Synthesis of New Proteomimetic Quinazolinone Alkaloids and Evaluation of Their Neuroprotective and Antitumor Effects

 ,
,  ,
,  ,
,  , ,
, ,  , ,
, ,  and
and 
Abstract
:
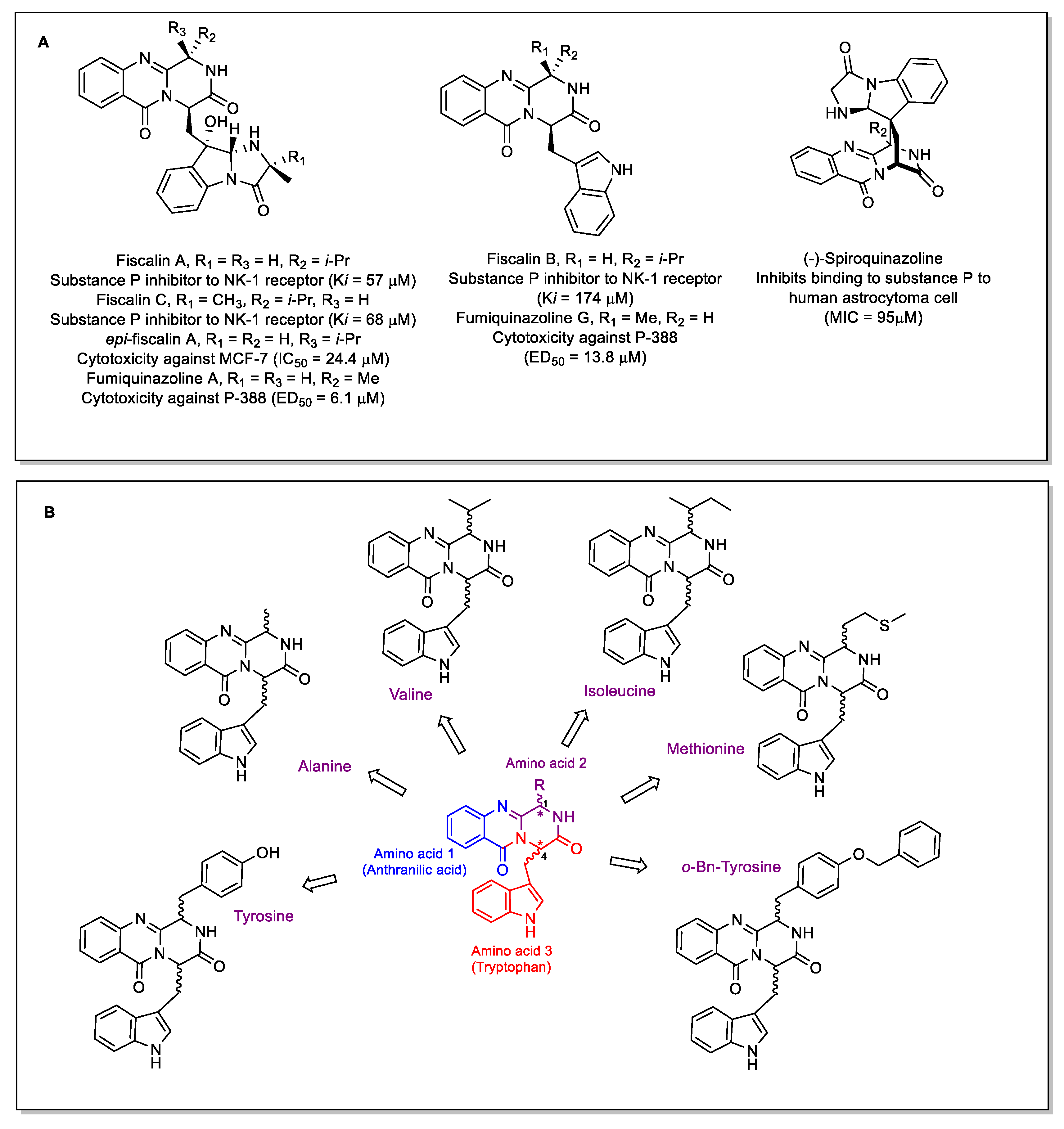
1. Introduction
2. Results
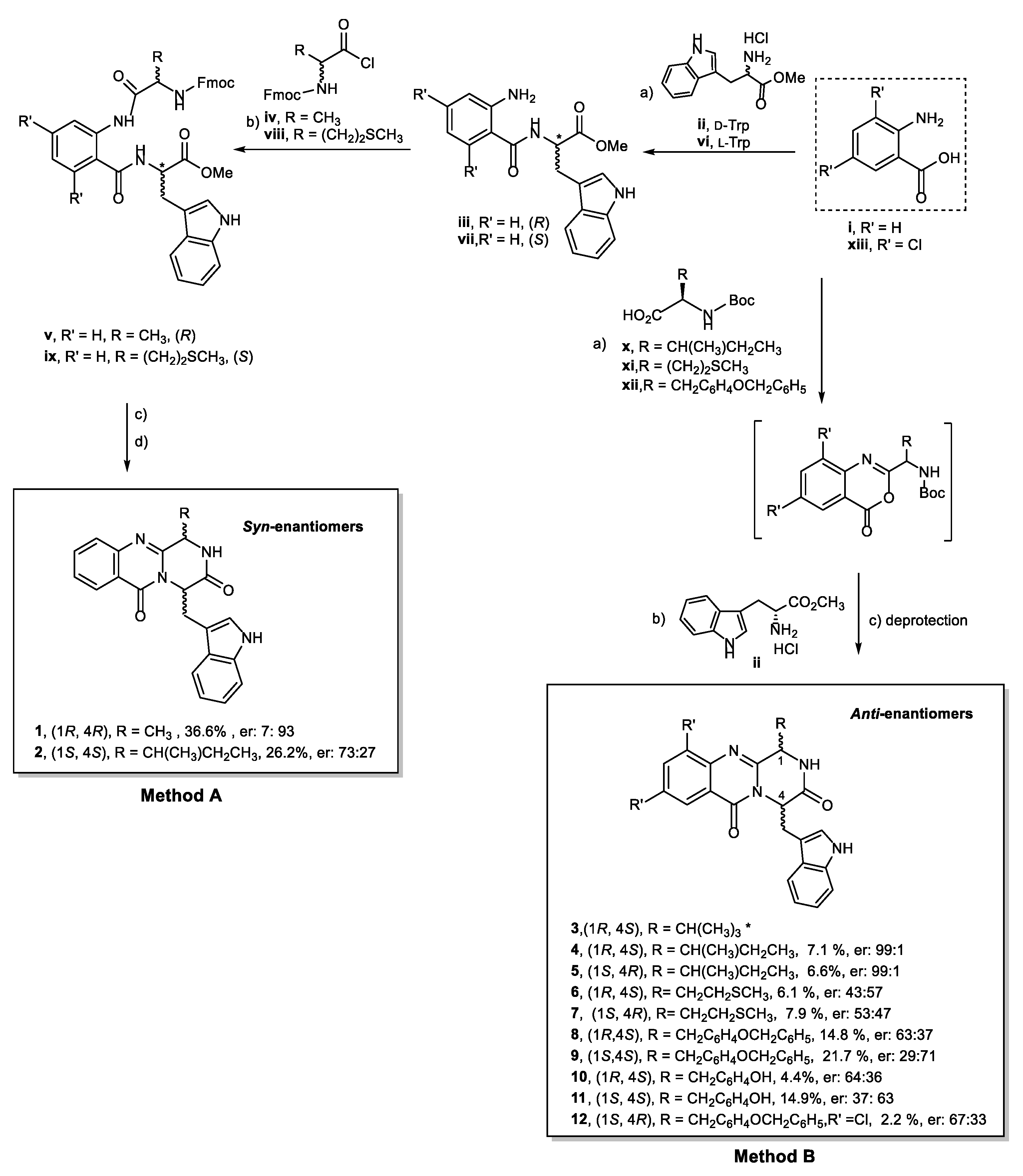
2.1. Chemistry
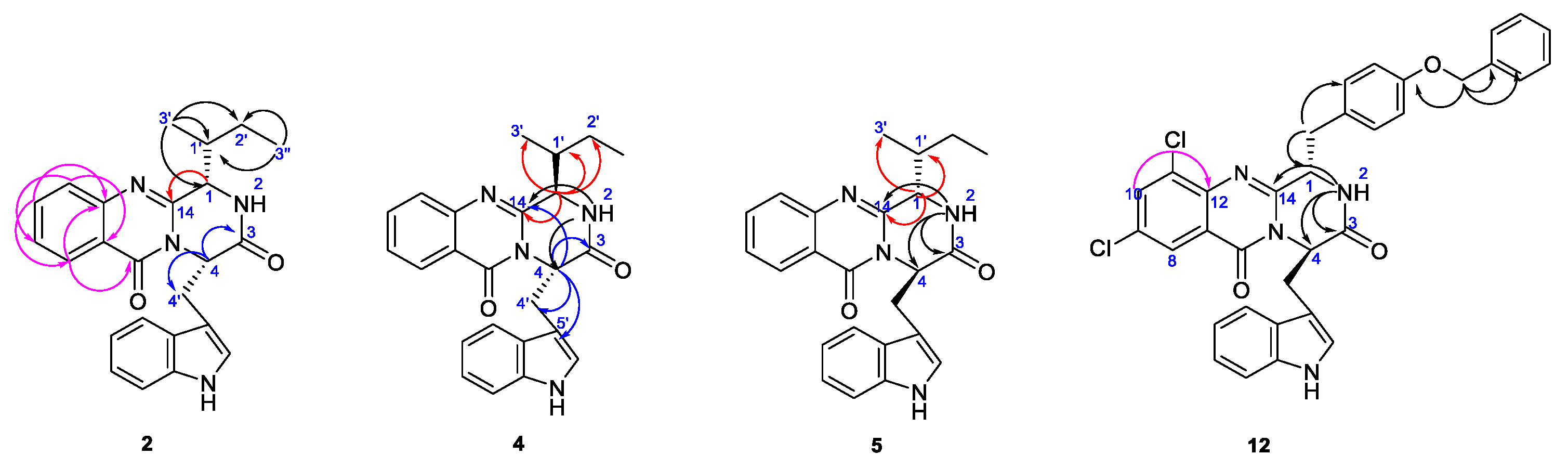
2.2. Structure Elucidation
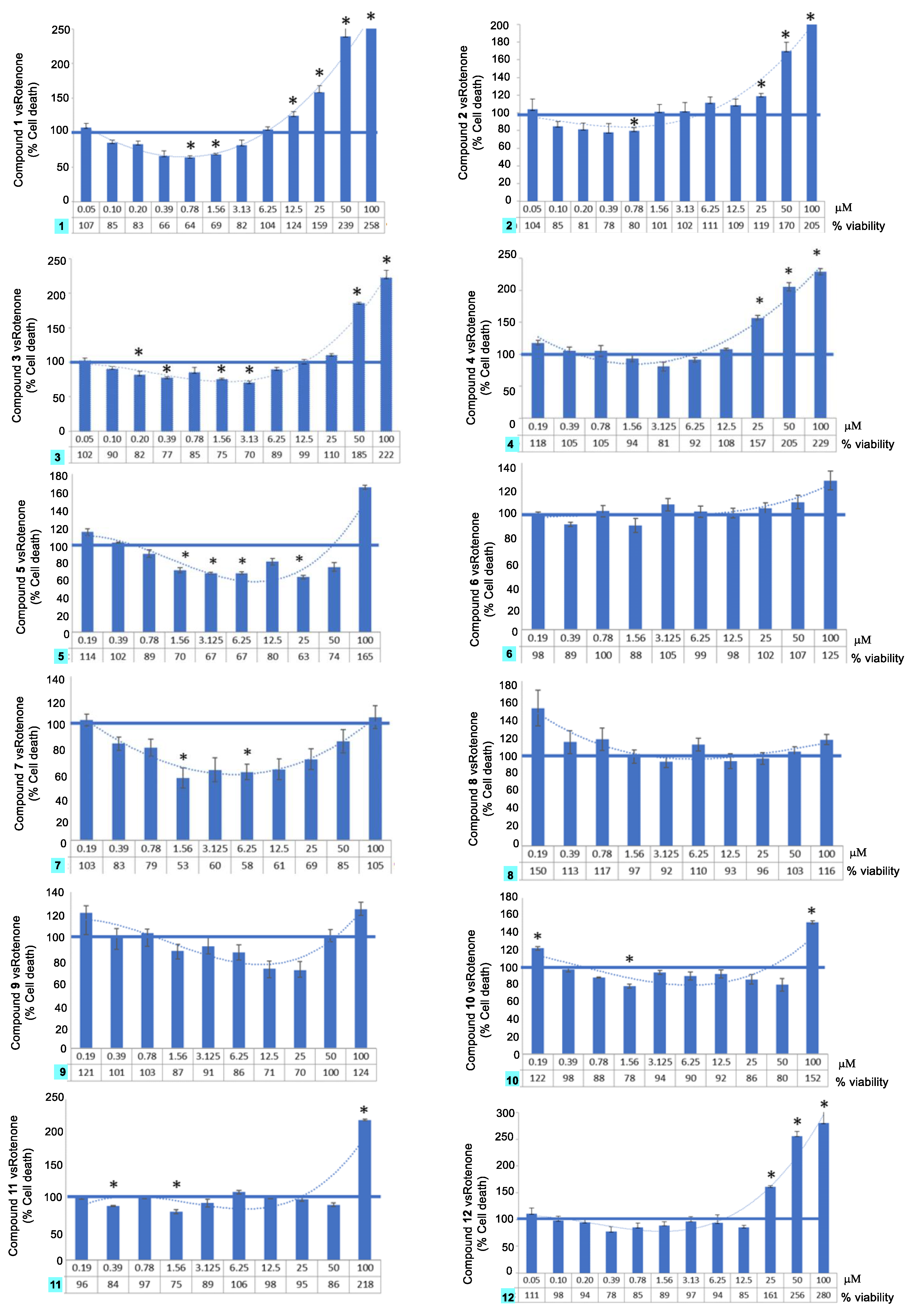
2.3. Neuroprotection Activity
2.4. Tumor Cell Growth Inhibitory Activity
2.5. Activity in Non-Tumor Cells
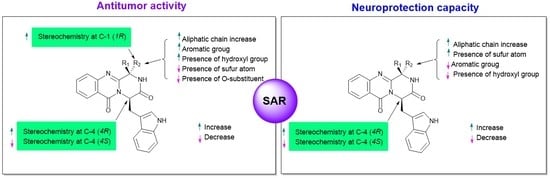
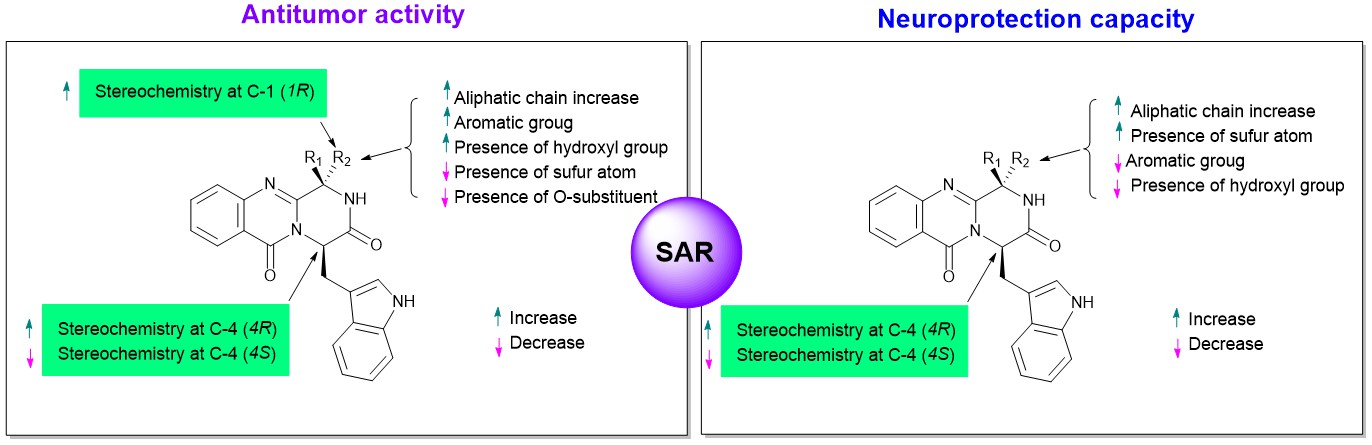
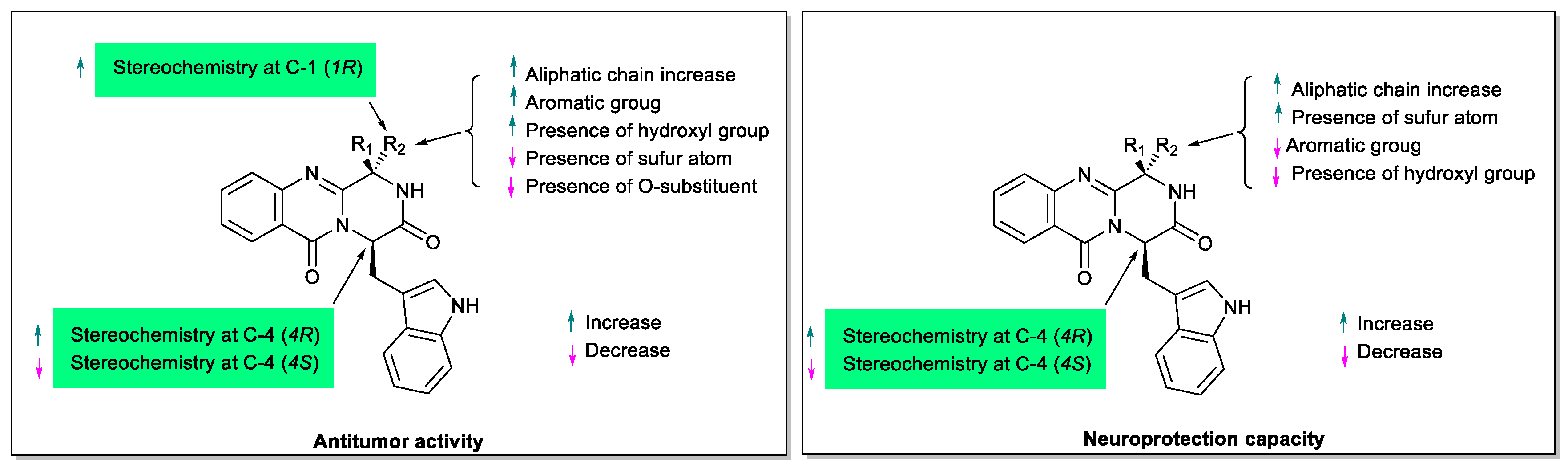
3. Structural-Activity Relationship (SAR)
4. Materials and Methods
4.1. General Procedure
4.2. General Conditions for the Synthesis of Compound (1R,4R)-4-((1H-indol-3-yl)methyl)-1-((R)-methyl)-1,2-dihydro-6H-pyrazino[2,1-b]quinazoline-3,6(4H)-dione (1)
4.3. General Conditions for the Synthesis of Compound (1S,4S)-4-((1H-indol-3-yl)methyl)-1-((S)-sec-butyl) -1,2-dihydro-6H-pyrazino[2,1-b]quinazoline-3,6(4H)-dione (2)
4.4. General Conditions for the Synthesis of Quinazolinone-3,6-(4H)-Diones Compound 4, 5, 6, 7, 8, and 9
4.5. General Conditions for the Synthesis of (1S)-4-((1H-indol-3-yl)methyl)-1-(4-hydroxybenzyl)-1,2-dihydro -6H-pyrazino[2,1-b]quinazoline-3,6(4H)-dione (10 and 11)
4.6. General Conditions for the Synthesis of (1S,4R)-4-((1H-indol-3-yl)methyl)-1-(4-(benzyloxy)benzyl)-8,10 -dichloro-1,2-dihydro-6H-pyrazino[2,1-b]quinazoline-3,6(4H)-dione (12)
