
Extent of Spin Contamination Errors in DFT/Plane-wave Calculation of Surfaces: A Case of Au Atom Aggregation on a MgO Surface


Abstract
:1. Introduction
2. Computational Procedure
2.1. Method
2.2. AP Scheme
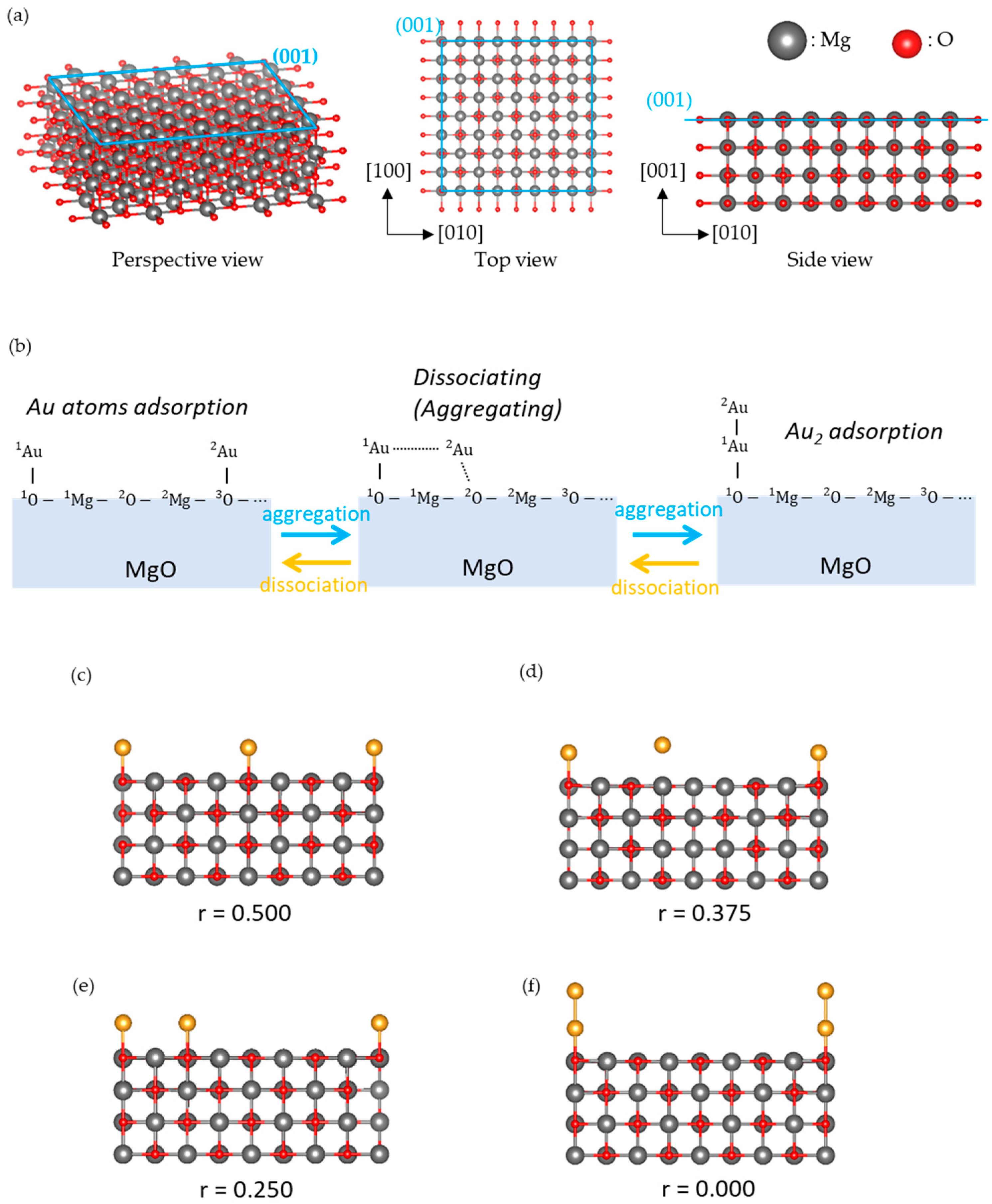
2.3. Model
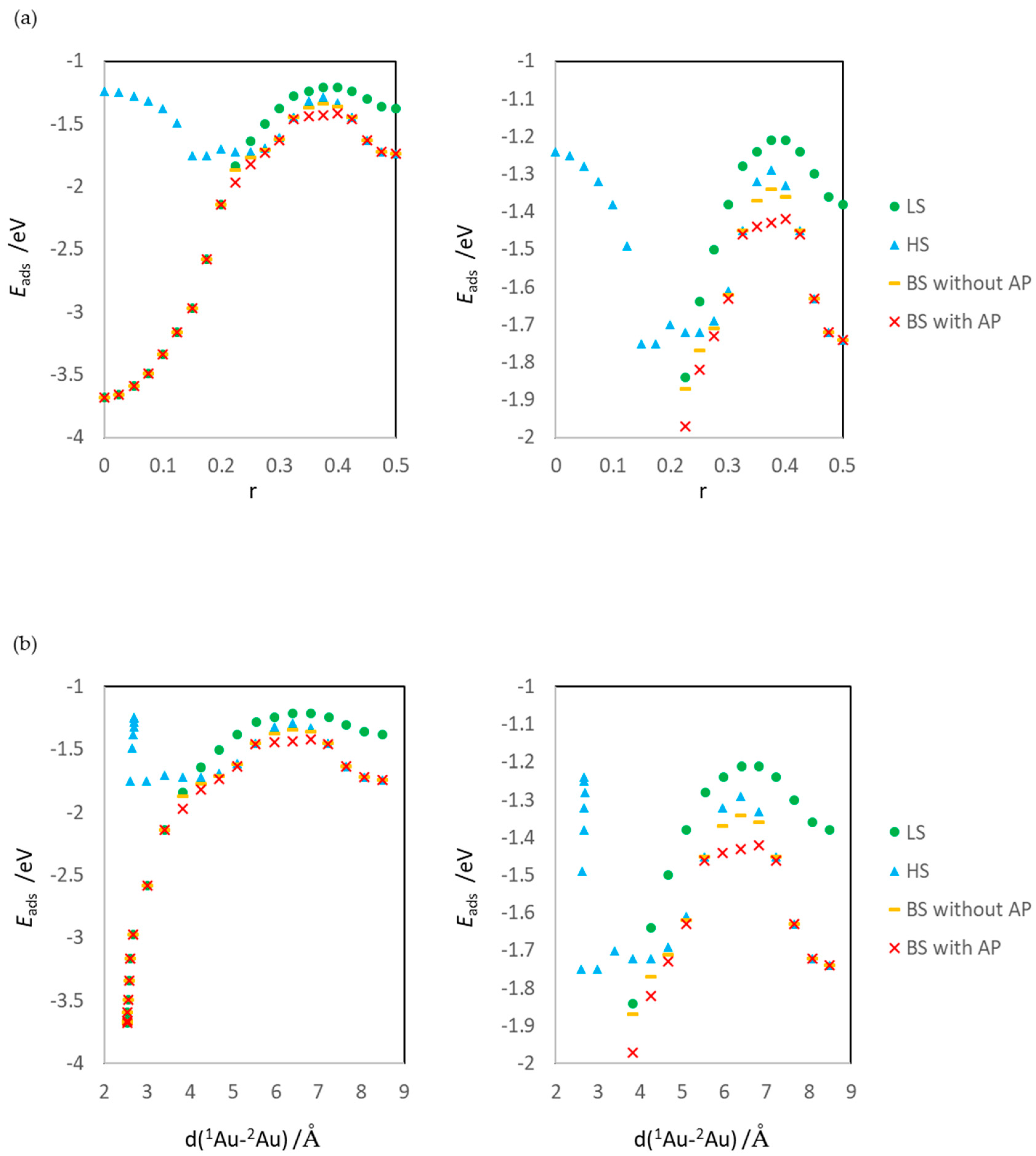
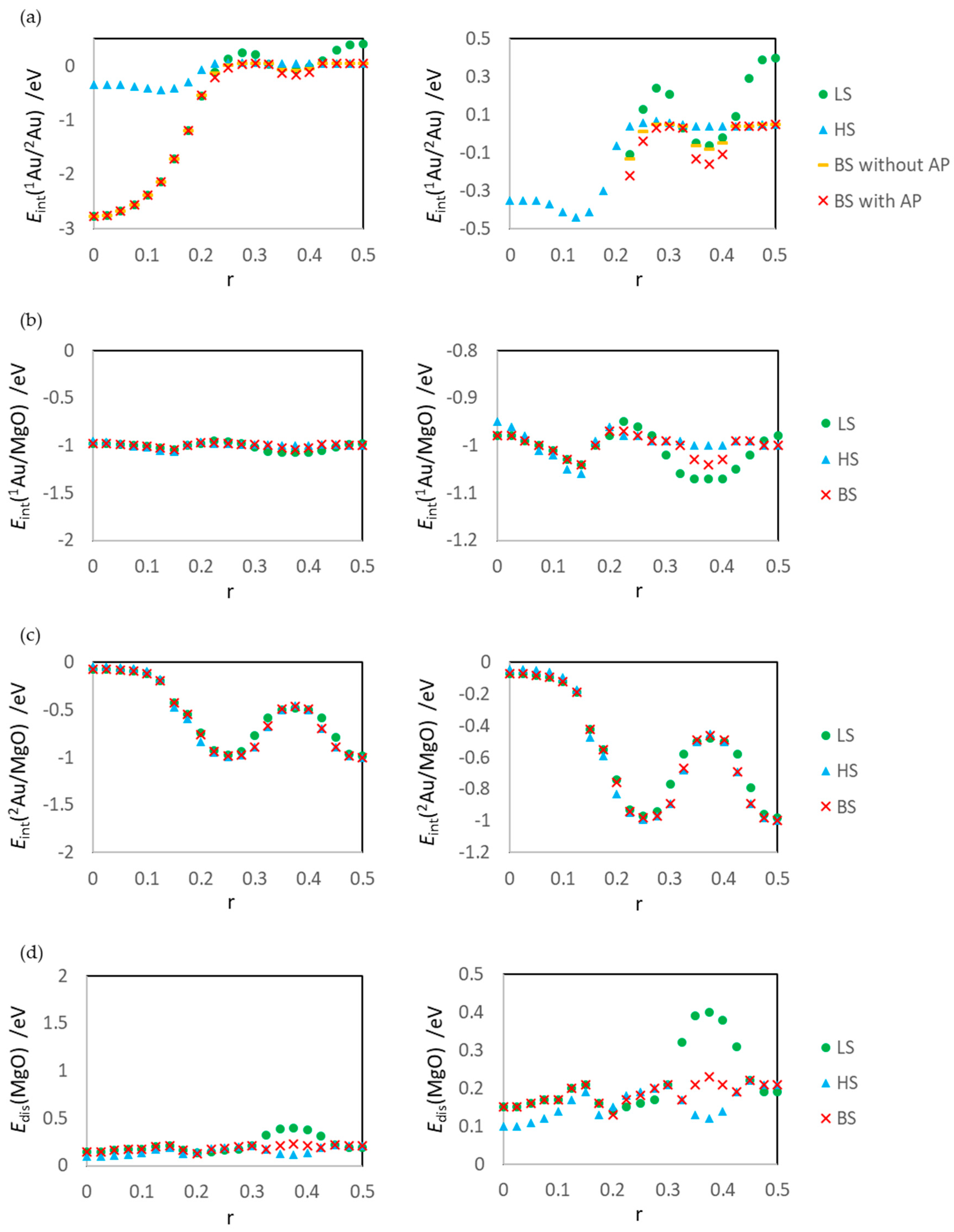
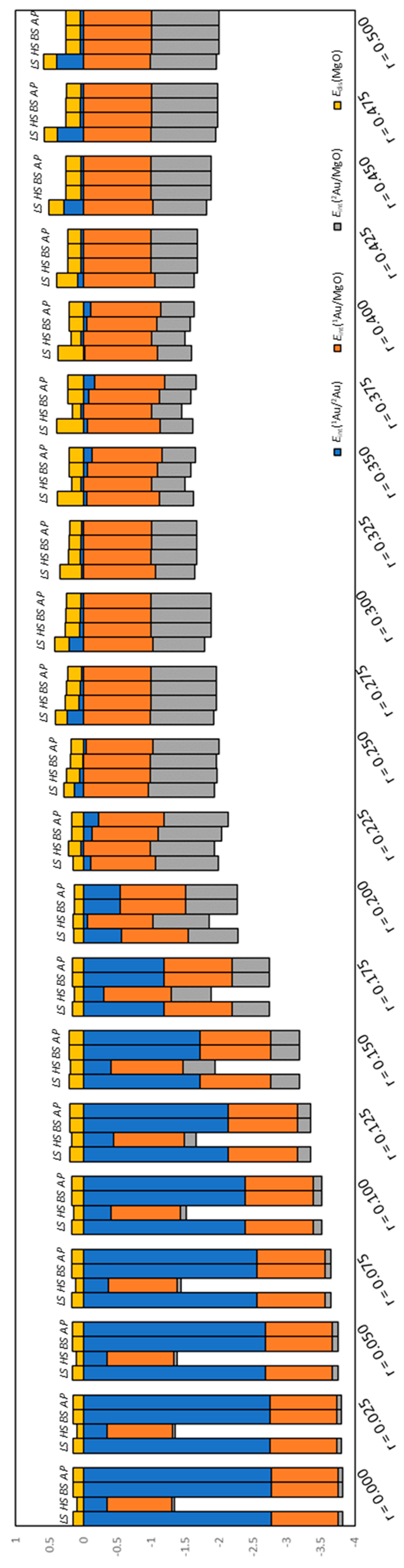
2.4. Definitions of Energies
3. Results and Discussion
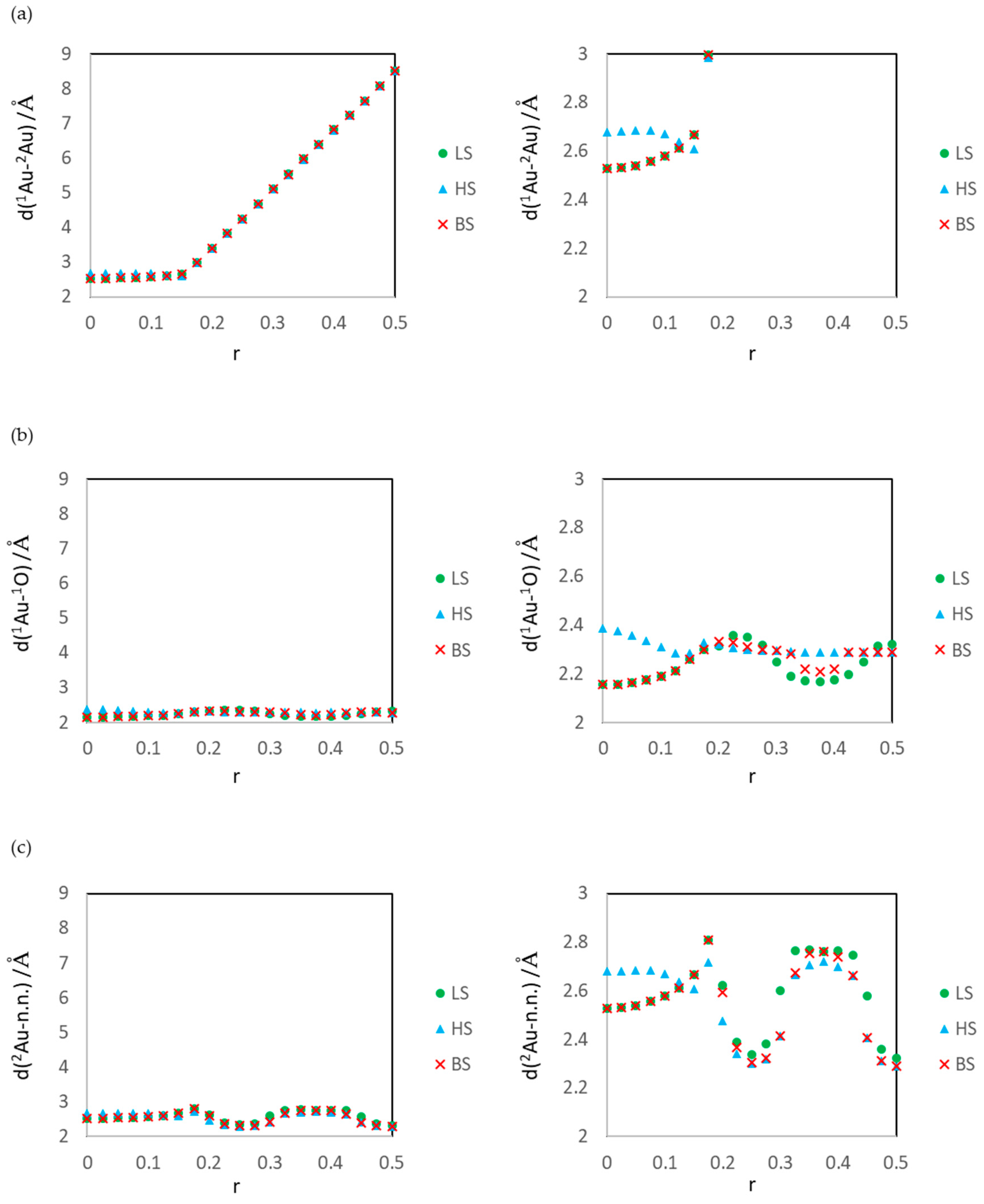
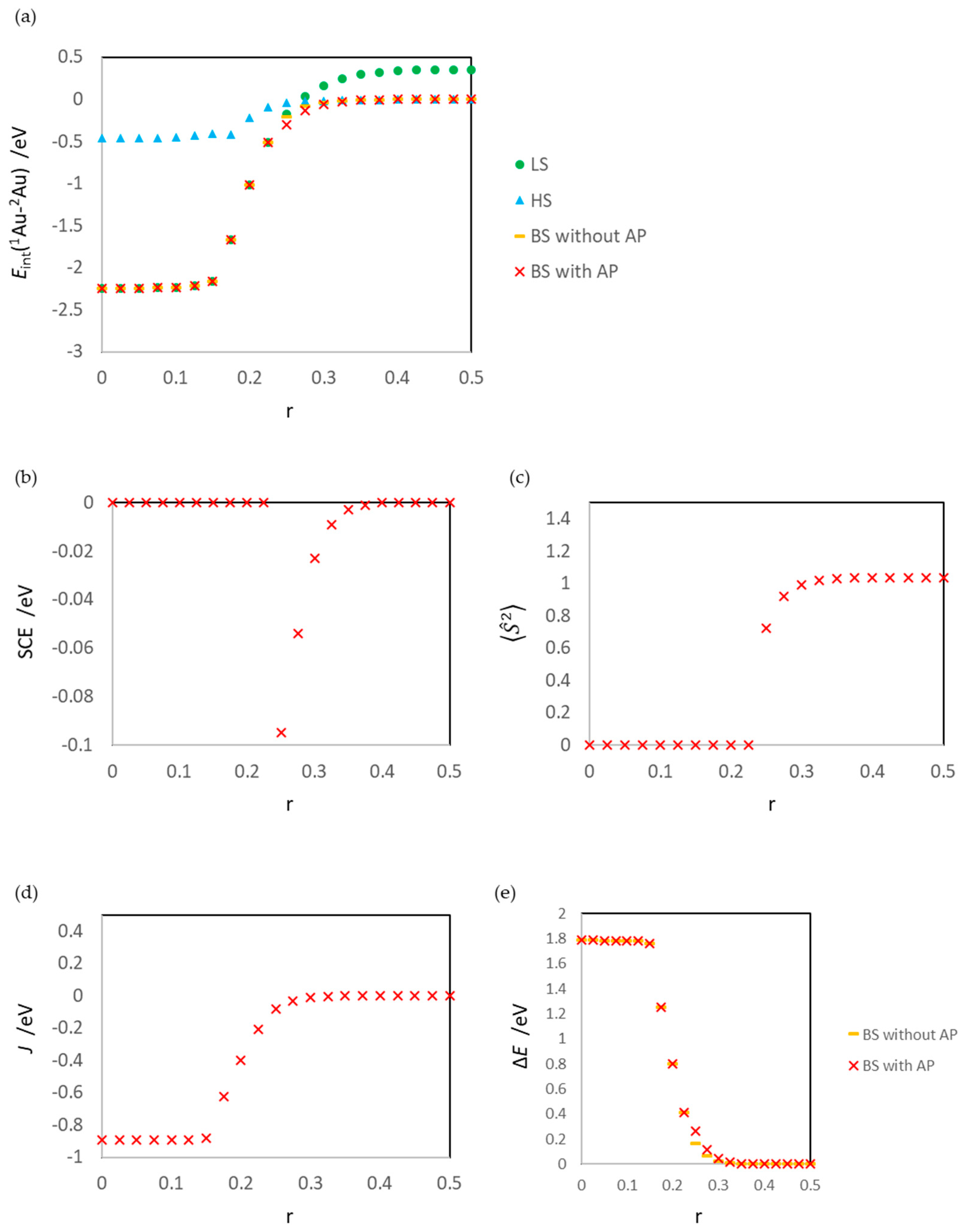
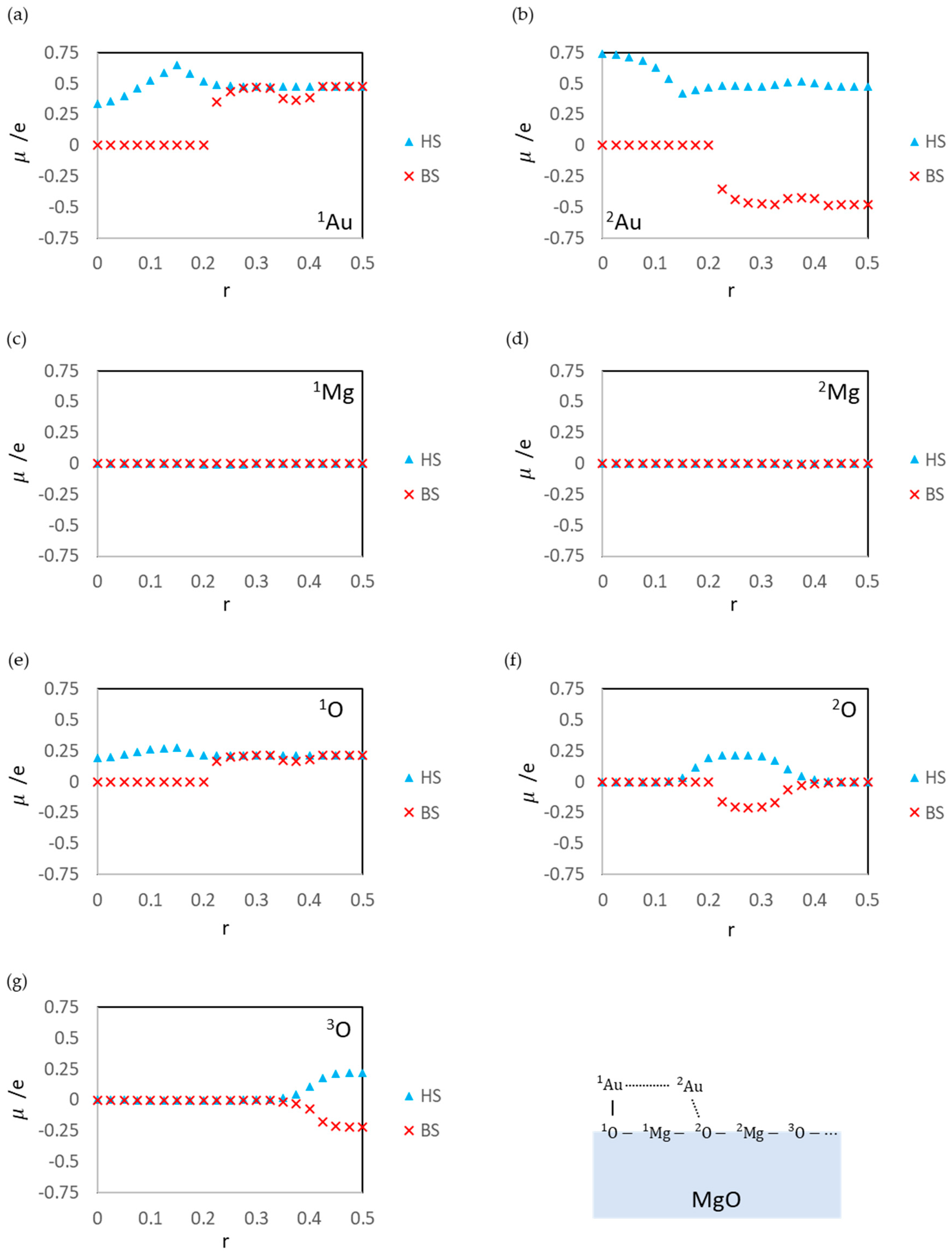
3.1. Dissociated Au2 Adsorption onto MgO: r = 0.500–0.450
3.2. Dissociating Au2 Adsorption onto MgO (1): r = 0.425–0.325
3.3. Dissociating Au2 Adsorption onto MgO (2): r = 0.300–0.200
3.4. Non-Dissociated Au2 Adsorption onto MgO: r = 0.175–0.000
4. Conclusions
- (1)
- Two isolated Au atoms are adsorbed onto the on-top of O site in MgO (001), which is the initial state of the aggregation of Au atoms
- (2)
- One Au atom interacts with Mg and the other Au atom interacts with O, and there is a slight electrostatic interaction between the Au atoms, which includes the transition state of the Au atom aggregation
- (3)
- Generation of Au-Au covalent interaction
- (4)
- Non-dissociated adsorption state of Au2 on MgO, which is the final structure of the Au atom aggregation.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nilsson, A.; Petersson, L.G.M.; Nørskov, J.K. Chemical Bonding at Surface and Interface; Elsevier: Amsterdam, The Netherlands, 2008; ISBN 978-0-444-52837-7. [Google Scholar]
- Greeley, J.; Jaramillo, T.F.; Bonde, J.; Chorkendorff, I.B.; Nørskov, J.K. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nature Mater. 2006, 5, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Honkala, K.; Hellman, A.; Remediakis, I.N.; Logadottir, A.; Carlsson, A.; Dohl, S.; Christensen, C.H.; Nørskov, J.K. Ammonia synthesis from first-principles calculations. Science 2005, 307, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Norskov, J.K.; Blligaard, T.; Logattir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 2005, 152, J23–J26. [Google Scholar] [CrossRef]
- Darling, G.R.; Holloway, S. The dissociation of diatomic molecules at surfaces. Rep. Prog. Phys. 1995, 58, 1595–1672. [Google Scholar] [CrossRef]
