
Optimisation of Protein Extraction from Medicinal Cannabis Mature Buds for Bottom-Up Proteomics



Abstract
:1. Introduction
2. Results and Discussion
2.1. Intact Protein Analysis
2.2. Tryptic Peptides Analysis
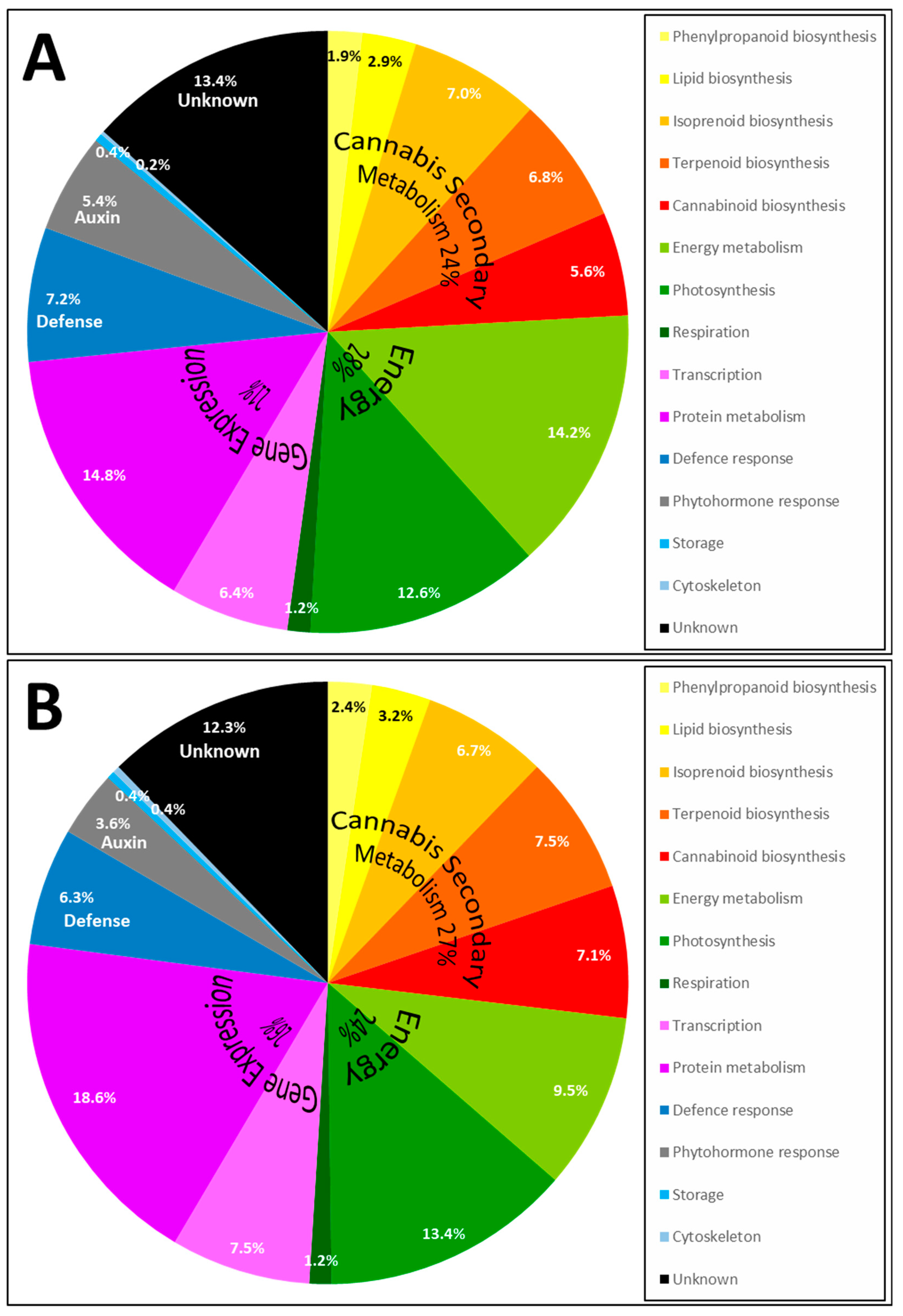
2.3. Proteins Identified by Bottom-Up Proteomics
2.4. Enzymes Involved in Phytocannabinoid Pathway
3. Materials and Methods
3.1. Plant Materials
3.1.1. Apical Bud Sampling and Grinding
3.1.2. Trichome Recovery
3.2. Protein Extraction Methods
3.2.1. Extraction 1: Resuspension in Urea Buffer
3.2.2. Extraction 2: Resuspension in Guanidine Hydrochloride Buffer
3.2.3. Extraction 3: TCA/Acetone Precipitation Followed by Resuspension in Urea Buffer
3.2.4. Extraction 4: TCA/Acetone Precipitation Followed by Resuspension in Guanidine Hydrochloride Buffer
3.2.5. Extraction 5: TCA/Ethanol Precipitation Followed by Resuspension in Urea Buffer
3.2.6. Extraction 6: TCA/Ethanol Precipitation Followed by Resuspension in Guanidine Hydrochloride Buffer
3.3. Alkylation and Protein Assay
3.4. Trypsin/LysC protein Digestion and Desalting
3.4.1. Protease Digestion
3.4.2. Desalting
3.5. Intact Protein Analysis by Ultraperformance Liquid Chromatography Mass Spectrometry
3.5.1. UPLC separation
3.5.2. MS acquisition
3.6. Peptide Digest Analysis by Nanoliquid Chromatography-Tandem Mass Spectrometry
3.7. Database Search for Protein Identification
3.8. Data Processing and Statistical Analyses
3.8.1. LC-MS and nLC-MS/MS Data Processing
3.8.2. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hand, A.; Blake, A.; Kerrigan, P.; Samuel, P.; Friedberg, J. History of medical cannabis. J. Pain Manage. 2016, 9, 387–394. [Google Scholar]
- Zuardi, A.W. History of cannabis as a medicine: A review. Rev. Bras. Psiquiatr. 2006, 28, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.; Hunt, M.; Clark, J.H. Structure of cannabidiol, a product isolated from the marihuana extract of Minnesota wild hemp. J. Am. Chem. Soc. 1940, 62, 196–200. [Google Scholar] [CrossRef]
- Mechoulam, R.; Gaoni, Y. Recent advances in the chemistry of hashish. Prog. Chem. Org. Nat. Prod. 1967, 25, 175–213. [Google Scholar]
- ElSohly, M.A.; Slade, D. Chemical constituents of marijuana: The complex mixture of natural cannabinoids. Life Sci. 2005, 78, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Sirikantaramas, S.; Taura, F.; Morimoto, S.; Shoyama, Y. Recent advances in Cannabis sativa research: Biosynthetic studies and its potential in biotechnology. Curr. Pharm. Biotechnol. 2007, 8, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Andre, C.M.; Hausman, J.-F.; Guerriero, G. Cannabis sativa: The Plant of the Thousand and One Molecules. Front. Plant Sci. 2016, 7, 19. [Google Scholar] [CrossRef]
- Bona, E.; Marsano, F.; Cavaletto, M.; Berta, G. Proteomic characterization of copper stress response in Cannabis sativa roots. Proteomics 2007, 7, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Behr, M.; Sergeant, K.; Leclercq, C.C.; Planchon, S.; Guignard, C.; Lenouvel, A.; Renaut, J.; Hausman, J.F.; Lutts, S.; Guerriero, G. Insights into the molecular regulation of monolignol-derived product biosynthesis in the growing hemp hypocotyl. BMC Plant Biol. 2018, 18, 1. [Google Scholar] [CrossRef]
- Aiello, G.; Fasoli, E.; Boschin, G.; Lammi, C.; Zanoni, C.; Citterio, A.; Arnoldi, A. Proteomic characterization of hempseed (Cannabis sativa L.). J. Proteomics 2016, 147, 187–196. [Google Scholar] [CrossRef]
- Park, S.K.; Seo, J.B.; Lee, M.Y. Proteomic profiling of hempseed proteins from Cheungsam. Biochim. Biophys. Acta 2012, 1824, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Raharjo, T.J.; Widjaja, I.; Roytrakul, S.; Verpoorte, R. Comparative proteomics of Cannabis sativa plant tissues. J. Biomol. Tech. 2004, 15, 97–106. [Google Scholar] [PubMed]
- Happyana, N. Metabolomics, Proteomics, and Transcriptomics of Cannabis sativa L. Trichomes. Ph.D. Thesis, TU Dortmund, Dortmund, Germany, 2014. [Google Scholar]