4.7. Neuroprotection Assay
4.8. Screening Test for Antitumor Activity
4.9. Testing Effect of Compounds on Non-malignant Breast Cells
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Plun-Favreau, H.; Lewis, P.A.; Hardy, J.; Martins, L.M.; Wood, N.W. Cancer and neurodegeneration: Between the devil and the deep blue sea. PLoS Genet. 2010, 6, e1001257. [Google Scholar] [CrossRef] [PubMed]
- Klus, P.; Cirillo, D.; Botta Orfila, T.; Tartaglia, G.G. Neurodegeneration and cancer: Where the disorder prevails. Sci. Rep. 2015, 5, 15390. [Google Scholar] [CrossRef] [PubMed]
- Resende, D.I.S.P.; Boonpothong, P.; Sousa, E.; Kijjoa, A.; Pinto, M.M.M. Chemistry of the fumiquinazolines and structurally related alkaloids. Nat. Prod. Rep. 2019, 36, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Buttachon, S.; Chandrapatya, A.; Manoch, L.; Silva, A.; Gales, L.; Bruyère, C.; Kiss, R.; Kijjoa, A. Sartorymensin, a new indole alkaloid, and new analogues of tryptoquivaline and fiscalins produced by neosartorya siamensis (kufc 6349). Tetrahedron 2012, 68, 3253–3262. [Google Scholar] [CrossRef]
- Tamiya, H.; Ochiai, E.; Kikuchi, K.; Yahiro, M.; Toyotome, T.; Watanabe, A.; Yaguchi, T.; Kamei, K. Secondary metabolite profiles and antifungal drug susceptibility of aspergillus fumigatus and closely related species, aspergillus lentulus, aspergillus udagawae, and aspergillus viridinutans. J. Infect. Chemother. 2015, 21, 385–391. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Sun, Y.-L.; Liu, K.-S.; Zhang, X.-Y.; Qian, P.-Y.; Wang, Y.-F.; Qi, S.-H. Indole alkaloids from marine-derived fungus aspergillus sydowii scsio 00305. J. Antibiot. 2012, 65, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, C.; Matsushita, T.; Doi, M.; Minoura, K.; Shingu, T.; Kumeda, Y.; Numata, A. Fumiquinazolines a–g, novel metabolites of a fungus separated from a pseudolabrus marine fish. J. Chem. Soc. Perkin Trans. 1 1995, 18, 2345–2353. [Google Scholar] [CrossRef]
- Numata, A.; Takahashi, C.; Matsushita, T.; Miyamoto, T.; Kawai, K.; Usami, Y.; Matsumura, E.; Inoue, M.; Ohishi, H.; Shingu, T. Fumiquinazolines, novel metabolites of a fungus isolated from a saltfish. Tetrahedron Lett. 1992, 33, 1621–1624. [Google Scholar] [CrossRef]
- Han, X.-x.; Xu, X.-y.; Cui, C.-b.; Gu, Q.-q. Alkaloidal compounds produced by a marine-derived fungus, aspergillus fumigatus h1-04, and their antitumor activities. Chinese J. Org. Chem. 2007, 17, 232–237. [Google Scholar]
- Cheng, Z.; Lou, L.; Liu, D.; Li, X.; Proksch, P.; Yin, S.; Lin, W. Versiquinazolines a–k, fumiquinazoline-type alkaloids from the gorgonian-derived fungus aspergillus versicolor lzd-14-1. J. Nat. Prod. 2016, 79, 2941–2952. [Google Scholar] [CrossRef]