- Gross, A. Reactions at surfaces studied by ab initio dynamics calculations. Surf. Sci. Rep. 1998, 32, 291–340. [Google Scholar] [CrossRef] [Green Version]
- Alducin, M.; Muino, R.D.; Juaristi, J.I. Non-adiabatic effects in elementary reaction processes at metal surfaces. Prog. Surf. Sci. 2017, 92, 317–340. [Google Scholar] [CrossRef]
- Dino, W.A.; Kasai, H.; Okiji, A. Orientational effects in dissociative adsorption/associative desorption dynamics of H2 (D2) on Cu and Pd. Prog. Surf. Sci. 2000, 63, 63–134. [Google Scholar] [CrossRef]
- Padama, A.A.B.; Kishi, H.; Arevalo, R.L.; Moreno, J.L.V.; Kasai, H.; Taniguchi, M.; Uenishi, M.; Tanaka, H.; Nishihata, Y. NO dissociation on Cu (111) and Cu2O (111) surfaces: A density functional theory based study. J. Phys. Condens. Matter 2012, 24, 175005. [Google Scholar] [CrossRef]
- Tada, K.; Sakata, K.; Kitagawa, Y.; Kawakami, T.; Yamanaka, S.; Okumura, M. DFT calculations for chlorine elimination from chlorine-adsorbed gold clusters by hydrogen. Chem. Phys. Lett. 2013, 579, 94–99. [Google Scholar] [CrossRef]
- Koga, H.; Tada, K.; Hayashi, A.; Ato, Y.; Okumura, M. Potential of Titania-covered Ag Catalysts for NOx Reduction: A DFT study. Chem. Lett. 2017, 46, 456–459. [Google Scholar] [CrossRef]
- Koga, H.; Tada, K.; Hayashi, A.; Ato, Y.; Okumura, M. High NOx Reduction Activity of an Ultrathin Zirconia Film Covering Cu Surface: A DFT Study. Catal. Lett. 2017, 147, 1827–1833. [Google Scholar] [CrossRef]
- Takei, T.; Akita, T.; Nakamura, I.; Fujitani, T.; Okumura, M.; Okazaki, K.; Huang, J.; Ishida, T.; Haruta, M. Heterogeneous catalysis by gold. Adv. Catal. 2012, 55, 1–126. [Google Scholar] [CrossRef]
- Taketoshi, A.; Haruta, M. Size- and structure- specificity in catalysis by gold clusters. Chem. Lett. 2014, 43, 380–387. [Google Scholar] [CrossRef]
- Hutchings, G.J. Nanocrystalline gold and gold palladium alloy catalysts for chemical synthesis. Chem. Commun. 2008, 10, 1148–1164. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.; Hakkinen, H.; Landman, U.; Worz, A.S.; Antonietti, J.