
Identification and Quantitation of Ursolic and Oleanolic Acids in Ilex aquifolium L. Leaf Extracts Using 13C and 1H-NMR Spectroscopy


Abstract
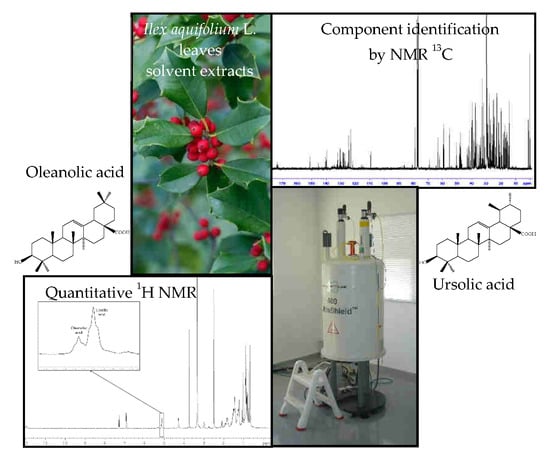
:
1. Introduction
- -
- HPLC (or reversed-phase RP-HPLC) with various detector modes (UV, evaporative light scattering detection, photodiode array detection) appeared as the most employed technique for quantitation of UA and/or OA in plant extracts from Lantana camara [19], Salvia chinensis [20], Thymus ssp [21], and Nyctanthes arbor-tristis [22].
- -
- -
- -
- -
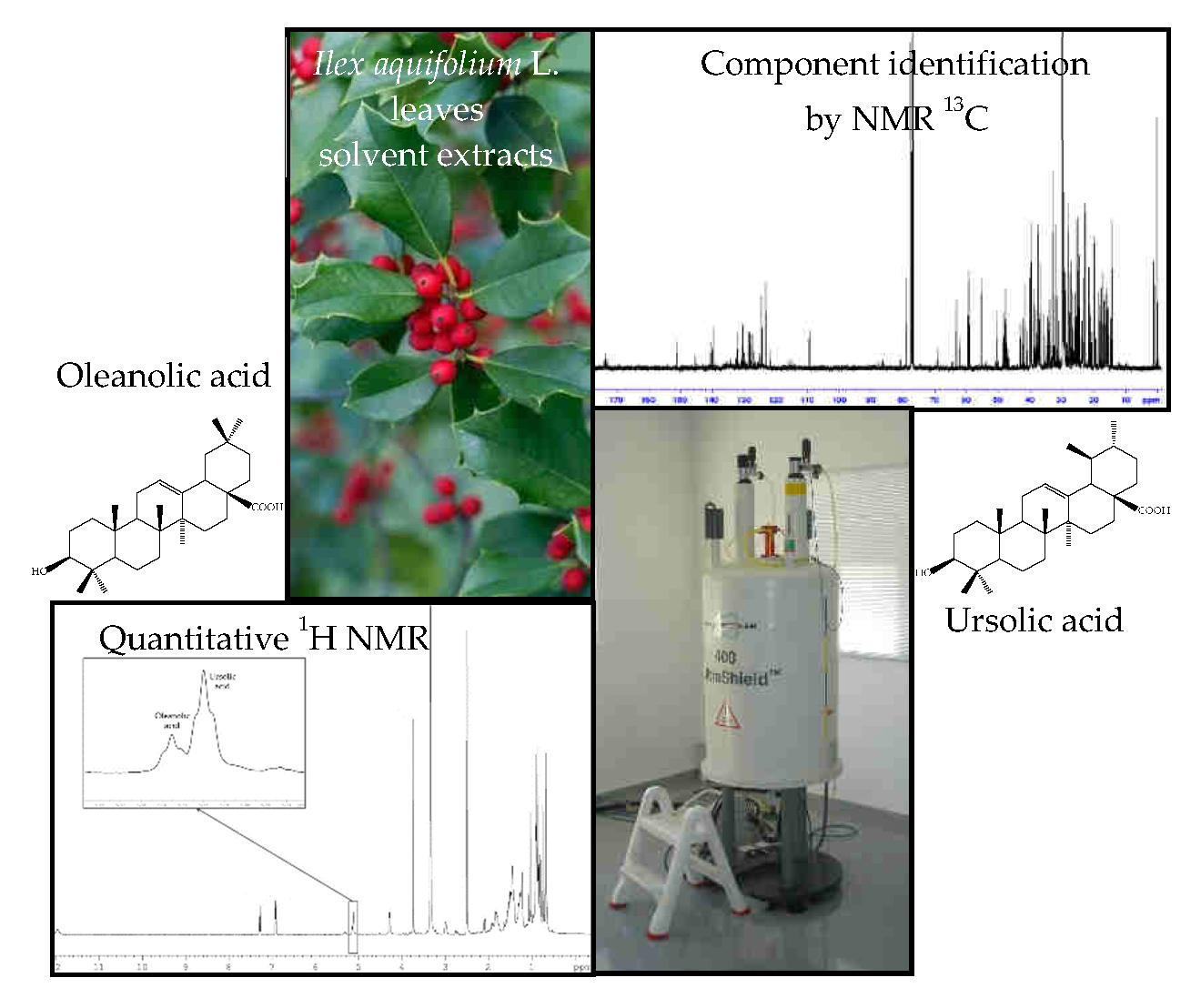
- Apart from chromatographic techniques, NMR has been punctually used for quantitation of UA and/or OA in plant extracts. OA has been quantified in a precipitate obtained during the industrial extraction of the leaves of Olea europaea using 13C-NMR spectroscopy [29]. Otherwise, the combined use of proton-carbon heteronuclear single-quantum coherence (HSQC) and proton-carbon heteronuclear multiple-bond correlation (HMBC), allowed the identification and quantitation of OA and UA in plant extracts of the Lamiaceae and Oleaceae family [30].