- Wong, S.M.; Musza, L.L.; Kydd, G.C.; Kullnig, R.; Gillum, A.M.; Cooper, R. Fiscalins: New sibstamce p inhibitors produced by the fungus neosartorya fischeri. Taxonomy, fermentation, structures, and biological properties. J. Antibiot. 1993, 46, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Barrow, C.J.; Sun, H.H. Spiroquinazoline, a novel substance p inhibitor with a new carbon skeleton, isolated from aspergillus flavipes. J. Nat. Prod. 1994, 57, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Thornton, E.; Vink, R. Treatment with a substance p receptor antagonist is neuroprotective in the intrastriatal 6-hydroxydopamine model of early parkinson’s disease. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Dunlop, R.A.; Cox, P.A.; Banack, S.A.; Rodgers, K.J. The non-protein amino acid bmaa is misincorporated into human proteins in place of l-serine causing protein misfolding and aggregation. PLoS ONE 2013, 8, e75376. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.J. Non-protein amino acids and neurodegeneration: The enemy within. Exp. Neurol. 2014, 253, 192–196. [Google Scholar] [CrossRef]
- Long, S.; Resende, D.I.S.P.; Kijjoa, A.; Silva, A.M.S.; Pina, A.; Fernández-Marcelo, T.; Vasconcelos, M.H.; Sousa, E.; Pinto, M.M.M. Antitumor activity of quinazolinone alkaloids inspired by marine natural products. Mar. Drugs 2018, 16, 261. [Google Scholar] [CrossRef]
- Wang, H.; Ganesan, A. Total synthesis of the fumiquinazoline alkaloids: Solution-phase studies. J. Org. Chem. 2000, 65, 1022–1030. [Google Scholar] [CrossRef]