-M.; Abbet, S.; Judai, K.; Heiz, U. Charging effects on bonding and catalyzed oxidation of CO on Au8 clusters on MgO. Science 2005, 307, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Tada, K.; Koga, H.; Hayashi, A.; Kondo, Y.; Kawakami, T.; Yamanaka, S.; Okumura, M. Theoretical clarification of the coexistence of Cl effects on Au/TiO2: The interaction between Au clusters and the TiO2 surface, and the aggregation of Au clusters on the TiO2 surface. Bull. Chem. Soc. Jpn. 2017, 90, 506–519. [Google Scholar] [CrossRef]
- Tada, K.; Koga, H.; Sakurai, H.; Tanaka, S.; Ato, Y.; Hayashi, A.; Kawakami, T.; Yamanaka, S.; Okumura, M. Theoretical investigation of the effect of phosphate doping on the aggregation of Au atoms on an Al2O3 (0001) surface. Appl. Surf. Sci. 2019, 465, 1003–1013. [Google Scholar] [CrossRef]
- Szabo, A.; Ostlund, N.S. Modern Quantum Chemistry—Introduction to Advanced Electronic Structure Theory; Dover Publications: New York, NY, USA, 1996. [Google Scholar]
- Kawalski, K.; Piecuch, P. New classes of non-iterative energy corrections to multi-reference coupled-cluster energies. Mol. Phys. 2004, 102, 2425–2449. [Google Scholar] [CrossRef]
- Evangelista, F.A.; Prochnow, E.; Gauss, J.; Schaefer, H.F. Perturbative triples corrections in state-specific multireference coupled cluster theory. J. Chem. Phys. 2010, 132, 074107. [Google Scholar] [CrossRef] [PubMed]
- Noodleman, L.; Lovell, T.; Han, W.G.; Li, J.; Himo, F. Quantum chemical studies of intermediates and reaction pathways in selected enzymes and catalytic synthetic systems. Chem. Rev. 2004, 104, 459–508. [Google Scholar] [CrossRef] [PubMed]
- Malrieu, J.P.; Coballol, R.; Calzado, C.J.; de Graaf, C.; Guihery, N. Magnetic interactions in molecules and highly correlated materials: Physical content, analytical derivation, and rigorous extraction of magnetic Hamiltonians. Chem. Rev. 2014, 114, 429–492. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Isobe, H.; Nakajima, T.; Shigeta, Y.; Suga, M.; Akita, F.; Shen, J.R.; Yamaguchi, K. Large-scale QM/MM calculations of the CaMn4O5 cluster in the S3 state of the oxygen evolving complex of photosystem II. Comparison between water-inserted and no water-inserted structures. Faraday Disc. 2017, 198, 83–106. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Saito, T.; Sharma, S.; Yamanaka, S.; Yamada, S.; Nakajima, T.; Okumura, M.; Yamaguchi, K. Full-valence density matrix renormalization group calculations on meta-benzyne based on unrestricted natural orbitals. Revisit of seamless continuation from broken-symmetry to symmetry adopted models for diradicals. Mol. Phys. 2017, 115, 2267–2284. [Google Scholar] [CrossRef]
- Muhammad, S.; Nakano, M.; Al-Sehemi, A.G.; Irfan, A.; Chaudhry, A.R.; Tonami, T.; Ito, S.; Kishi, R.; Kitagawa, Y. Exploring the novel donor-nanotube archetype as an efficient third-order nonlinear optical material: Asymmetric open-shell carbon nanotubes. Nanoscale 2018, 10, 16345–16944. [Google Scholar] [CrossRef] [PubMed]
- David, G.; Wennmohs, F.; Neese, F.; Ferre, N. Chemical tuning of magnetic exchange couplings using broken symmetry density functional theory. Inorg. Chem. 2018, 57, 12769–12776. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Jensen, F.; Dorigo, A.; Houk, K.N. A spin correction procedure for unrestricted Hartree-Fock and Møller-Plesset wave functions for singlet diradicals and polyradicals. Chem. Phys. Lett. 1988, 149, 537–542. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Saito, T.; Ito, M.; Shoji, M.; Koizumi, K.; Yamanaka, S.; Kawakami, T.; Okumura, M.; Yamaguchi, K. Approximately spin-projected geometry optimization method and its application to di-chromium systems. Chem. Phys. Lett. 2007, 442, 445–450. [Google Scholar] [CrossRef]
- Saito, T.; Thiel, W. Analytical gradients for density functional calculations with approximate spin projection. J. Phys. Chem. A 2012, 116, 10864–10869. [Google Scholar] [CrossRef]
- Schlegel, H.B. Moller-Plesset perturbation theory with spin projection. J. Phys. Chem. 1988, 92, 3075–3078. [Google Scholar] [CrossRef]
- Sosa, C.; Schlegel, H.B. An ab initio study of the reaction pathways for OH + C2H4 → HOCH2CH2 → products. J. Am. Chem. Soc. 1987, 109, 7007–7015. [Google Scholar] [CrossRef]