2. Results and Discussion
2.1. Phytochemicals Contained in I. aquifolium Leaf Extracts
2.1.1. Hexane Extract from Leaves of I. aquifolium
- -
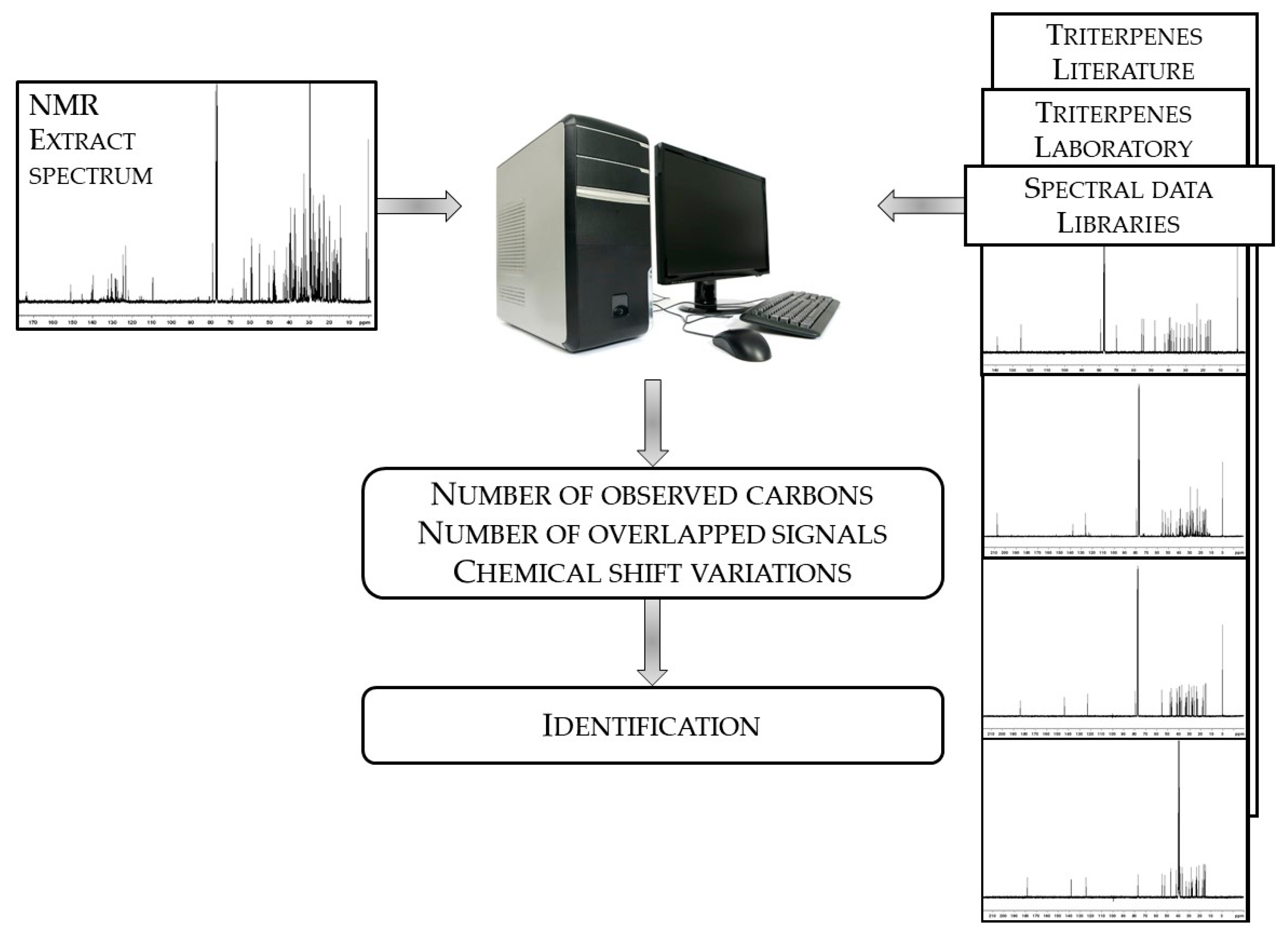
- NMR spectra of fractions H2–H4 displayed the signals of all carbons of α-amyrin and β-amyrin as well as those of saturated and unsaturated fatty acid chains. In order to evaluate the nature of these fatty acid chains, a fraction H2 of CC was submitted to trans-esterification. Basic hydrolysis was followed by methylation (BF3-Methanol). The organic layer was submitted to GC (Retention indices) and GC-MS analyses. Five methyl esters have been identified with characteristic chains of palmitic acid (C16:0; 54.1%), linolenic acid (C18:3; 13.2%), oleic acid (C18:1; 11.3%), linoleic acid (C18:2; 10.0%), and stearic acid (C18:0; 3.0%).