- Liu, J.F.; Ye, P.; Zhang, B.; Bi, G.; Sargent, K.; Yu, L.; Yohannes, D.; Baldino, C.M. Three-component one-pot total syntheses of glyantrypine, fumiquinazoline f, and fiscalin b promoted by microwave irradiation. J. Org. Chem. 2005, 70, 6339–6345. [Google Scholar] [CrossRef] [PubMed]
- Okaya, S.; Okuyama, K.; Okano, K.; Tokuyama, H. Trichloroboron-promoted deprotection of phenolic benzyl ether using pentamethylbenzene as a non lewis-basic cation scavenger. In Organic Syntheses; Wiley: Hoboken, NJ, USA, 2016; Volume 93, pp. 63–74. [Google Scholar]
- Hernández, F.; Buenadicha, F.L.; Avendao, C.; Söllhuber, M. 1-alkyl-2,4-dihydro-1h-pyrazino[2,1-b]quinazoline-3,6-diones as glycine templates. Synthesis of fiscalin b. Tetrahedron Asymmetr. 2002, 12, 3387–3398. [Google Scholar] [CrossRef]
- Campos, A.C.; Fogaça, M.V.; Sonego, A.B.; Guimarães, F.S. Cannabidiol, neuroprotection and neuropsychiatric disorders. Pharmacol. Res. 2016, 112, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Bagli, E.; Goussia, A.; Moschos, M.M.; Agnantis, N.; Kitsos, G. Natural compounds and neuroprotection: Mechanisms of action and novel delivery systems. In Vivo 2016, 30, 535–547. [Google Scholar] [PubMed]
- Cannon, J.R.; Tapias, V.; Na, H.M.; Honick, A.S.; Drolet, R.E.; Greenamyre, J.T. A highly reproducible rotenone model of parkinson’s disease. Neurobiol. Dis. 2009, 34, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Condello, S.; Currò, M.; Ferlazzo, N.; Caccamo, D.; Satriano, J.; Ientile, R. Agmatine effects on mitochondrial membrane potential andnf-κb activation protect against rotenone-induced cell damage in human neuronal-like sh-sy5y cells. J. Neurochem. 