- Saito, T.; Nishihara, S.; Kataoka, Y.; Nakanishi, Y.; Kitagawa, Y.; Kawakami, T.; Yamanaka, S.; Okumura, M.; Yamaguchi, K. Reinvestigation of the reaction of ethylene and singlet oxygen by the approximate spin projection method. comparison with multireference coupled-cluster calculations. J. Phys. Chem. A 2010, 114, 7967–7974. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Yasuda, N.; Kataoka, Y.; Nakanishi, Y.; Kitagawa, Y.; Kawakami, T.; Yamanaka, S.; Okumura, M.; Yamaguchi, K. Potential energy curve for ring-opening reactions: Comparison between broken-symmetry and multireference coupled cluster methods. J. Phys. Chem. A 2011, 115, 5625–5631. [Google Scholar] [CrossRef] [PubMed]
- Tada, K.; Koga, H.; Okumura, M.; Tanaka, S. Estimation of spin contamination error in dissociative adsorption of Au2 onto MgO (001) surface: First application of approximate spin projection (AP) method to plane wave basis. Chem. Phys. Lett. 2018, 701, 103–108. [Google Scholar] [CrossRef]
- Tada, K.; Koga, H.; Ato, Y.; Hayashi, A.; Okumura, M.; Tanaka, S. Effect of spin contamination error on surface catalytic reaction: NO reduction by core-shell catalysts. Mol. Phys. 2018. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effect. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoftpseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Fukui, H.; Fueno, T. Molecular-orbital (mo) theory for magnetically interacting organic-compounds-abinitio mo calculations of the effictive exchange integrals for cyclophane-type carbene dimers. Chem. Lett. 1986, 15, 625–628. [Google Scholar] [CrossRef]
- Barcaro, G.; Fortunelli, A. The interaction of coinage metal clusters with the MgO (100) surface. J. Chem. Theor. Comp. 2005, 1, 972–985. [Google Scholar] [CrossRef]
- Del Vitto, A.; Pacchioni, G.; Delbecq, F.O.; Sautet, P. Au atoms and dimers on the MgO (100) surface: A DFT study of nucleation at defects. J. Phys. Chem. B 2005, 109, 8040–8048. [Google Scholar] [CrossRef] [PubMed]
- Caballero, R.; Quintanar, C.; Koster, A.M.; Khana, S.N.; Reveles, J.U. Structural and electronic properties of Au and Au2 on an MgO (100) surface: A DFT cluster embedding approach. J. Phys. Chem. C 2008, 112, 14919–14928. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef] [Green Version]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. An initio molecular-dynamics simulation of the liquid-metal-amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient interactive schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comp. Nater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Henkelman, G.; Arnaldsson, A.; Jonsson, H. A fast and robust algorithm for Bader decomposition of charge density. Comp. Mater. Sci. 2006, 36, 354–360. [Google Scholar] [CrossRef]