- McCarthy, J.; Hopwood, F.; Oxley, D.; Laver, M.; Castagna, A.; Righetti, P.G.; Williams, K.; Herbert, B. Carbamylation of proteins in 2-D electrophoresis--myth or reality? J. Proteome Res. 2003, 2, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, J.W.; Madsen, C.T.; Young, C.; Poulsen, F.M.; Nielsen, M.L. Using guanidine-hydrochloride for fast and efficient protein digestion and single-step affinity-purification mass spectrometry. J. Proteome Res. 2013, 12, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Takakura, D.; Hashii, N.; Kawasaki, N. An improved in-gel digestion method for efficient identification of protein and glycosylation analysis of glycoproteins using guanidine hydrochloride. Proteomics 2014, 14, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Raynes, J.K.; Vincent, D.; Zawadzki, J.L.; Savin, K.; Mertens, D.; Logan, A.; Williams, R.P.W. Investigation of Age Gelation in UHT Milk. Beverages 2018, 4, 95. [Google Scholar] [CrossRef]
- Vincent, D.; Elkins, A.; Condina, M.R.; Ezernieks, V.; Rochfort, S. Quantitation and Identification of Intact Major Milk Proteins for High-Throughput LC-ESI-Q-TOF MS Analyses. PLoS ONE 2016, 11, e0163471. [Google Scholar] [CrossRef]
- Vincent, D.; Mertens, D.; Rochfort, S. Optimisation of Milk Protein Top-Down Sequencing Using In-Source Collision-Induced Dissociation in the Maxis Quadrupole Time-of-Flight Mass Spectrometer. Molecules 2018, 23, 2777. [Google Scholar] [CrossRef]
- Booth, J.K.; Page, J.E.; Bohlmann, J. Terpene synthases from Cannabis sativa. PLoS ONE 2017, 12, e0173911. [Google Scholar] [CrossRef]
- van Bakel, H.; Stout, J.M.; Cote, A.G.; Tallon, C.M.; Sharpe, A.G.; Hughes, T.R.; Page, J.E. The draft genome and transcriptome of Cannabis sativa. Genome Biol. 2011, 12, R102. [Google Scholar] [CrossRef]
- Laverty, K.U.; Stout, J.M.; Sullivan, M.J.; Shah, H.; Gill, N.; Holbrook, L.; Deikus, G.; Sebra, R.; Hughes, T.R.; Page, J.E.; et al. A physical and genetic map of Cannabis sativa identifies extensive rearrangement at the THC/CBD acid synthase locus. Genome Res. 2018. [Google Scholar] [CrossRef]
- Grassa, C.J.; Wenger, J.P.; Dabney, C.; Poplawski, S.G.; Motley, S.T.; Michael, T.P.; Schwartz, C.J.; Weiblen, G.D. A complete Cannabis chromosome assembly and adaptive admixture for elevated cannabidiol (CBD) content. bioRxiv 2018. [Google Scholar] [CrossRef]
- Bridgeman, M.B.; Abazia, D.T. Medicinal Cannabis: History, Pharmacology, And Implications for the Acute Care Setting. P T. 2017, 42, 180–188. [Google Scholar] [PubMed]
- Yerger, E.H.; Grazzini, R.A.; Hesk, D.; Cox-Foster, D.L.; Craig, R.; Mumma, R.O. A rapid method for isolating glandular trichomes. Plant Physiol. 1992, 99, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Vincent, D.; Ezernieks, V.; Elkins, A.; Nguyen, N.; Moate, P.J.; Cocks, B.G.; Rochfort, S. Milk Bottom-Up Proteomics: Method Optimization. Front Genet 2015, 6, 360. [Google Scholar] [CrossRef] [PubMed]
- McPartland, J.M. Cannabis Systematics at the Levels of Family, Genus, and Species. Cannabis Cannabinoid Res. 2018, 3.1, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Page, J.; Boubakir, Z. Aromatic Prenyltransferase from Cannabis. Patent WO 2011/017798 Al, 4 October 2010. [Google Scholar]
Sample Availability: Samples are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue | Extraction Number | Extraction Method | Extraction Code | Protein Concentration (mg/mL) | Number of Proteins | ||
|---|---|---|---|---|---|---|---|
| Average (SD 1) | Average (SD 1) | CV 2 | Percent | ||||
| apical bud | extraction 1 | Urea | AB1 | 6.58 (± 0.89) | 254 (± 12) | 4.80 | 44.51 |
| apical bud | extraction 2 | Gnd-HCl | AB2 | 3.50 (± 0.99) | 335 (± 15) | 4.47 | 58.58 |
| apical bud | extraction 3 | TCA-A/urea | AB3 | 0.63 (± 0.15) | 247 (± 21) | 8.69 | 43.23 |
| apical bud | extraction 4 | TCA-A/Gnd-HCl | AB4 | 1.50 (± 0.28) | 314 (± 16) | 5.13 | 54.90 |
| apical bud | extraction 5 | TCA-E/urea | AB5 | 0.60 (± 0.11) | 201 (± 5) | 2.64 | 35.11 |
| apical bud | extraction 6 | TCA-E/Gnd-HCl | AB6 | 0.76 (± 0.48) | 264 (± 18) | 6.84 | 46.18 |