- -
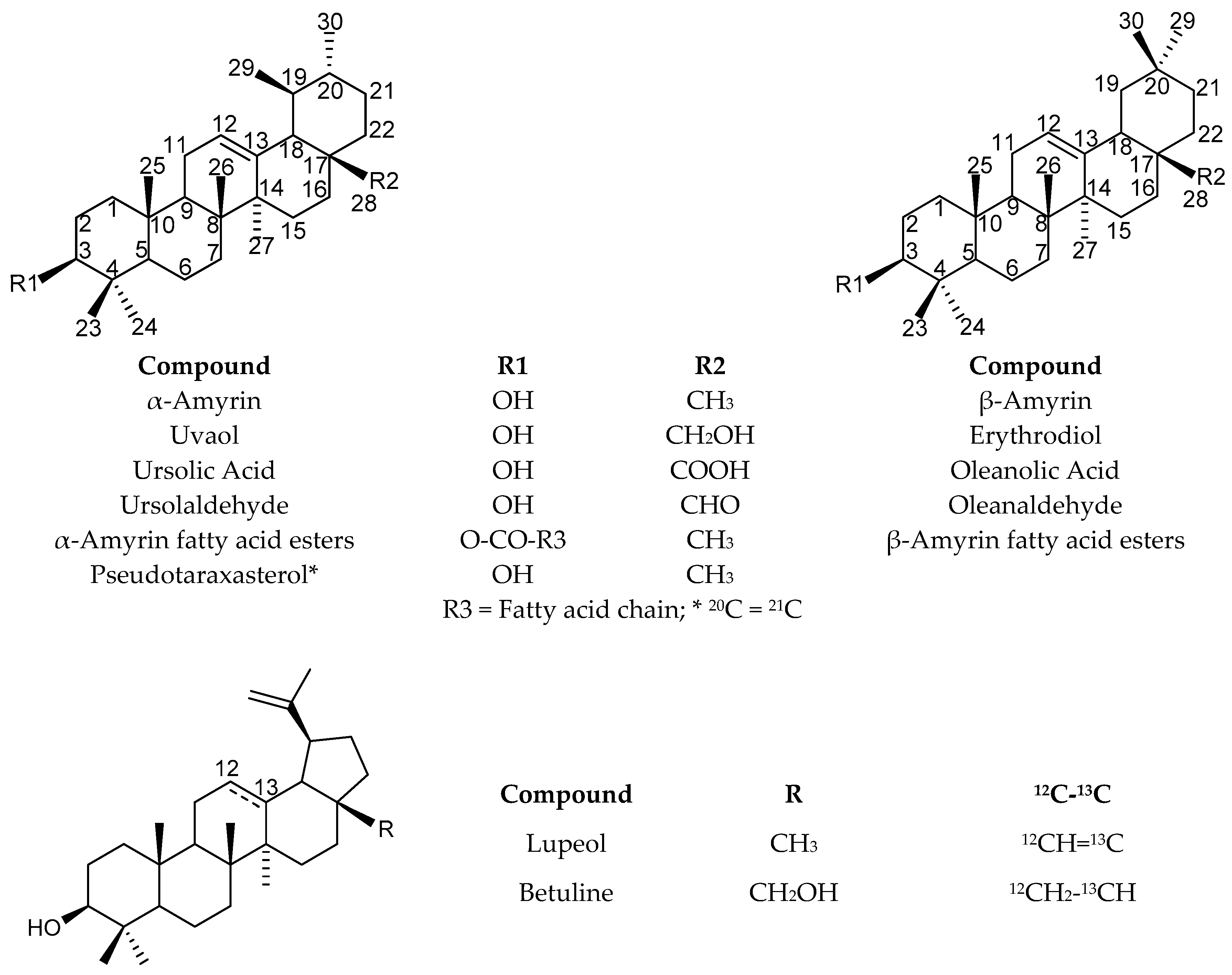
- Fraction H5 contained α-amyrin, β-amyrin, and lupeol previously identified in the crude extract as well as pseudotaraxasterol, which is present at a lower extent.
- -
- Fraction H7 contained betuline, erythrodiol, and uvaol where the latter is already identified in the crude extract and also present in fraction H8 where it constituted the major component.
