [CrossRef]
- Huang, Y.; Mao, Y.; Zong, C.; Lin, C.; Boons, G.J.; Zaia, J. Discovery of a Heparan Sulfate 3-O-Sulfation Specific Peeling Reaction. Anal. Chem. 2015, 87, 592–600. [Google Scholar] [CrossRef] [PubMed]
- Bisio, A.; Urso, E.; Guerrini, M.; Witt, P.; Torri, G.; Naggi, A. Structural Characterization of the Low-Molecular-Weight Heparin Dalteparin by Combining Different Analytical Strategies. Molecules 2017, 22, 1051. [Google Scholar] [CrossRef] [PubMed]
- Langeslay, D.J.; Urso, E.; Gardini, C.; Naggi, A.; Torri, G.; Larive, C.K. Reversed-phase ion-pair ultra-high-performance-liquid chromatography–mass spectrometry for fingerprinting low-molecular-weight heparins. J. Chromatogr. A 2013, 1292, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Guo, Z.; Yu, M.; Lin, C.; Sheng, A.; Wang, Z.; Linhardt, R.J.; Chi, L. Hydrophilic interaction chromatography-multiple reaction monitoring mass spectrometry method for basic building block analysis of low molecular weight heparins prepared through nitrous acid depolymerization. J. Chromatogr. A 2017, 1479, 121–128. [Google Scholar] [CrossRef]
- Li, G.; Steppich, J.; Wang, Z.; Sun, Y.; Xue, C.; Linhardt, R.J.; Li, L. Bottom-Up Low Molecular Weight Heparin Analysis Using Liquid Chromatography-Fourier Transform Mass Spectrometry for Extensive Characterization. Anal. Chem. 2014, 86, 6626–6632. [Google Scholar] [CrossRef]
- Chen, Y.; Lin, L.A.; Zhang, X.; Ange, K. St.; Yu, Y.; Zhang, F.; Liu, J.; Amster, I.J.; Linhardt, R.J. Structural analysis of heparin-derived 3-O-sulfated tetrasaccharides: antithrombin binding site variants. Carbohydr. Polym. 2017, 157, 244–250. [Google Scholar] [CrossRef]
- Xiao, Z.; Tappen, B.R.; Ly, M.; Zhao, W.; Canova, L.P.; Guan, H.; Linhardt, R. Heparin mapping using heparin lyases and the generation of a novel low molecular weight heparin. J. Med. Chem. 2011, 54, 603–610. [Google Scholar] [CrossRef]
- Sommers, D.C.; Ye, H.; Kolinski, R.E.; Nasr, M.; Buhse, L.F.; Al-Halim, A.; Keire, D. Characterization of currently marketed heparin products: Analysis of molecular weight and heparinase-I digest patterns. Anal. Bioanal. Chem. 2011, 401, 2445–2454. [Google Scholar] [CrossRef]
- Wang, B.; Buhse, L.; Al-Hakim, A.; Boyne Ii, M.T.; Keire, D.A. Characterization of currently marketed heparin products: Analysis of heparin digests by RPIP-UHPLC-QTOF-MS. J. Pharm. Biomed. Anal. 2012, 67, 42–50. [Google Scholar] [CrossRef]
- Mourier, P.; Anger, P.; Martinez, C.; Herman, F.; Viskov, C. Quantitative compositional analysis of heparin using exhaustive heparinase digestion and strong anion exchange chromatography. Anal. Chem. Res. 2015, 3, 46–53. [Google Scholar] [CrossRef]
- Lee, S.; Raw, A.; Yu, L.; Lionberger, R.; Ya, N.; Verthelyi, D.; Rosenberg, A.; Kozlowski, S.; Webber, K.; Woodcock, J. Scientific considerations in the review and approval of generic enoxaparin in the United States. Nat. Biotechnol. 2013, 31, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Fairbanks, M.B.; Mildner, A.M.; Leone, J.W.; Cavey, G.S.; Mathews, W.R.; Drong, R.F.; Slightom, J.L.; Bienkowski, M.J.; Smith, C.W.; Bannow, C.A.; et al. Processing of the human heparanase precursor and evidence that the active enzyme is a heterodimer. J. Biol. Chem. 1999, 274, 29587–29590. [Google Scholar] [CrossRef] [PubMed]