| trychome | extraction 1 | Urea | T1 | 3.67 (± 0.39) | 170 (± 5) | 2.97 | 29.83 |
| trychome | extraction 2 | Gnd-HCl | T2 | 2.28 (± 1.17) | 249 (± 45) | 18.12 | 43.61 |
| TOTAL | 571 | ||||||
| Tissue | Extraction Number | Extraction Method | Extraction Code | Number of Hits | ||
|---|---|---|---|---|---|---|
| Average (SD 1) | CV 2 | Percent | ||||
| apical bud | extraction 1 | Urea | AB1 | 211 (± 34) | 16.09 | 43.24 |
| apical bud | extraction 2 | Gnd-HCl | AB2 | 356 (± 20) | 5.51 | 72.88 |
| apical bud | extraction 3 | TCA-A/urea | AB3 | 265 (± 55) | 20.70 | 54.23 |
| apical bud | extraction 4 | TCA-A/Gnd-HCl | AB4 | 435 (± 9) | 2.09 | 89.07 |
| apical bud | extraction 5 | TCA-E/urea | AB5 | 41 (± 15) | 35.71 | 8.33 |
| apical bud | extraction 6 | TCA-E/Gnd-HCl | AB6 | 429 (± 6) | 1.33 | 87.91 |
| trychome | extraction 1 | Urea | T1 | 97 (± 22) | 22.27 | 19.88 |
| trychome | extraction 2 | Gnd-HCl | T2 | 102 (± 23) | 22.78 | 20.83 |
| TOTAL | 488 | |||||
| Protein Annotation | Abbreviation | Uniprot Accession or Patent Number | Species | Length (AA) | Number of Peptides Identified | EC Number | Function | Pathway |
|---|---|---|---|---|---|---|---|---|
| Small auxin upregulated protein | SAUR03 | A0A172J1 × 8 | Boehmeria nivea | 93 | 1 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR20 | A0A172J1Z7 | Boehmeria nivea | 147 | 1 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR23 | A0A172J212 | Boehmeria nivea | 99 | 1 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR24 | A0A172J211 | Boehmeria nivea | 102 | 1 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR28 | A0A172J206 | Boehmeria nivea | 108 | 1 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR30 | A0A172J210 | Boehmeria nivea | 100 | 1 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR31 | A0A172J276 | Boehmeria nivea | 152 | 1 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR40 | A0A172J219 | Boehmeria nivea | 105 | 1 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR44 | A0A172J227 | Boehmeria nivea | 152 | 4 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR48 | A0A172J226 | Boehmeria nivea | 133 | 1 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR54 | A0A172J237 | Boehmeria nivea | 118 | 5 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR55 | A0A172J229 | Boehmeria nivea | 97 | 3 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR58 | A0A172J236 | Boehmeria nivea | 97 | 1 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR59 | A0A172J243 | Boehmeria nivea | 106 | 5 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR60 | A0A172J238 | Boehmeria nivea | 105 | 1 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR70 | A0A172J249 | Boehmeria nivea | 183 | 1 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR71 | A0A172J2A4 | Boehmeria nivea | 183 | 1 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR51 | A0A172J290 | Boehmeria nivea | 97 | 1 | response to auxin | Phytohormone response | |
| Small auxin upregulated protein | SAUR52 | A0A172J241 | Boehmeria nivea | 149 | 1 | response to auxin | Phytohormone response | |
| Cannabidiolic acid synthase | CBDAS | A6P6V9 | Cannabis sativa | 544 | 8 | 1.21.3.8 | oxidative cyclisation of CBGA, producing CBDA | Cannabinoid biosynthesis |
| Geranylpyrophosphate:olivetolate geranyltransferase | GOT | WO 2011/017798 Al | Cannabis sativa | 395 | 4 | alkylation of OLA with geranyldiphosphate to form CBGA | Cannabinoid biosynthesis | |
| Olivetolic acid cyclase | OAC | I1V0C9 | Cannabis sativa | 545 | 1 | 4.4.1.26 | functions in concert with OLS/TKS to form OLA | Cannabinoid biosynthesis |
| Olivetolic acid cyclase | OAC | I6WU39 | Cannabis sativa | 101 | 5 | 4.4.1.26 | functions in concert with OLS/TKS to form OLA | Cannabinoid biosynthesis |
| 3,5,7-trioxododecanoyl-CoA synthase | OLS | B1Q2B6 | Cannabis sativa | 385 | 7 | 2.3.1.206 | olivetol biosynthesis | Cannabinoid biosynthesis |
| Tetrahydrocannabinolic acid synthase | THCAS | A0A0H3UZT7 | Cannabis sativa | 325 | 1 | 1.21.3.7 | oxidative cyclisation of CBGA, producing THCA | Cannabinoid biosynthesis |