- -
- Fraction H6 contained α-amyrin, β-amyrin, and lupeol as well as β-sitosterol not yet identified. The 13C-NMR spectrum of that fraction exhibited a series of signals with appreciable intensities including the aldehydic signal (207.5 ppm). Therefore, the fraction H6 has been chromatographed once again, which leads to sub-fractions H6.1-H6.7. Our attention was focused on fraction H6.3. Its 13C-NMR spectrum displayed two series of 30 signals. The chemical shifts of the major component were similar to those of α-amyrin with a few exceptions: one signal belonged to a carbon linked to a hydroxyl group while one methyl signal was missing and replaced by the aldehydic signal. Search in the literature led us to ursolaldehyde whose 13C-NMR spectrum has been reported by Kim et al. [34]. The second series of 30 signals displayed higher intensities on the spectrum of sub-fraction H6.2. Using the same procedure as above, oleanaldehyde was identified by comparison with the chemical shift values reported by Zhang et al. [35]. All the signals belonging to both molecules were observed. The chemical shift differences (Δδ) measured on the spectrum and those reported in the literature are inferior or equal to 0.12 ppm for most carbons, except for those of aldehydic carbons (about 0.20 ppm). It could be pointed out that, despite the similarities of the structures, only four signals out of 30 of oleanaldehyde overlapped with those of ursolaldehyde.
2.1.2. Dichloromethane Extract from Leaves of I. aquifolium
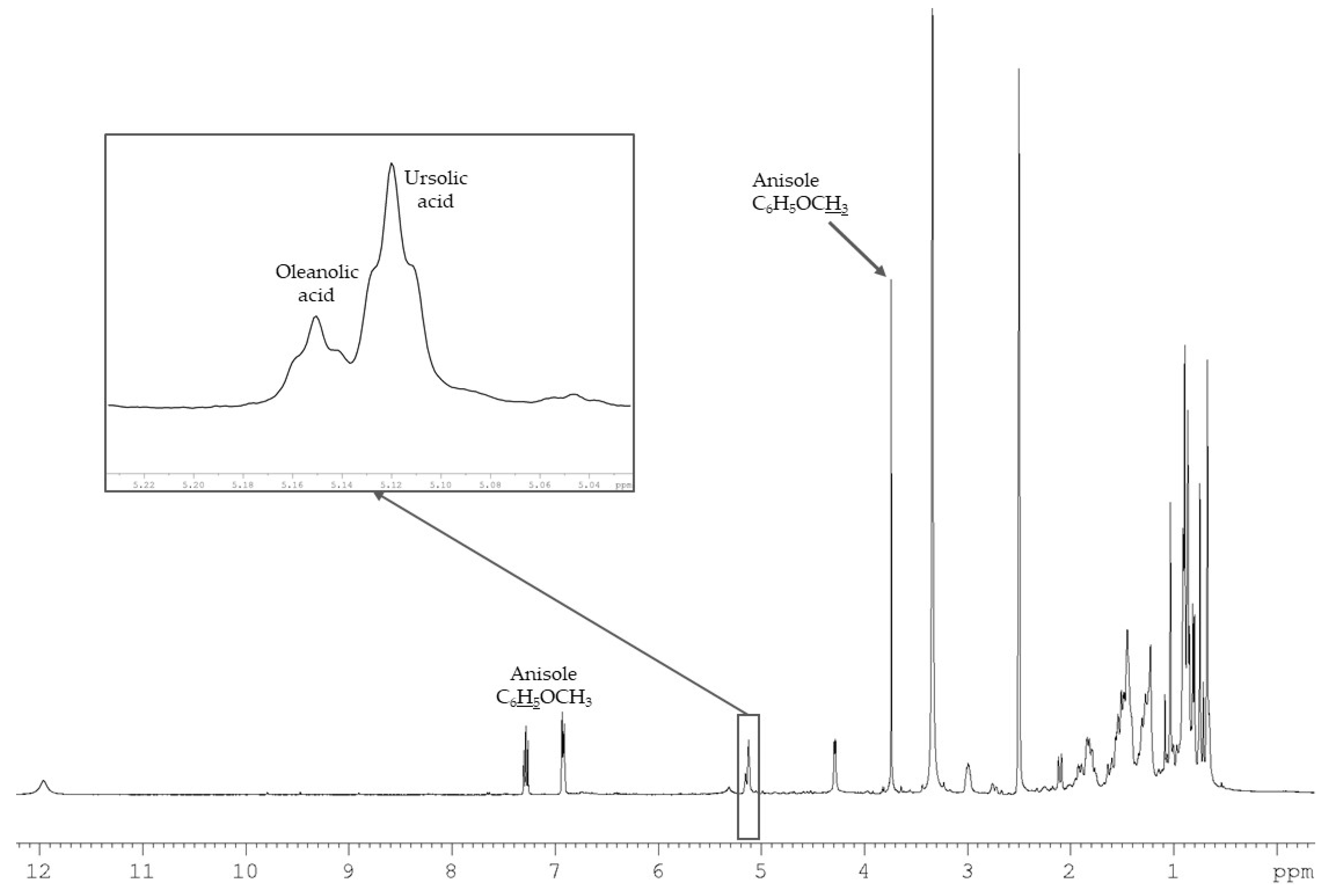
2.2. Quantitative Analysis of Ursolic Acid and Oleanolic Acid in I. aquifolium Dichloromethane Extract Using 1H-NMR
- -
- OA is widely distributed in food and plants. Medicinal plants such as Lantana camara are rich sources of OA (0.21–0.58% in flowers, 1.14–1.67% in roots) [26]. Common culinary spices (thyme, clove plants) and fruit plants (apple, loquat, elderberry, and sage) are also sources of OA [40]. Overall, OA can easily be obtained in high yield (up to 3.1%) from olive tree leaves, which is its main commercial source [40].