2011, 116, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine b colorimetric assay for cytotoxicity screening. Nat. protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Zhou, Y.; Debbab, A.; Mándi, A.; Wray, V.; Schulz, B.; Müller, W.E.G.; Kassack, M.; Lin, W.; Kurtán, T.; Proksch, P.; et al. Alkaloids from the sponge-associated fungus aspergillus sp. Eur. J. Org. Chem. 2013, 5, 894–906. [Google Scholar] [CrossRef]
- Rodrigues, B.S.F.; Sahm, B.D.B.; Jimenez, P.C.; Pinto, F.C.L.; Mafezoli, J.; Mattos, M.C.; Rodrigues-Filho, E.; Pfenning, L.H.; Abreu, L.M.; Costa-Lotufo, L.V.; et al. Bioprospection of cytotoxic compounds in fungal strains recovered from sediments of the brazilian coast. Chem. Biodivers. 2015, 12, 432–442. [Google Scholar] [CrossRef]
- Fujimoto, H.; Negishi, E.; Yamaguchi, K.; Nishi, N.; Yamazaki, M. Isolation of new tremorgenic metabolites from an ascomycete, corynascus setosus. Chem. Pharm. Bull. 1996, 40, 1843–1848. [Google Scholar] [CrossRef]
- Wu, B.; Chen, G.; Liu, Z.-g.; Pei, Y. Two new alkaloids from a marine-derived fungus neosartorya fischeri. Rec. Nat. Prod. 2015, 9, 271–275. [Google Scholar]
- Kantharaju, B.S.P.; Suresh Babu, V.V. Synthesis of fmoc-amino acid chlorides assisted by ultrasonication, a rapid approach. Int. J. Pept. Res. Ther. 2002, 9, 227–229. [Google Scholar] [CrossRef]