| Tetrahydrocannabinolic acid synthase | THCAS | Q33DP7 | Cannabis sativa | 545 | 1 | 1.21.3.7 | oxidative cyclisation of CBGA, producing THCA | Cannabinoid biosynthesis |
| Tetrahydrocannabinolic acid synthase | THCAS | Q8GTB6 | Cannabis sativa | 545 | 4 | 1.21.3.7 | oxidative cyclisation of CBGA, producing THCA | Cannabinoid biosynthesis |
| Putative kinesin heavy chain | kin | Q5TIP9 | Cannabis sativa | 145 | 1 | microtubule-based movement | Cytoskeleton | |
| Betv1-like protein | Betv1 | I6XT51 | Cannabis sativa | 161 | 38 | Defence response | ||
| ATP synthase subunit alpha | atp1 | A0A0M5M1Z3 | Cannabis sativa | 509 | 12 | Produces ATP from ADP | Energy metabolism | |
| ATP synthase subunit alpha | atp1 | E5DK51 | Cannabis sativa | 349 | 1 | Produces ATP from ADP | Energy metabolism | |
| ATP synthase subunit 4 | atp4 | A0A0M4S8F3 | Cannabis sativa | 198 | 7 | Produces ATP from ADP | Energy metabolism | |
| ATP synthase subunit alpha | atpA | A0A0C5ARX6 | Cannabis sativa | 507 | 9 | Produces ATP from ADP | Energy metabolism | |
| ATP synthase subunit beta | atpB | F8TR83 | Cannabis sativa | 413 | 1 | 3.6.3.14 | Produces ATP from ADP | Energy metabolism |
| ATP synthase CF1 epsilon subunit | atpE | A0A0C5AUH9 | Cannabis sativa | 133 | 1 | Produces ATP from ADP | Energy metabolism | |
| ATP synthase subunit beta, chloroplastic | atpF | A0A0C5AUE9 | Cannabis sativa | 189 | 2 | Component of the F(0) channel | Energy metabolism | |
| NADH-ubiquinone oxidoreductase chain 1 | nad1 | A0A0M4S8G1 | Cannabis sativa | 324 | 1 | 7.1.1.2 | Energy metabolism | |
| NADH-ubiquinone oxidoreductase chain 5 | nad5 | A0A0M4RVP1 | Cannabis sativa | 669 | 1 | 7.1.1.2 | Energy metabolism | |
| NADH dehydrogenase subunit 7 | nad7 | A0A0M4S7M8 | Cannabis sativa | 394 | 1 | 7.1.1.2 | Energy metabolism | |
| NADH dehydrogenase subunit 9 | nad9 | A0A0M4R4N3 | Cannabis sativa | 190 | 2 | 7.1.1.2 | Energy metabolism | |
| NADH dehydrogenase subunit 7 | nadhd7 | A0A0X8GLG5 | Cannabis sativa | 394 | 1 | Energy metabolism | ||
| NADH-quinone oxidoreductase subunit H | ndhA | A0A0C5APZ2 | Cannabis sativa | 363 | 1 | 1.6.5.11 | NDH-1 shuttles electrons from NADH to quinones | Energy metabolism |
| NADH-quinone oxidoreductase subunit N | ndhB | A0A0C5B2K5 | Cannabis sativa | 510 | 1 | 1.6.5.11 | NDH-1 shuttles electrons from NADH to quinones | Energy metabolism |
| NADH-quinone oxidoreductase subunit K | ndhE | A0A0C5AUJ8 | Cannabis sativa | 101 | 4 | 1.6.5.11 | NDH-1 shuttles electrons from NADH to quinones | Energy metabolism |
| NADH-quinone oxidoreductase subunit C | ndhJ | A0A0C5B2I2 | Cannabis sativa | 158 | 2 | 1.6.5.11 | NDH-1 shuttles electrons from NADH to quinones | Energy metabolism |
| 1-deoxy-D-xylulose-5-phosphate reductoisomerase | DXR | A0A1V0QSG8 | Cannabis sativa | 472 | 2 | Converts 2-C-methyl-D-erythritol 4P into 1-deoxy-D-xylulose 5P | Isoprenoid biosynthesis | |
| Transferase FPPS1 | FPPS1 | A0A1V0QSH0 | Cannabis sativa | 341 | 1 | Isoprenoid biosynthesis | ||
| Transferase FPPS2 | FPPS2 | A0A1V0QSH7 | Cannabis sativa | 340 | 3 | Isoprenoid biosynthesis | ||
| Transferase GPPS large subunit | GPPS | A0A1V0QSH4 | Cannabis sativa | 393 | 2 | Isoprenoid biosynthesis | ||
| Transferase GPPS small subunit | GPPS | A0A1V0QSG9 | Cannabis sativa | 326 | 1 | Isoprenoid biosynthesis | ||
| Transferase GPPS small subunit2 | GPPS | A0A1V0QSI1 | Cannabis sativa | 278 | 1 | Isoprenoid biosynthesis | ||
| 4-hydroxy-3-methylbut-2-en-1-yl diphosphate reductase | HDR | A0A1V0QSH9 | Cannabis sativa | 408 | 6 | Converts (E)-4-hydroxy-3-methylbut-2-en-1-yl-2P into isopentenyl-2P | Isoprenoid biosynthesis | |
| Isopentenyl-diphosphate delta-isomerase | IDI | A0A1V0QSG5 | Cannabis sativa | 304 | 7 | Converts isopentenyl diphosphate into dimethylallyl diphosphate | Isoprenoid biosynthesis | |
| Mevalonate kinase | MK | A0A1V0QSI0 | Cannabis sativa | 416 | 3 | 2.7.1.36 | Converts (R)-mevalonate into (R)-5-phosphomevalonate | Isoprenoid biosynthesis |