- Sanville, E.; Kenny, S.D.; Smith, R.; Henkelman, G. Improved grid-based algorithm for Bader charge allocation. J. Comp. Chem. 2007, 28, 899–908. [Google Scholar] [CrossRef]
- Tang, W.; Sanvile, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Trinkle, D.R. Accurate and efficient algorithm for Bader charge integration. J. Chem. Phys. 2011, 134, 064111. [Google Scholar] [CrossRef] [PubMed]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Wang, J.H.; Becke, A.D.; Smith, V.H. Evaluation of in restricted, unrestricted Hartree-Fock, and density functional theories. J. Chem. Phys. 1995, 102, 3477–3480. [Google Scholar] [CrossRef]
- Tada, K.; Koga, H.; Sakata, K.; Oguni, A.; Saito, T.; Kawakami, T.; Yamanaka, S.; Okumura, M. Theoretical investigation on the Au-anchor site in phosphate-doped Au/Al2O3 catalysts. e-J. Surf. Sci. Nanotech. 2015, 13, 380–384. [Google Scholar] [CrossRef]
- Tada, K.; Sakata, K.; Yamada, S.; Okazaki, K.; Kitagawa, Y.; Kawakami, T.; Yamanaka, S.; Okumura, M. DFT calculations for Au adsorption onto a reduced TiO2 (110) surface with coexistence of Cl. Mol. Phys. 2014, 112, 365–378. [Google Scholar] [CrossRef]
- Okazaki, K.; Morikawa, Y.; Tanaka, S.; Tanaka, K.; Kohyama, M. Electronic structures of Au on TiO2 (110) by first-principles calculations. Phys. Rev. B 2004, 69, 235404. [Google Scholar] [CrossRef]
- Vittodini, A.; Selloni, A. Small golds clusters on stoichiometric and defected TiO2 anatase (101) and their interaction with CO: A density functional theory. J. Chem. Phys. 2002, 117. [Google Scholar] [CrossRef]
- Tada, K.; Koga, H.; Okumura, M.; Tanaka, S. Clarification of the interaction between Au atoms and the anatase TiO2 (112) surface using density functional theory. Surf. Sci. 2018, 670, 23–32. [Google Scholar] [CrossRef]
- Di Valentin, C.; Scagnelli, A.; Pacchioni, G.; Risse, T.; Freund, H.J. EPR properties of Au atoms on various-sites of the MgO (100) surface from relativistic DFT calculations. Surf. Sci. 2006, 600, 2434–2442. [Google Scholar] [CrossRef]