- -
- In parallel, UA is widely distributed especially in higher plants [41]. For instance, it is present in bearberry leaves (1.24%) [40], in dry flowers of loquat tree (0.22–0.27%) [42], in apple peels (1.43% [40]; 0.71% [43], at around 50 mg per medium sized fruit) [44]). UA has been also found in the leaves of Ilex paraguariensis (0.26%) [45]. Concerning I. aquifolium, leaves collected in winter near Camaiore (Lucca, Italy) were extracted successively with light petroleum ether and acetone. UA and OA were the principal constituents of the acetone extract and their relative amounts (%) were evaluated by GC of the corresponding methyl esters trimethylsilyl ethers. UA and OA accounted for 3.34% and 0.067%, respectively, with respect to the dried leaves. However, true quantitation was not performed [7].
3. Material and Methods
3.1. Plant Material and Solvent Extractions
3.2. Column Chromatography on Silica Gel (CC)
3.2.1. Hexane Extract
3.2.2. Dichloromethane Extract
3.3. Hydrolysis of α-Amyrin and β-Amyrin Esters. Synthesis of Fatty Acid Methyl Esters
3.4. Analytical GC
3.5. GC-MS Analysis
3.6. NMR Spectroscopy
3.7. Identification of Individual Components
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alikaridis, F. Natural constituents of Ilex species. J. Ethnopharmacol. 1987, 20, 121–144. [Google Scholar] [CrossRef]
- Erdemoglu, N.; Iscan, G.; Sener, B.; Palittapongarnpim, P. Antibacterial, antifungal, and antimycobacterial activity of Ilex aquifolium leaves. Pharm. Biol. 2009, 47, 697–700. [Google Scholar] [CrossRef]
- Nahar, L.; Russell, W.R.; Middleton, M.; Shoeb, M.; Sarker, S.D. Antioxidant phenylacetic acid derivatives from the seeds of Ilex aquifolium. Acta Pharm. 2005, 55, 187–193. [Google Scholar] [PubMed]
- Jalali, H.; Mozaffari Nejad, A.S.; Ebadi, A.G.; Laey, G. Ethnobotany and folk pharmaceutical properties of major trees or shrubs in northeast of Iran. Asian J. Chem. 2009, 21, 5632–5638. [Google Scholar]
- Willems, M. A cyanogenic glucoside from Ilex aquifolium. Phytochemistry 1988, 27, 1852–1853. [Google Scholar] [CrossRef]
- Hao, D.; Gu, X.; Xiao, P.; Liang, Z.; Xu, L.; Peng, Y. Research progress in the phytochemistry and biology of Ilex pharmaceutical resources. Acta Pharmacol. Sin. 2013, 3, 8–19. [Google Scholar] [CrossRef] [Green Version]
- Catalano, S.; Marsili, A.; Morelli, I.; Pistelli, L.; Scartoni, V. Constituents of the leaves of Ilex aquifolium L. Planta Med. 1978, 33, 416–417. [Google Scholar] [CrossRef]
- Bate-Smith, E.C. The phenolic constituents of plants and their taxonomic significance. J. Linn. Soc. (Bot.) 1962, 58, 95–173. [Google Scholar]
- Ishikura, N. Paper chromatographic analysis of anthocyanins in the red epicarp of Ilex aquifolium. Bot. Mag. Tokyo 1971, 84, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Liu, J. Pharmacology of oleanolic acid and ursolic acid. J. Ethnopharmacol. 1995, 49, 57–68. [Google Scholar] [CrossRef]
- Fontanay, S.; Grare, M.; Mayer, J.; Finance, C.; Duval, R.E. Ursolic, oleanolic and betulinic acids: Antibacterial spectra and selectivity indexes. J. Ethnopharmacol. 2008, 120, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Murakami, A.; Ohigashi, H. Ursolic acid: An anti- and pro-inflammatory triterpenoid. Mol. Nutr. Food Res. 2008, 52, 26–42. [Google Scholar] [CrossRef]