| Diphosphomevalonate decarboxylase | MPDC | A0A1V0QSG4 | Cannabis sativa | 455 | 4 | Isoprenoid biosynthesis | ||
| Phosphomevalonate kinase | PMK | A0A1V0QSH8 | Cannabis sativa | 486 | 4 | Converts (R)-5-phosphomevalonate into (R)-5-diphosphomevalonate | Isoprenoid biosynthesis | |
| Non-specific lipid-transfer protein | ltp | P86838 | Cannabis sativa | 20 | 3 | transfer lipids across membranes | Lipid biosynthesis | |
| Non-specific lipid-transfer protein | ltp | W0U0V5 | Cannabis sativa | 91 | 9 | transfer lipids across membranes | Lipid biosynthesis | |
| 4-coumarate:CoA ligase | 4CL | A0A142EGJ1 | Cannabis sativa | 544 | 1 | 6.2.1.12 | forms 4-coumaroyl-CoA from 4-coumarate | Phenylpropanoid biosynthesis |
| 4-coumarate:CoA ligase | 4CL | V5KXG5 | Cannabis sativa | 550 | 3 | 6.2.1.12 | forms 4-coumaroyl-CoA from 4-coumarate | Phenylpropanoid biosynthesis |
| Phenylalanine ammonia-lyase | PAL | V5KWZ6 | Cannabis sativa | 707 | 4 | 4.3.1.24 | Catalyses L-phenylalanine = trans-cinnamate + ammonia | Phenylpropanoid biosynthesis |
| NAD(P)H-quinone oxidoreductase subunit 5, chloroplastic | ndhF | A0A0C5AUJ6 | Cannabis sativa | 755 | 1 | 1.6.5.- | NDH shuttles electrons from NAD(P)H:plastoquinone to quinones | Photosynthesis |
| Photosystem I P700 chlorophyll a apoprotein A1 | pasA | A0A0U2DTB0 | Cannabis sativa | 750 | 2 | 1.97.1.12 | bind P700, the primary electron donor of PSI | Photosynthesis |
| Photosystem I P700 chlorophyll a apoprotein A2 | psaB | A0A0C5APY0 | Cannabis sativa | 734 | 2 | 1.97.1.12 | bind P700, the primary electron donor of PSI | Photosynthesis |
| Photosystem I iron-sulfur center | psaC | A0A0C5AS17 | Cannabis sativa | 81 | 10 | 1.97.1.12 | assembly of the PSI complex | Photosynthesis |
| Photosystem II CP47 reaction center protein | psbB | A9XV91 | Cannabis sativa | 488 | 1 | binds chlorophyll in PSII | Photosynthesis | |
| Ribulose bisphosphate carboxylase large chain | rbcL | A0A0B4SX31 | Cannabis sativa | 312 | 15 | 4.1.1.39 | carboxylation of D-ribulose 1,5-bisphosphate | Photosynthesis |
| Small ubiquitin-related modifier | smt3 | Q5TIQ0 | Cannabis sativa | 76 | 2 | protein sumoylation | Protein metabolism | |
| Cytochrome c biogenesis FC | ccmFc | A0A0M4RVN1 | Cannabis sativa | 447 | 1 | Mitochondrial electron carrier protein | Respiration | |
| Cytochrome c biogenesis FN | ccmFn | A0A0M3UM18 | Cannabis sativa | 575 | 2 | Mitochondrial electron carrier protein | Respiration | |
| Cytochrome c biogenesis protein CcsA | ccsA | A0A0C5B2L0 | Cannabis sativa | 320 | 1 | biogenesis of c-type cytochromes | Respiration | |
| Cytochrome c | cytC | P00053 | Cannabis sativa | 111 | 2 | Mitochondrial electron carrier protein | Respiration | |
| 7S vicilin-like protein | Cs7S | A0A219D1T7 | Cannabis sativa | 493 | 2 | nutrient reservoir activity | Storage | |
| Edestin 1 | ede1D | A0A090CXP5 | Cannabis sativa | 511 | 1 | Seed storage protein | Storage | |
| 4-(cytidine 5’-diphospho)-2-C-methyl-D-erythritol kinase | CMK | A0A1V0QSI2 | Cannabis sativa | 408 | 4 | Adds 2-phosphate to 4-CDP-2-C-methyl-d-erythritol | Terpenoid biosynthesis | |
| 1-deoxy-D-xylulose-5-phosphate synthase | DXPS1 | A0A1V0QSH6 | Cannabis sativa | 730 | 2 | Converts d-glyceraldehyde 3P into 1-deoxy-d-xylulose 5P | Terpenoid biosynthesis | |
| 1-deoxy-D-xylulose-5-phosphate synthase | DXS2 | A0A1V0QSH5 | Cannabis sativa | 606 | 5 | Converts d-glyceraldehyde 3P into 1-deoxy-d-xylulose 5P | Terpenoid biosynthesis | |
| 4-hydroxy-3-methylbut-2-en-1-yl diphosphate synthase | HDS | A0A1V0QSG3 | Cannabis sativa | 748 | 3 | Converts (E)-4-hydroxy-3-methylbut-2-en-1-yl-2P into 2-C-methyl-d-erythritol 2,4-cyclo-2P | Terpenoid biosynthesis | |
| 3-hydroxy-3-methylglutaryl coenzyme A reductase | hmgR | A0A1V0QSF5 | Cannabis sativa | 588 | 5 | 1.1.1.34 | synthesizes (R)-mevalonate from acetyl-CoA | Terpenoid biosynthesis |
| 3-hydroxy-3-methylglutaryl coenzyme A reductase | hmgR | A0A1V0QSG7 | Cannabis sativa | 572 | 2 | 1.1.1.34 | synthesizes (R)-mevalonate from acetyl-CoA | Terpenoid biosynthesis |