- Yulikov, M.; Sterrer, M.; Heyde, M.; Rust, H.P.; Risse, T.; Freund, H.J.; Pacchioni, G.; Scagnelli, A. Binding of single gold atoms on thin MgO (001) films. Phys. Rev. Lett. 2006, 96, 146804. [Google Scholar] [CrossRef] [PubMed]
- Puigdollers, A.R.; Schlexer, P.; Pacchioni, G. Gold and silver clusters on TiO2 and ZrO2 (101) surfaces: Role of dispersion forces. J. Phys. Chem. C 2015, 119, 15381–15389. [Google Scholar] [CrossRef]
- Schlexer, P.; Puigdollers, A.R.; Pacchioni, G. Tuning the charge state of Ag and Au atoms and clusters deposited on oxide surfaces by doping: A DFT study of the adsorption properties of nitrogen- and niobium- doped TiO2 and ZrO2. Phys. Chem. Chem. Phys. 2015, 17, 22342–22360. [Google Scholar] [CrossRef]
- Okazaki, K.; Morikawa, Y.; Tanaka, S.; Tanaka, K.; Kohyama, M. Effects of stoichiometry on electronic states of Au and Pt supported on TiO2 (110). J. Mater. Sci. 2005, 40, 3075–3080. [Google Scholar] [CrossRef]
- Nasluzov, V.A.; Shulimovich, T.V.; Shor, A.M.; Bukhtiyarov, V.I.; Rosch, N. Small gold species supported on alumina. A computational study of α-Al2O3 (0001) and γ-Al2O3 (001) using an embedded-cluster approach. Phys. Status Solidi B 2010, 247, 1023–1031. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
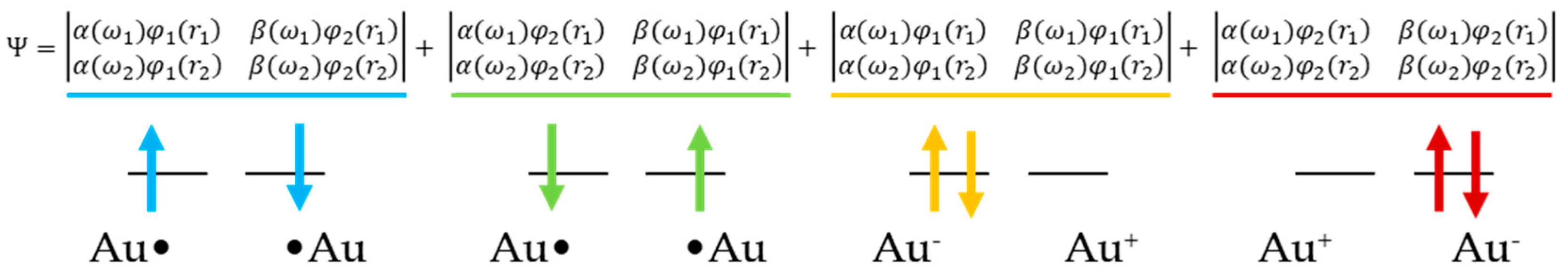{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | Aggregation of Au Atoms | Au2 Dissociation |
|---|---|---|
| LS | 0.17 eV (0.53 eV 1) | 2.47 eV |
| HS | 0.45 eV | 0.00 eV (2.39 eV 2) |
| BS | 0.40 eV | 2.34 eV |
| AP | 0.32 eV | 2.26 eV |
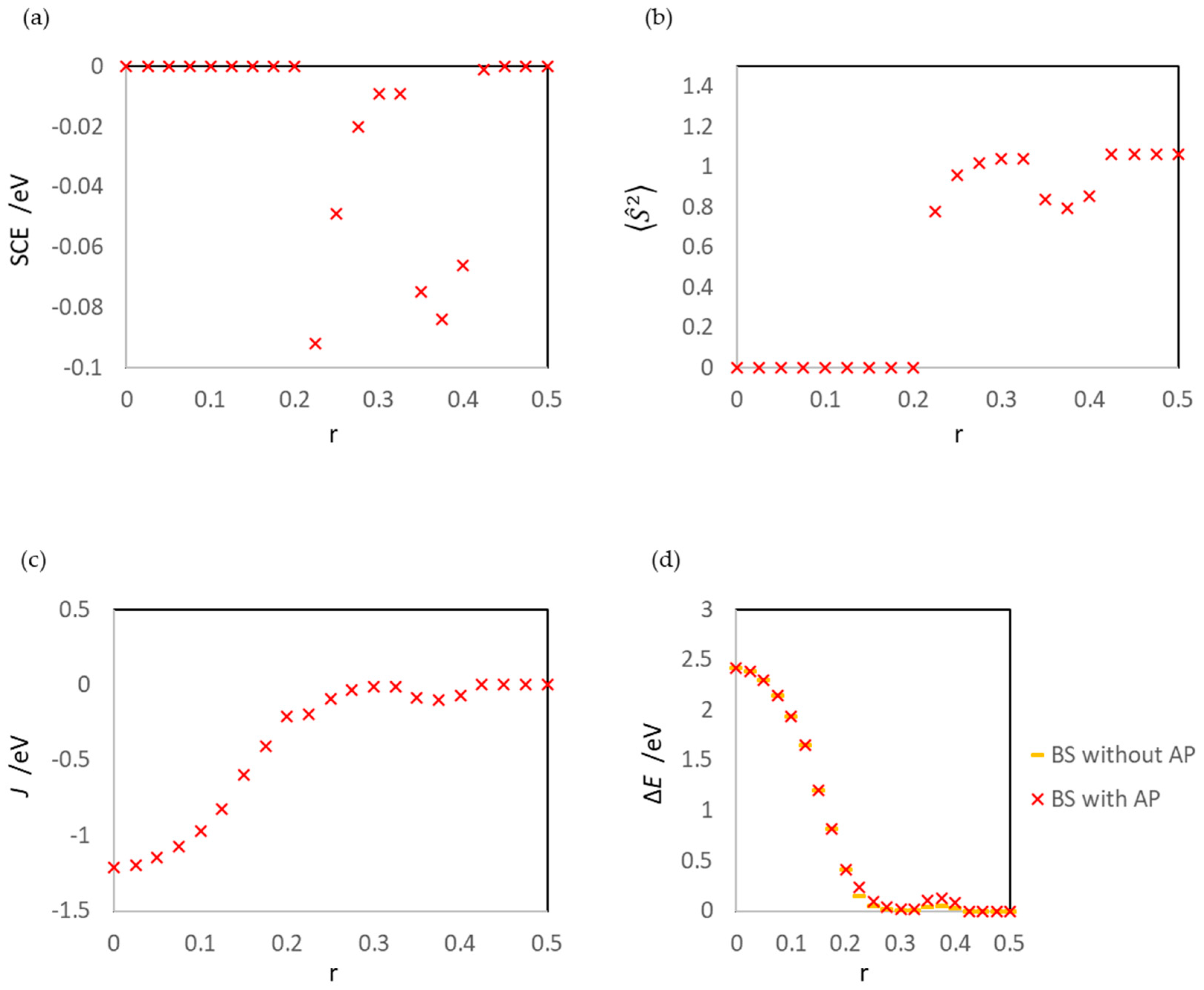
| r | SCE/eV | J/eV | d(1Au-2Au)/Å | ||||
|---|---|---|---|---|---|---|---|
| Surface | Gas | Surface | Gas | Surface | Gas | ||
| 0.000 | 0.000 | 0.000 | 0.0000 | 0.0000 | −1.208 1 | −0.894 1 | 2.52917 |
| 0.025 | 0.000 | 0.000 | 0.0000 | 0.0000 | −1.193 1 | −0.894 1 | 2.53112 |
| 0.050 | 0.000 | 0.000 | 0.0000 | 0.0000 | −1.148 1 | −0.893 1 | 2.54011 |
| 0.075 | 0.000 | 0.000 | 0.0000 | 0.0000 | −1.073 1 | −0.891 1 | 2.55653 |
| 0.100 | 0.000 | 0.000 | 0.0000 | 0.0000 | −0.966 1 | −0.891 1 | 2.57930 |
| 0.125 | 0.000 | 0.000 | 0.0000 | 0.0000 | −0.823 1 | −0.893 1 | 2.61054 |