- Sultana, N. Clinically useful anticancer, antitumor, and antiwrinkle agent, ursolic acid and related derivatives as medicinally important natural product. J. Enzyme Inhib. Med. Chem. 2011, 26, 616–642. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, L.; Skapska, S.; Marszalek, K. Ursolic acid - A pentacyclic triterpenoid with a wide spectrum of pharmacological activities. Molecules 2015, 20, 20614–20641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayeleso, T.B.; Matumba, M.G.; Mukwevho, E. Oleanolic acid and its derivatives: Biological activities and therapeutic potential in chronic diseases. Molecules 2017, 22, 1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Both, D.M.; Goodtzova, K.; Yarosh, D.B.; Brown, D.A. Liposome-encapsulated ursolic acid increases ceramides and collagen in human skin cells. Arch. Dermatol. Res. 2002, 293, 569–575. [Google Scholar] [CrossRef] [PubMed]
- Seebacher, W.; Simic, N.; Weis, R.; Saf, R.; Kunert, O. Complete assignments of 1H and 13C-NMR resonances of oleanolic acid, 18α-oleanolic acid, ursolic acid and their 11-oxo derivatives. Magn. Reson. Chem. 2003, 41, 636–638. [Google Scholar] [CrossRef]
- Xu, C.; Wang, B.; Pu, Y.; Tao, J.; Zhang, T. Techniques for the analysis of pentacyclic triterpenoids in medicinal plants. J. Sep. Sci. 2018, 41, 6–19. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.C.; Jain, C.L.; Nigami, S.; Padhi, M.M. Rapid extraction, isolation, and quantification of oleanolic acid from Lantana camara L. roots using microwave and HPLC–PDA techniques. Acta Chromatogr. 2013, 25, 181–199. [Google Scholar] [CrossRef]
- Jiang, Q.; Zou, S.Q. Determination of five triterpenic acids from Salvia chinensis of different parts by RP-HPLC-PDA. China J. Chin. Mater. Med. 2014, 39, 4379–4382. [Google Scholar]
- Mirjalili, M.H.; Ayyari, M.; Bakhtiar, Z.; Moridi Farimani, M.; Sonboli, A. Quantification of betulinic, oleanolic and ursolic acids as medicinally important triterpenoids in some Thymus species from Iran. Res. J. Pharmacognosy 2016, 3, 23–28. [Google Scholar]
- Vaidya, V.V.; Pradhan, P.M.; Shinde, M.A. High performance liquid chromatography method for simultaneous quantification of oleanolic acid, lupeol and β-sitosterol from Nyctanthes arbor-tristis and its marketed formulation. Int. J. Res. Pharm. Chem. 2017, 7, 387–393. [Google Scholar]
- Shen, D.; Pan, M.H.; Wu, Q.L.; Park, C.H.; Juliani, H.R.; Ho, C.T.; Simon, J.E. LC-MS method for the simultaneous quantitation of the anti-inflammatory constituents in oregano (Origanum species). J. Agric. Food Chem. 2010, 58, 7119–7125. [Google Scholar] [PubMed]
- Olmo-García, L.; Bajoub, A.; Monasterio, R.P.; Fernández-Gutiérrez, A.; Carrasco-Pancorbo, A. Development and validation of LC-MS-based alternative methodologies to GC-MS for the simultaneous determination of triterpenic acids and dialcohols in virgin olive oil. Food Chem. 2018, 239, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, S.; Scott, W.A. Analysis of four pentacyclic triterpenoid acids in several bioactive botanicals with gas and liquid chromatography and mass spectrometry detection. J. Sep. Sci. 2016, 39, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.K.; Malik, T.; Banik, R.M. Validated HPTLC method for quantification of variability in content of oleanolic acid in different variety of Lantana camara. Pharmacologia 2013, 4, 126–131. [Google Scholar]
- Gupta, A.; Maheta, P.; Chauhan, R.; Pandey, S.; Singh Yadav, J.; Shah, S. Simultaneous quantification of bioactive triterpene acids (ursolic acid and oleanolic acid) in different extracts of Eucalyptus globulus (L.) by HPTLC method. Pharmacogn. J. 2018, 10, 179–185. [Google Scholar]