| Terpene synthase | TPS | A0A1V0QSF2 | Cannabis sativa | 567 | 1 | formation of cyclic terpenes through the cyclisation of linear terpenes | Terpenoid biosynthesis | |
| Terpene synthase | TPS | A0A1V0QSF3 | Cannabis sativa | 551 | 3 | formation of cyclic terpenes through the cyclisation of linear terpenes | Terpenoid biosynthesis | |
| Terpene synthase | TPS | A0A1V0QSF4 | Cannabis sativa | 613 | 1 | formation of cyclic terpenes through the cyclisation of linear terpenes | Terpenoid biosynthesis | |
| Terpene synthase | TPS | A0A1V0QSF6 | Cannabis sativa | 551 | 1 | formation of cyclic terpenes through the cyclisation of linear terpenes | Terpenoid biosynthesis | |
| Terpene synthase | TPS | A0A1V0QSF8 | Cannabis sativa | 629 | 2 | formation of cyclic terpenes through the cyclisation of linear terpenes | Terpenoid biosynthesis | |
| Terpene synthase | TPS | A0A1V0QSF9 | Cannabis sativa | 624 | 2 | formation of cyclic terpenes through the cyclisation of linear terpenes | Terpenoid biosynthesis | |
| Terpene synthase | TPS | A0A1V0QSG0 | Cannabis sativa | 573 | 1 | formation of cyclic terpenes through the cyclisation of linear terpenes | Terpenoid biosynthesis | |
| Terpene synthase | TPS | A0A1V0QSG1 | Cannabis sativa | 640 | 1 | formation of cyclic terpenes through the cyclisation of linear terpenes | Terpenoid biosynthesis | |
| Terpene synthase | TPS | A0A1V0QSG6 | Cannabis sativa | 556 | 3 | formation of cyclic terpenes through the cyclisation of linear terpenes | Terpenoid biosynthesis | |
| Terpene synthase | TPS | A0A1V0QSH1 | Cannabis sativa | 594 | 1 | formation of cyclic terpenes through the cyclisation of linear terpenes | Terpenoid biosynthesis | |
| (-)-limonene synthase, chloroplastic | TPS1 | A7IZZ1 | Cannabis sativa | 622 | 2 | 4.2.3.16 | monoterpene (C10) olefins biosynthesis | Terpenoid biosynthesis |
| Maturase K | matK | A0A1V0IS32 | Cannabis sativa | 509 | 1 | assists in splicing its own and other chloroplast group II intron | Transcription | |
| Maturase K | matK | Q95BY0 | Cannabis sativa | 507 | 2 | assists in splicing its own and other chloroplast group II intron | Transcription | |
| Maturase R | matR | A0A0M5M254 | Cannabis sativa | 651 | 1 | assists in splicing introns | Transcription | |
| DNA-directed RNA polymerase subunit beta | rpoB | A0A0C5ARQ8 | Cannabis sativa | 1070 | 3 | 2.7.7.6 | transcription of DNA into RNA | Transcription |
| DNA-directed RNA polymerase subunit beta | rpoB | A0A0C5ARX9 | Cannabis sativa | 1393 | 4 | 2.7.7.6 | transcription of DNA into RNA | Transcription |
| DNA-directed RNA polymerase subunit beta | rpoB | A0A0U2H5U7 | Cannabis sativa | 1070 | 1 | 2.7.7.6 | transcription of DNA into RNA | Transcription |
| DNA-directed RNA polymerase subunit beta | rpoC1 | A0A0C5AUF5 | Cannabis sativa | 683 | 6 | 2.7.7.6 | transcription of DNA into RNA | Transcription |
| DNA-directed RNA polymerase subunit beta | rpoC2 | A0A0H3W6G1 | Cannabis sativa | 1389 | 1 | 2.7.7.6 | transcription of DNA into RNA | Transcription |
| DNA-directed RNA polymerase subunit beta | rpoC2 | A0A0X8GKF1 | Cannabis sativa | 1391 | 1 | 2.7.7.6 | transcription of DNA into RNA | Transcription |
| DNA-directed RNA polymerase subunit beta | rpoC2 | A0A1V0IS28 | Cannabis sativa | 1393 | 1 | 2.7.7.7 | transcription of DNA into RNA | Transcription |
| Ribosomal protein L14 | rpl14 | A0A0C5AS10 | Cannabis sativa | 122 | 2 | assembly of the ribosome | Protein metabolism | |
| 50S ribosomal protein L16, chloroplastic | rpl16 | A0A0C5AUJ2 | Cannabis sativa | 119 | 2 | assembly of the 50S ribosomal subunit | Protein metabolism | |
| Ribosomal protein L2 | rpl2 | A0A0M3ULW5 | Cannabis sativa | 337 | 2 | assembly of the ribosome | Protein metabolism | |
| 50S ribosomal protein L20 | rpl20 | A0A0C5B2J3 | Cannabis sativa | 120 | 1 | Binds directly to 23S rRNA to assemble the 50S ribosomal subunit | Protein metabolism | |
| Ribosomal protein S11 | rps11 | A0A0C5ART4 | Cannabis sativa | 138 | 1 | assembly of the ribosome | Protein metabolism | |