| 0.150 | 0.000 | 0.000 | 0.0000 | 0.0000 | −0.600 1 | −0.880 1 | 2.66684 |
| 0.175 | 0.000 | 0.000 | 0.0000 | 0.0000 | −0.407 1 | −0.626 1 | 2.99674 |
| 0.200 | 0.000 | 0.000 | 0.0000 | 0.0001 | −0.207 | −0.399 1 | 3.40169 |
| 0.225 | −0.092 | 0.000 | 0.7747 | 0.0001 | −0.195 | −0.207 | 3.82430 |
| 0.250 | −0.049 | −0.095 | 0.9567 | 0.7240 | −0.093 | −0.083 | 4.24920 |
| 0.275 | −0.020 | −0.054 | 1.0168 | 0.9215 | −0.037 | −0.031 | 4.67412 |
| 0.300 | −0.009 | −0.023 | 1.0394 | 0.9915 | −0.017 | −0.012 | 5.09924 |
| 0.325 | −0.009 | −0.009 | 1.0394 | 1.0178 | −0.015 | −0.004 | 5.52656 |
| 0.350 | −0.075 | −0.003 | 0.8399 | 1.0283 | −0.088 | −0.001 | 5.96645 |
| 0.375 | −0.084 | −0.001 | 0.7947 | 1.0325 | −0.101 | 0.000 | 6.39464 |
| 0.400 | −0.066 | 0.000 | 0.8554 | 1.0344 | −0.075 | 0.000 | 6.81369 |
| 0.425 | −0.001 | 0.000 | 1.0616 | 1.0353 | −0.001 | 0.000 | 7.22546 |
| 0.450 | 0.000 | 0.000 | 1.0619 | 1.0358 | −0.001 | 0.000 | 7.64864 |
| 0.475 | 0.000 | 0.000 | 1.0605 | 1.0360 | 0.000 | 0.000 | 8.07348 |
| 0.500 | 0.000 | 0.000 | 1.0599 | 1.0360 | 0.000 | 0.000 | 8.49840 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tada, K.; Maruyama, T.; Koga, H.; Okumura, M.; Tanaka, S. Extent of Spin Contamination Errors in DFT/Plane-wave Calculation of Surfaces: A Case of Au Atom Aggregation on a MgO Surface. Molecules 2019, 24, 505. https://doi.org/10.3390/molecules24030505
Tada K, Maruyama T, Koga H, Okumura M, Tanaka S. Extent of Spin Contamination Errors in DFT/Plane-wave Calculation of Surfaces: A Case of Au Atom Aggregation on a MgO Surface. Molecules. 2019; 24(3):505. https://doi.org/10.3390/molecules24030505
Chicago/Turabian StyleTada, Kohei, Tomohiro Maruyama, Hiroaki Koga, Mitsutaka Okumura, and Shingo Tanaka. 2019. "Extent of Spin Contamination Errors in DFT/Plane-wave Calculation of Surfaces: A Case of Au Atom Aggregation on a MgO Surface" Molecules 24, no. 3: 505. https://doi.org/10.3390/molecules24030505
APA StyleTada, K., Maruyama, T., Koga, H., Okumura, M., & Tanaka, S. (2019). Extent of Spin Contamination Errors in DFT/Plane-wave Calculation of Surfaces: A Case of Au Atom Aggregation on a MgO Surface. Molecules, 24(3), 505. https://doi.org/10.3390/molecules24030505