- Caligiani, A.; Malavasi, G.; Palla, G.; Marseglia, A.; Tognolini, M.; Bruni, R. A simple GC-MS method for the screening of betulinic, corosolic, maslinic oleanolic and ursolic acid contents in commercial botanicals used as food supplement ingredients. Food Chem. 2013, 136, 735–741. [Google Scholar] [CrossRef]
- Duquesnoy, E.; Castola, V.; Casanova, J. Identification and quantitative determination of triterpenes in the hexane extract of Olea europaea L. leaves using 13C-NMR spectroscopy. Phytochem. Anal. 2007, 18, 347–353. [Google Scholar] [CrossRef]
- Kontogianni, V.G.; Exarchou, V.; Troganis, A.; Gerothanassis, I.P. Rapid and novel discrimination and quantification of oleanolic and ursolic acids in complex plant extracts using two-dimensional nuclear magnetic resonance spectroscopy-Comparison with HPLC methods. Anal. Chim. Acta 2009, 635, 188–195. [Google Scholar]
- Castola, V.; Bighelli, A.; Casanova, J. Direct qualitative and quantitative analysis of triterpenes using 13C-NMR spectroscopy exemplified by dichloromethanic extracts of cork. Appl. Spectrosc. 1999, 53, 344–350. [Google Scholar] [CrossRef]
- Gunstone, F.D.; Pollard, M.R.; Scrimgeour, C.M.; Vedanayagam, H.S. Fatty acids. Part 50. 13C Nuclear Magnetic Resonance studies of olefinic fatty acids and esters. Chem. Phys. Lipids 1977, 18, 115–129. [Google Scholar] [CrossRef]
- Alexandri, E.; Ahmed, R.; Siddiqui, H.; Choudhary, M.I.; Tsiafoulis, C.G.; Gerothanassis, I.P. High resolution NMR spectroscopy as a structural and analytical tool for unsaturated lipids in solution. Molecules 2017, 22, 1663. [Google Scholar] [CrossRef]
- Kim, D.H.; Han, K.M.; Chung, I.S.; Kim, D.K.; Kim, S.H.; Kwon, B.M.; Jeong, T.S.; Park, M.H.; Ahn, E.M.; Baek, N.I. Triterpenoids from the flower of Campsis grandiflora K. Schum. as human AcyI-CoA: Cholesterol acyltransferase inhibitors. Arch. Pharm. Res. 2005, 28, 550–556. [Google Scholar] [CrossRef]
- Zhang, Y.; Jayaprakasam, B.; Seeram, N.P.; Olson, L.K.; DeWitt, D.; Nair, M.G. Insulin secretion and cyclooxygenase enzyme inhibition by cabernet sauvignon grape skin compounds. J. Agric. Food Chem. 2004, 52, 228–233. [Google Scholar] [CrossRef]
- Niemann, G.J.; Baas, W.J. The composition of the lipid constituents of Ilex aquifolium L. (Aquifoliaceae) in relation to the age of the leaf. I. The leaf wax. J. Plant. Physiol. 1985, 118, 219–226. [Google Scholar] [CrossRef]
- Van Genderen, H.H.; Jaarsma, J. Triterpenes and alkanes in developing variegated and albino leaves of Ilex aquifolium L. (Aquifoliaceae). Plant. Sci. 1990, 72, 165–172. [Google Scholar] [CrossRef]
- Nam, A.M.; Paoli, M.; Castola, V.; Casanova, J.; Bighelli, A. Identification and quantitative determination of lignans in Cedrus atlantica resins using 13C-NMR spectroscopy. Nat. Prod. Commun. 2011, 6, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Becker, E.D.; Ferretti, J.A.; Gambhir, P.N. Selection of optimum parameters for pulse Fourier transform nuclear magnetic resonance. Anal. Chem. 1979, 51, 1413–1420. [Google Scholar] [CrossRef]
- Jäger, S.; Trojan, H.; Kopp, T.; Laszczyk, M.N.; Scheffler, A. Pentacyclic triterpene distribution in various plants–rich sources for a new group of multi-potent plant extracts. Molecules 2009, 14, 2016–2031. [Google Scholar] [CrossRef] [Green Version]