| 30S ribosomal protein S12, chloroplastic | rps12 | A0A0C5APY5 | Cannabis sativa | 132 | 1 | translational accuracy | Protein metabolism | |
| 30S ribosomal protein S12, chloroplastic | rps12 | A0A0C5B2L8 | Cannabis sativa | 125 | 1 | translational accuracy | Protein metabolism | |
| Ribosomal protein S13 | rps13 | A0A0M5M201 | Cannabis sativa | 116 | 1 | assembly of the ribosome | Protein metabolism | |
| Ribosomal protein S19 | rps19 | A0A0M3ULW7 | Cannabis sativa | 94 | 1 | assembly of the ribosome | Protein metabolism | |
| Ribosomal protein S2 | rps2 | A0A0C5APX8 | Cannabis sativa | 236 | 1 | assembly of the ribosome | Protein metabolism | |
| 30S ribosomal protein S3, chloroplastic | rps3 | A0A0C5ART6 | Cannabis sativa | 155 | 3 | assembly of the 30S ribosomal subunit | Protein metabolism | |
| Ribosomal protein S3 | rps3 | A0A0M3UM22 | Cannabis sativa | 548 | 1 | assembly of the ribosome | Protein metabolism | |
| Ribosomal protein S3 | rps3 | A0A110BC84 | Cannabis sativa | 548 | 1 | assembly of the ribosome | Protein metabolism | |
| Ribosomal protein S4 | rps4 | A0A0M4RG21 | Cannabis sativa | 352 | 1 | assembly of the ribosome | Protein metabolism | |
| Ribosomal protein S7 | rps7 | A0A0C5ARU3 | Cannabis sativa | 155 | 2 | assembly of the ribosome | Protein metabolism | |
| Ribosomal protein S7 | rps7 | A0A0M4R6T5 | Cannabis sativa | 148 | 1 | assembly of the ribosome | Protein metabolism | |
| Protein TIC 214 | ycf1 | A0A0C5AS14 | Cannabis sativa | 356 | 2 | protein precursor import into chloroplasts | Protein metabolism | |
| Protein TIC 214 | ycf1 | A0A0H3W815 | Cannabis sativa | 1878 | 21 | protein precursor import into chloroplasts | Protein metabolism | |
| Acyl-activating enzyme 1 | aae1 | H9A1V3 | Cannabis sativa | 720 | 1 | Unknown | ||
| Acyl-activating enzyme 10 | aae10 | H9A1W2 | Cannabis sativa | 564 | 1 | Unknown | ||
| Acyl-activating enzyme 12 | aae12 | H9A8L1 | Cannabis sativa | 757 | 2 | Unknown | ||
| Acyl-activating enzyme 13 | aae13 | H9A8L2 | Cannabis sativa | 715 | 3 | Unknown | ||
| Acyl-activating enzyme 2 | aae2 | H9A1V4 | Cannabis sativa | 662 | 3 | Unknown | ||
| Acyl-activating enzyme 3 | aae3 | H9A1V5 | Cannabis sativa | 543 | 7 | Unknown | ||
| Acyl-activating enzyme 4 | aae4 | H9A1V6 | Cannabis sativa | 723 | 3 | Unknown | ||
| Acyl-activating enzyme 5 | aae5 | H9A1V7 | Cannabis sativa | 575 | 1 | Unknown | ||
| Acyl-activating enzyme 6 | aae6 | H9A1V8 | Cannabis sativa | 569 | 1 | Unknown | ||
| Acyl-activating enzyme 8 | aae8 | H9A1W0 | Cannabis sativa | 526 | 3 | Unknown | ||
| Cannabidiolic acid synthase-like 2 | CBDAS3 | A6P6W1 | Cannabis sativa | 545 | 1 | Has no cannabidiolic acid synthase activity | Unknown | |
| Putative LOV domain-containing protein | LOV | A0A126WVX7 | Cannabis sativa | 664 | 8 | Unknown | ||
| Putative LOV domain-containing protein | LOV | A0A126WVX8 | Cannabis sativa | 1063 | 7 | Unknown | ||
| Putative LOV domain-containing protein | LOV | A0A126WZD3 | Cannabis sativa | 574 | 1 | Unknown | ||
| Putative LOV domain-containing protein | LOV | A0A126X0M1 | Cannabis sativa | 725 | 4 | Unknown | ||
| Putative LOV domain-containing protein | LOV | A0A126X1H2 | Cannabis sativa | 910 | 6 | Unknown | ||
| Putative LysM domain containing receptor kinase | lyk2 | U6EFF4 | Cannabis sativa | 599 | 1 | Unknown | ||
| Uncharacterised protein | unknown | A0A1V0IS79 | Cannabis sativa | 1525 | 2 | Unknown | ||
| Uncharacterised protein | unknown | L0N5C8 | Cannabis sativa | 543 | 1 | Unknown | ||
| Protein Ycf2 | ycf2 | A0A0C5APZ4 | Cannabis sativa | 2302 | 9 | ATPase of unknown function | Unknown | |
| Protein translocase subunit | secA | A0A0N9ZJA6 | Cannabis sativa | 158 | 7 | Binds ATP | Protein metabolism | |
| ATP synthase subunit beta, chloroplastic | atpB | A0A0U2DTF2 | Cannabis sativa subsp. sativa | 498 | 20 | 3.6.3.14 | Produces ATP from ADP | Energy metabolism |