- Levy-Adam, F.; Miao, H.; Heinrikson, R.L.; Vlodavsky, I.; Ilan, N. Heterodimer formation is essential for heparanase enzymatic activity. Biochem. Biophys. Res. Commun. 2003, 308, 885–891. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Friedmann, Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J. Clin. Invest. 2001, 108, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Pisano, C.; Vlodavsky, I.; Ilan, N.; Zunino, F. The potential of heparanase as a therapeutic target in cancer. Biochem. Pharmacol. 2014, 89, 12–19. [Google Scholar] [CrossRef]
- Okada, Y.; Yamada, S.; Toyoshima, M.; Dong, J.; Nakajima, M.; Sugahara, K. Structural recognition by recombinant human heparanase that plays critical roles in tumor metastasis. Hierarchical sulfate groups with different effects and the essential target disulfated trisaccharide sequence. J. Biol. Chem. 2002, 277, 42488–42495. [Google Scholar] [CrossRef]
- Peterson, S.B.; Liu, J. Multi-faceted substrate specificity of heparanase. Matrix Biol. 2013, 32, 223–227. [Google Scholar] [CrossRef]
- Mao, Y.; Huang, Y.; Buczek-Thomas, J.A.; Ethen, C.M.; Nugent, M.A.; Wu, Z.L.; Zaia, J. A liquid chromatography-mass spectrometry-based approach to characterize the substrate specificity of mammalian heparanase. JBC 2014, 289, 34141–34151. [Google Scholar] [CrossRef]
- Bisio, A.; Mantegazza, A.; Urso, E.; Naggi, A.; Torri, G.; Viskov, C.; Casu, B. High-Performance Liquid Chromatographic/Mass Spectrometric Studies on the Susceptibility of Heparin Species to Cleavage by Heparanase. Seminars Thrombosis Hemostasis 2007, 33, 488–495. [Google Scholar] [CrossRef]
- Chase, C.; Elaine, G.; Paulo, M. Diversifying the global heparin supply chain: reintroduction of bovine heparin in the United States? Pharm. Technol. 2015, 39, 28–35. [Google Scholar]
- Loganathan, D.; Wang, H.M.; Mallis, L.M.; Linhardt, R.J. Structural variation in the antithrombin III binding site region and its occurrence in heparin from different sources. Biochemistry 1990, 29, 4362–4368. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Yoshida, K.; Sugiura, M.; Sugahara, K.; Khoo, K.H.; Morris, H.R.; Dell, A. Structural studies on the bacterial lyase-resistant tetrasaccharides derived from the antithrombin III-binding site of porcine intestinal heparin. J. Biol. Chem. 1993, 268, 4780–4787. [Google Scholar] [PubMed]
- Li, G.; Yang, B.; Zhang, F.; Xue, C.; Linhardt, R.J. Analysis of 3-O-sulfo group-containing heparin tetrasaccharides in heparin by liquid chromatography-mass spectrometry. J. Biochem. 2014, 449, 3–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Zhang, T.; Xie, S.; Liu, X.; Li, H.; Linhardt, R.J.; Chi, L. Sequencing of oligosaccharide pool in the low molecular weight heparin dalteparin with offline HPLC and ESI-MS/MS. Carbohydr. Polym. 2018, 183, 81–90. [Google Scholar] [CrossRef]
- Alekseeva, A.; Casu, B.; Cassinelli, G.; Guerrini, M.; Torri, G.; Naggi, A. Structural features of glycol-split low-molecular-weight heparins and their heparin lyase generated fragments. Anal. Bioanal. Chem. 2014, 406, 249–265. [Google Scholar] [CrossRef] [Green Version]
- Linhardt, R.J.; Loganathan, D.; Al-Hakim, A.; Wang, H.M.; Walenga, J.M.; Hoppensteadt, D.; Fareed, J. Oligosaccharide mapping of low molecular weight heparins: structure and activity differences. J. Med. Chem. 1990, 33, 1639–1645. [Google Scholar] [CrossRef]