- Wang, H.; Ganesan, A. Total synthesis of the quinazoline alkaloids (−)-fumiquinazoline g and (−)-fiscalin b. J. Org. Chem. 1998, 63, 2432–2433. [Google Scholar] [CrossRef]
- Lopes-Rodrigues, V.; Oliveira, A.; Correia-da-Silva, M.; Pinto, M.; Lima, R.T.; Sousa, E.; Vasconcelos, M.H. A novel curcumin derivative which inhibits p-glycoprotein, arrests cell cycle and induces apoptosis in multidrug resistance cells. Bioorg. Med. Chem. 2017, 25, 581–596. [Google Scholar] [CrossRef] [PubMed]
- Kijjoa, A.; Santos, S.; Dethoup, T.; Manoch, L.; Almeida, A.P.; Vasconcelos, M.H.; Silva, A.; Gales, L.; Herz, W. Sartoryglabrins, analogs of ardeemins, from neosartorya glabra. Nat. Prod. Commun. 2011, 6, 807–812. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds 1–12 are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | GI50 (µM) | ||
|---|---|---|---|
| NCI-H460 | BxPC3 | PANC1 | |
| 1 | 7.62 ± 0.7 | 17.34 ± 1.7 | 10.06 ± 0.8 |
| 2 | 65.38 ± 4.0 | 104.77 ± 10.9 | 86.30 ± 11.1 |
| 4 | 69.26 ± 2.9 | 88.81 ± 9.6 | 78.17 ± 8.4 |
| 5 | 40.11 ± 5.0 | 57.44 ± 4.7 | 60.68 ± 4.8 |
| 6 | 151.07 ± 2.9 | 126.06 ± 21.5 | 112.44 ± 12.7 |
| 7 | 61.37 ± 2.3 | 99.10 ±5.8 | 76.65 ± 4.8 |
| 8 | 72.68 ± 6.2 | 115.42 ± 12.1 | 74.90 ± 7.2 |
| 10 | 38.00 ± 1.5 | 50.59 ± 2.3 | 34.13 ± 1.8 |
| 11 | 46.25 ± 5.8 | 29.87 ± 3.7 | 27.93 ± 0.8 |
| Gemcitabine | - | 0.20 ± 0.08 | 0.73 ± 0.22 |
| Doxorubicin | 0.0124 ± 0.0018 | - | - |
| Compounds | Concentration (μM) * | % Cell Growth Inhibition (Relative to the Control) |
|---|---|---|
| 5 | 65 | 71.69 ± 7.9 |