| Acetyl-coenzyme A carboxylase carboxyl transferase subunit beta, chloroplastic | accD | A0A0U2DTG7 | Cannabis sativa subsp. sativa | 497 | 3 | 2.1.3.15 | acetyl coenzyme A carboxylase complex | Lipid biosynthesis |
| NAD(P)H-quinone oxidoreductase subunit K, chloroplastic | ndhK | A0A0U2DTF9 | Cannabis sativa subsp. sativa | 226 | 1 | 1.6.5.- | NDH shuttles electrons from NAD(P)H:plastoquinone to quinones | Photosynthesis |
| Cytochrome f | petA | A0A0U2DW83 | Cannabis sativa subsp. sativa | 320 | 1 | mediates electron transfer between PSII and PSI | Photosynthesis | |
| Photosystem II protein D1 | psbA | A0A0U2DTE4 | Cannabis sativa subsp. sativa | 353 | 2 | 1.10.3.9 | assembly of the PSII complex | Photosynthesis |
| Photosystem II CP43 reaction center protein | psbC | A0A0U2DTE2 | Cannabis sativa subsp. sativa | 473 | 5 | core complex of PSII | Photosynthesis | |
| Photosystem II D2 protein | psbD | A0A0U2DVP6 | Cannabis sativa subsp. sativa | 353 | 3 | 1.10.3.9 | assembly of the PSII complex | Photosynthesis |
| Cytochrome b559 subunit alpha | psbE | A0A0U2DTH9 | Cannabis sativa subsp. sativa | 83 | 2 | reaction center of PSII | Photosynthesis | |
| Ribulose bisphosphate carboxylase large chain | rbcL | A0A0U2DW50 | Cannabis sativa subsp. sativa | 475 | 13 | 4.1.1.39 | carboxylation of D-ribulose 1,5-bisphosphate | Photosynthesis |
| Photosystem I assembly protein Ycf4 | ycf4 | A0A0U2DVM4 | Cannabis sativa subsp. sativa | 184 | 1 | assembly of the PSI complex | Photosynthesis | |
| 30S ribosomal protein S14, chloroplastic | rps14 | A0A0U2DTI4 | Cannabis sativa subsp. sativa | 100 | 2 | Binds 16S rRNA, required for the assembly of 30S particles | Protein metabolism | |
| 30S ribosomal protein S15, chloroplastic | rps15 | A0A0U2DW79 | Cannabis sativa subsp. sativa | 90 | 1 | assembly of the 30S ribosomal subunit | Protein metabolism | |
| ATP synthase subunit beta, chloroplastic | atpB | A0A0U2H0U7 | Humulus lupulus | 498 | 2 | 3.6.3.14 | Produces ATP from ADP | Energy metabolism |
| ATP synthase subunit beta, chloroplastic | atpB | A0A0U2H587 | Humulus lupulus | 191 | 1 | Component of the F(0) channel | Energy metabolism | |
| NAD(P)H-quinone oxidoreductase subunit I, chloroplastic | ndhI | A0A0U2GY49 | Humulus lupulus | 171 | 2 | 1.6.5.- | NDH shuttles electrons from NAD(P)H:plastoquinone to quinones | Photosynthesis |
| DNA-directed RNA polymerase subunit beta | rpoC2 | A0A0U2H146 | Humulus lupulus | 1398 | 1 | 2.7.7.6 | transcription of DNA into RNA | Transcription |
| 50S ribosomal protein L20, chloroplastic | rpl20 | A0A0U2H0V8 | Humulus lupulus | 120 | 1 | Binds directly to 23S rRNA to assemble the 50S ribosomal subunit | Protein metabolism | |
| 30S ribosomal protein S4, chloroplastic | rps4 | A0A0U2H5A0 | Humulus lupulus | 202 | 1 | binds directly to 16S rRNA to assemble the 30S subunit | Protein metabolism | |
| 30S ribosomal protein S8, chloroplastic | rps8 | A0A0U2GZU5 | Humulus lupulus | 134 | 2 | binds directly to 16S rRNA to assemble the 30S subunit | Protein metabolism | |
| Protein Ycf2 | ycf2 | A0A0U2H6B6 | Humulus lupulus | 2287 | 1 | ATPase of unknown function | Unknown |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vincent, D.; Rochfort, S.; Spangenberg, G. Optimisation of Protein Extraction from Medicinal Cannabis Mature Buds for Bottom-Up Proteomics. Molecules 2019, 24, 659. https://doi.org/10.3390/molecules24040659
Vincent D, Rochfort S, Spangenberg G. Optimisation of Protein Extraction from Medicinal Cannabis Mature Buds for Bottom-Up Proteomics. Molecules. 2019; 24(4):659. https://doi.org/10.3390/molecules24040659
Chicago/Turabian StyleVincent, Delphine, Simone Rochfort, and German Spangenberg. 2019. "Optimisation of Protein Extraction from Medicinal Cannabis Mature Buds for Bottom-Up Proteomics" Molecules 24, no. 4: 659. https://doi.org/10.3390/molecules24040659
APA StyleVincent, D., Rochfort, S., & Spangenberg, G. (2019). Optimisation of Protein Extraction from Medicinal Cannabis Mature Buds for Bottom-Up Proteomics. Molecules, 24(4), 659. https://doi.org/10.3390/molecules24040659