- Lemoine, J.; Fournet, B.; Despeyroux, D.; Jennings, K.R.; Rosenberg, R.; De Hoffmann, E. Collision-Induced Dissociation of Alkali Metal Cationized and Permethylated Oligosaccharides: Influence of the Collision Energy and of the Collision Gas for the Assignment of Linkage Position. J. Am. Soc. Mass Spectrom. 1993, 4, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Spina, E.; Cozzolino, R.; Ryan, E.; Garozzo, D.J. Sequencing of Oligosaccharides by Collision Induced Dissociation Matrix Assisted Laser Desorption/Ionization Mass Spectrometry. Mass Spectrom. 2000, 35, 1042–1048. [Google Scholar] [CrossRef]
- Shi, X.; Huang, Y.; Mao, Y.; Naimy, H.; Zaia, J. Tandem mass spectrometryof heparan sulfate negative ions: sulfate loss patterns and chemical modification methods for improvement of product ion profiles. J. Am. Soc. Mass Spectrom. 2012, 23, 1498–1511. [Google Scholar] [CrossRef] [Green Version]
- Peterson, S.B.; Liu, J. Deciphering mode of action of heparanase using structurally defined oligosaccharides. J. Biol. Chem. 2012, 287, 34836–34843. [Google Scholar] [CrossRef] [Green Version]
- Turnbull, J.E.; Hopwood, J.J.; Gallagher, J.T. A strategy for rapid sequencing of heparan sulphate and heparin saccharides. Proc. Natl. Acad. Sci. USA 1999, 96, 2698–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nardella, C.; Lahm, A.; Pallaoro, M.; Brunetti, M.; Vannini, A.; Steinkuhler, C. Mechanism of Activation of Human Heparanase Investigated by Protein Engineering. Biochemistry 2004, 43, 1862–1873. [Google Scholar] [CrossRef] [PubMed]
- Bitter, T.; Muir, H.M. A modified uronic acid carbazole reaction. Anal. Biochem. 1962, 4, 330–334. [Google Scholar] [CrossRef]
- Domon, B.; Costello, C.E. A Systematic Nomenclature for Carbohydrate Fragmentations in FAB-MS/MS Spectra of Glycoconjugates. Glycoconjugate J. 1988, 5, 397–409. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| m/z | z | Fragmentation Ion | |
|---|---|---|---|
| Isomer No. 1 (U–ANS3S6S) | Isomer No. 2 (U2S–ANS6S) | ||
| 137.99 | −1 | 0,2X2 (+1 SO3) | 0,2X2 (+1 SO3) |
| 168.49 | −2 | Y2 (+2 SO3) | Y2 (+2 SO3) |
| 175.03 | −1 | B1 | B1 |
| 198.99 | −1 | 0,2A2 (+1 SO3) | - |
| 210.99 | −1 | - | B1 – CO2 (+1 SO3) |
| 222.01 | −1 | - | Z2 – H2O (+1 SO3) |
| Oligosaccharide Identification by Current Nomenclature | Structure |
|---|---|
| ∆U4,3,1 * | ΔU–ANAc6S–G–ANS3S |
| ∆U4,4,1(1) | ΔU–ANAc6S–G–ANS3S6S |
| ∆U4,4,1 (2)* | ΔU2S–ANAc6S–G–ANS3S $ |
| ∆U4,4,0 (1) * | ΔU–ANS6S–G–ANS3S $ |
| ∆U4,4,0 (2) * | ΔU2S–ANS–G–ANS3S $ |
| ∆U4,5,0(1) | ΔU–ANS6S–G–ANS3S6S |
| ∆U4,5,0 (2) * | ΔU2S–ANS–G–ANS3S6S |
| ∆U4,5,0 (3) * | ΔU2S–ANS6S–G–ANS3S $ |
| ∆U4,5,1 | ΔU2S–ANAc6S–G–ANS3S6S |
| ∆U4,6,0 * | ΔU2S–ANS6S–G–ANS3S6S |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alekseeva, A.; Urso, E.; Mazzini, G.; Naggi, A. Heparanase as an Additional Tool for Detecting Structural Peculiarities of Heparin Oligosaccharides. Molecules 2019, 24, 4403. https://doi.org/10.3390/molecules24234403
Alekseeva A, Urso E, Mazzini G, Naggi A. Heparanase as an Additional Tool for Detecting Structural Peculiarities of Heparin Oligosaccharides. Molecules. 2019; 24(23):4403. https://doi.org/10.3390/molecules24234403
Chicago/Turabian StyleAlekseeva, Anna, Elena Urso, Giulia Mazzini, and Annamaria Naggi. 2019. "Heparanase as an Additional Tool for Detecting Structural Peculiarities of Heparin Oligosaccharides" Molecules 24, no. 23: 4403. https://doi.org/10.3390/molecules24234403
APA StyleAlekseeva, A., Urso, E., Mazzini, G., & Naggi, A. (2019). Heparanase as an Additional Tool for Detecting Structural Peculiarities of Heparin Oligosaccharides. Molecules, 24(23), 4403. https://doi.org/10.3390/molecules24234403