- Babalola, I.T.; Shode, F.O. Ubiquitous ursolic acid: A potential pentacyclic triterpene natural product. J. Pharmacogn. Phytochem. 2013, 2, 214–222. [Google Scholar]
- Zhou, C.; Chen, K.; Sun, C.; Chen, Q.; Zhang, W.; Li, X. Determination of oleanolic acid, ursolic acid and amygdalin in the flower of Eriobotrya japonica Lindl. by HPLC. Biomed. Chromatogr. 2007, 21, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Noshita, T.; Kidachi, Y.; Umetsu, H.; Hayashi, M.; Komiyama, K.; Funayama, S.; Ryoyama, K. Isolation of ursolic acid from apple peels and its specific efficacy as a potent antitumor agent. J. Health Sci. 2008, 54, 654–660. [Google Scholar] [CrossRef] [Green Version]
- Frighetto, R.T.S.; Welendorf, R.M.; Nigro, E.N.; Frighetto, N.; Siani, A.C. Isolation of ursolic acid from apple peels by high speed counter-current chromatography. Food Chem. 2008, 106, 767–771. [Google Scholar] [CrossRef]
- Gnoatto, S.C.; Dassonville-Klimpt, A.; Da Nascimento, S.; Galéra, P.; Boumediene, K.; Gosmann, G.; Sonnet, P.; Moslemi, S. Evaluation of ursolic acid isolated from Ilex paraguariensis and derivatives on aromatase inhibition. Eur. J. Med. Chem. 2008, 43, 1865–1877. [Google Scholar] [CrossRef]
- Cerceau, C.I.; Barbosa, L.C.A.; Alvarenga, E.S.; Ferreira, A.G.; Thomasi, S.S. A validated 1H-NMR method for quantitative analysis of α-bisabolol in essential oils of Eremanthus erythropappus. Talanta 2016, 161, 71–79. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
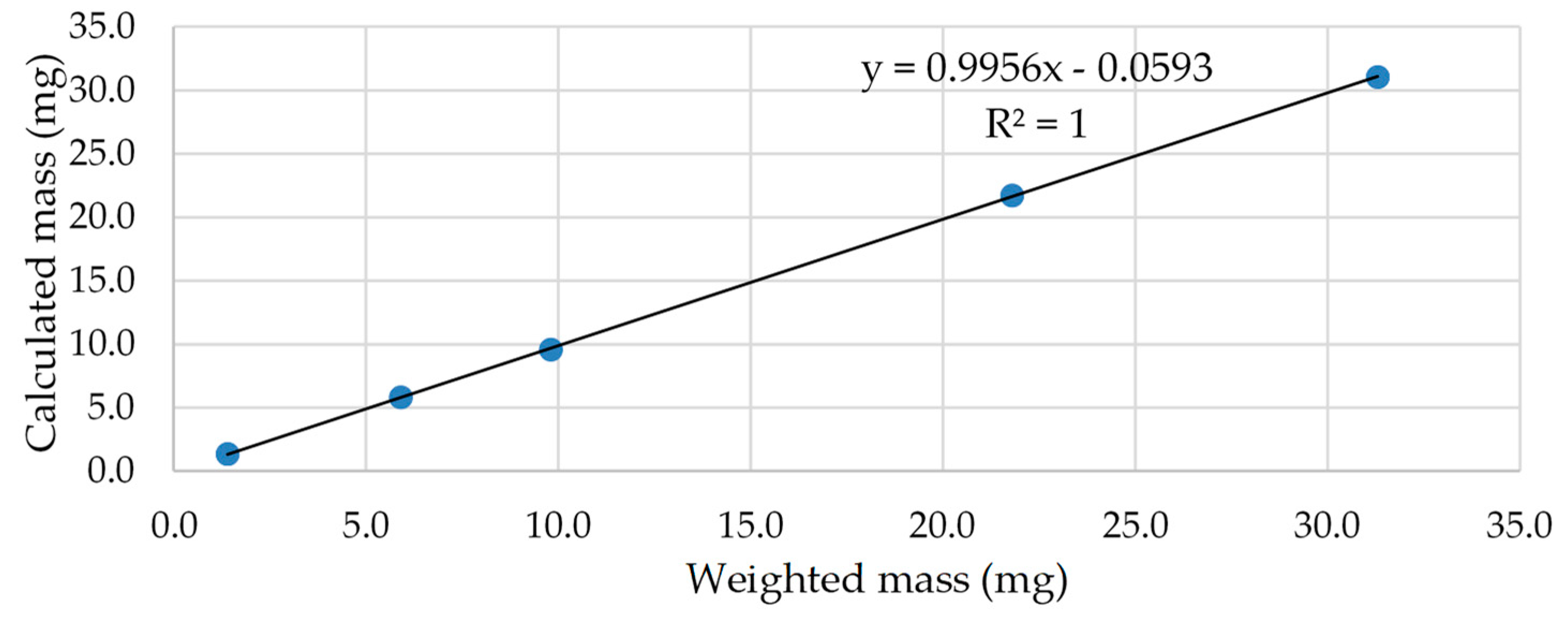
| Exp n° | Anisole Area | Mass of Anisole (mg) | UA Area | Weighted Mass (mg) | Calculated Mass (mg) | RE% |
|---|---|---|---|---|---|---|
| 1 | 1.0000 | 1.2 | 0.0857 | 1.4 | 1.4 | 0.0 |
| 2 | 1.0000 | 1.2 | 0.3782 | 5.9 | 6.0 | 1.7 |
| 3 | 1.0000 | 1.2 | 0.6187 | 9.8 | 9.8 | 0.0 |
| 4 | 1.0000 | 1.2 | 1.3006 | 20.8 | 20.6 | −1.0 |
| 5 | 1.0000 | 1.2 | 1.9297 | 30.9 | 30.6 | −1.0 |
| Test 1 | Test 2 | Test 3 | |
|---|---|---|---|
| Weighted mass of UA (mg) | 20.8 | 20.8 | 20.8 |
| UA Area | 1.3006 | 1.3151 | 1.3077 |
| Calculated mass of UA (mg) | 20.6 | 20.8 | 20.7 |
| RE (%) | −1.0 | 0.0 | −0.5 |
| Ursolic Acid | Oleanolic Acid | ||
|---|---|---|---|
| AUA | 0.4380 | AOA | 0.1658 |
| mUA (mg) | 8.7 | mOA (mg) | 3.3 |
| UA content (%) | 55.3 | OA content (%) | 20.8 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palu, D.; Bighelli, A.; Casanova, J.; Paoli, M. Identification and Quantitation of Ursolic and Oleanolic Acids in Ilex aquifolium L. Leaf Extracts Using 13C and 1H-NMR Spectroscopy. Molecules 2019, 24, 4413. https://doi.org/10.3390/molecules24234413
Palu D, Bighelli A, Casanova J, Paoli M. Identification and Quantitation of Ursolic and Oleanolic Acids in Ilex aquifolium L. Leaf Extracts Using 13C and 1H-NMR Spectroscopy. Molecules. 2019; 24(23):4413. https://doi.org/10.3390/molecules24234413
Chicago/Turabian StylePalu, Doreen, Ange Bighelli, Joseph Casanova, and Mathieu Paoli. 2019. "Identification and Quantitation of Ursolic and Oleanolic Acids in Ilex aquifolium L. Leaf Extracts Using 13C and 1H-NMR Spectroscopy" Molecules 24, no. 23: 4413. https://doi.org/10.3390/molecules24234413
APA StylePalu, D., Bighelli, A., Casanova, J., & Paoli, M. (2019). Identification and Quantitation of Ursolic and Oleanolic Acids in Ilex aquifolium L. Leaf Extracts Using 13C and 1H-NMR Spectroscopy. Molecules, 24(23), 4413. https://doi.org/10.3390/molecules24234413