| 7 | 100 | 89.56 ± 3.7 |
| 10 | 50 | 75.63 ± 4.7 |
| 11 | 50 | 71.15 ± 2.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Long, S.; Resende, D.I.S.P.; Kijjoa, A.; Silva, A.M.S.; Fernandes, R.; Xavier, C.P.R.; Vasconcelos, M.H.; Sousa, E.; Pinto, M.M.M. Synthesis of New Proteomimetic Quinazolinone Alkaloids and Evaluation of Their Neuroprotective and Antitumor Effects. Molecules 2019, 24, 534. https://doi.org/10.3390/molecules24030534
Long S, Resende DISP, Kijjoa A, Silva AMS, Fernandes R, Xavier CPR, Vasconcelos MH, Sousa E, Pinto MMM. Synthesis of New Proteomimetic Quinazolinone Alkaloids and Evaluation of Their Neuroprotective and Antitumor Effects. Molecules. 2019; 24(3):534. https://doi.org/10.3390/molecules24030534
Chicago/Turabian StyleLong, Solida, Diana I. S. P. Resende, Anake Kijjoa, Artur M. S. Silva, Ricardo Fernandes, Cristina P. R. Xavier, M. Helena Vasconcelos, Emília Sousa, and Madalena M. M. Pinto. 2019. "Synthesis of New Proteomimetic Quinazolinone Alkaloids and Evaluation of Their Neuroprotective and Antitumor Effects" Molecules 24, no. 3: 534. https://doi.org/10.3390/molecules24030534
APA StyleLong, S., Resende, D. I. S. P., Kijjoa, A., Silva, A. M. S., Fernandes, R., Xavier, C. P. R., Vasconcelos, M. H., Sousa, E., & Pinto, M. M. M. (2019). Synthesis of New Proteomimetic Quinazolinone Alkaloids and Evaluation of Their Neuroprotective and Antitumor Effects. Molecules, 24(3), 534. https://doi.org/10.3390/molecules24030534