
Analysis of 27 β-Blockers and Metabolites in Milk Powder by High Performance Liquid Chromatography Coupled to Quadrupole Orbitrap High-Resolution Mass Spectrometry

Abstract
:1. Introduction
2. Results and Discussion
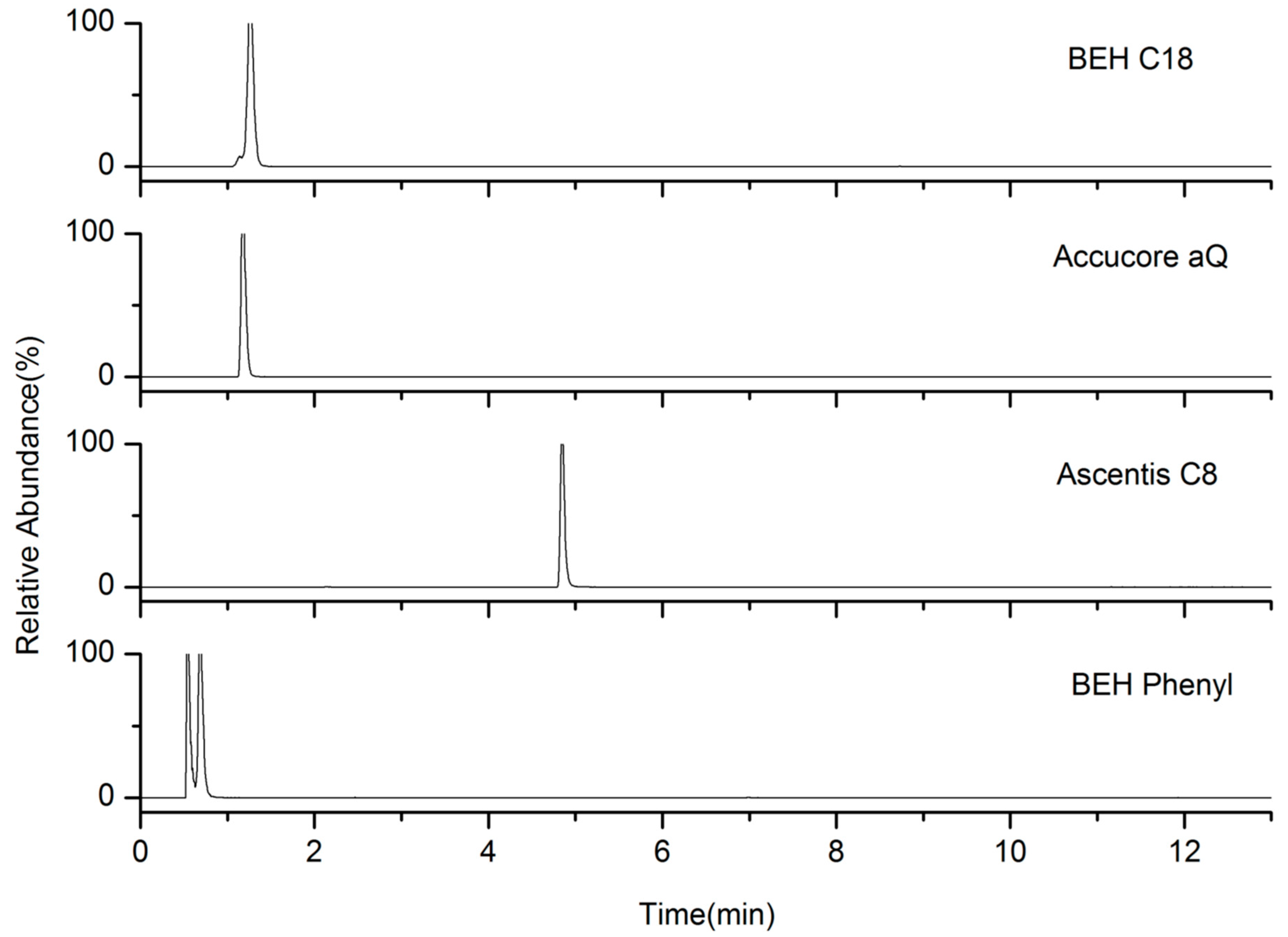
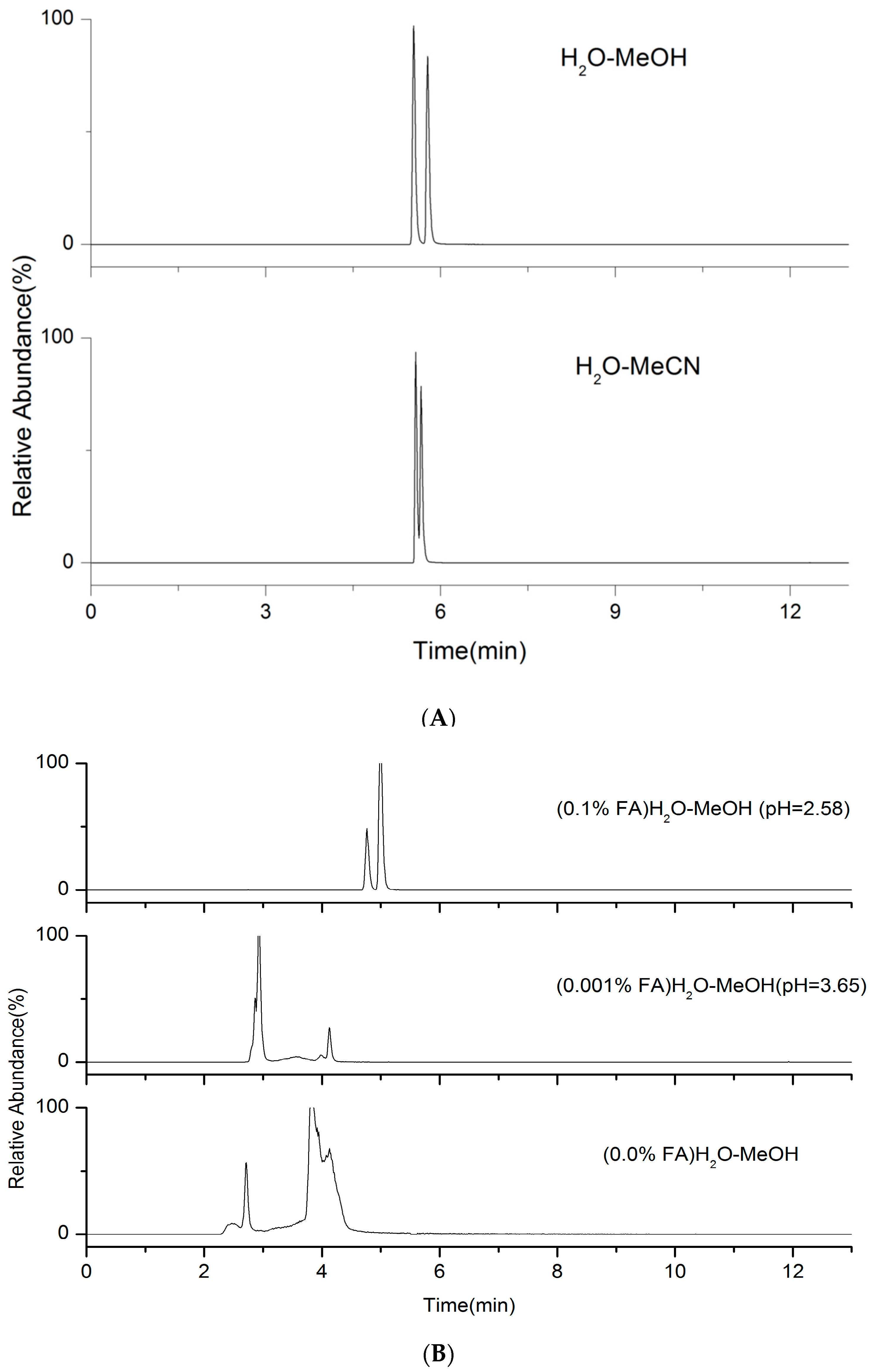
2.1. LC Parameters Optimization
2.2. Optimization of the Mass Spectrometric Parameters
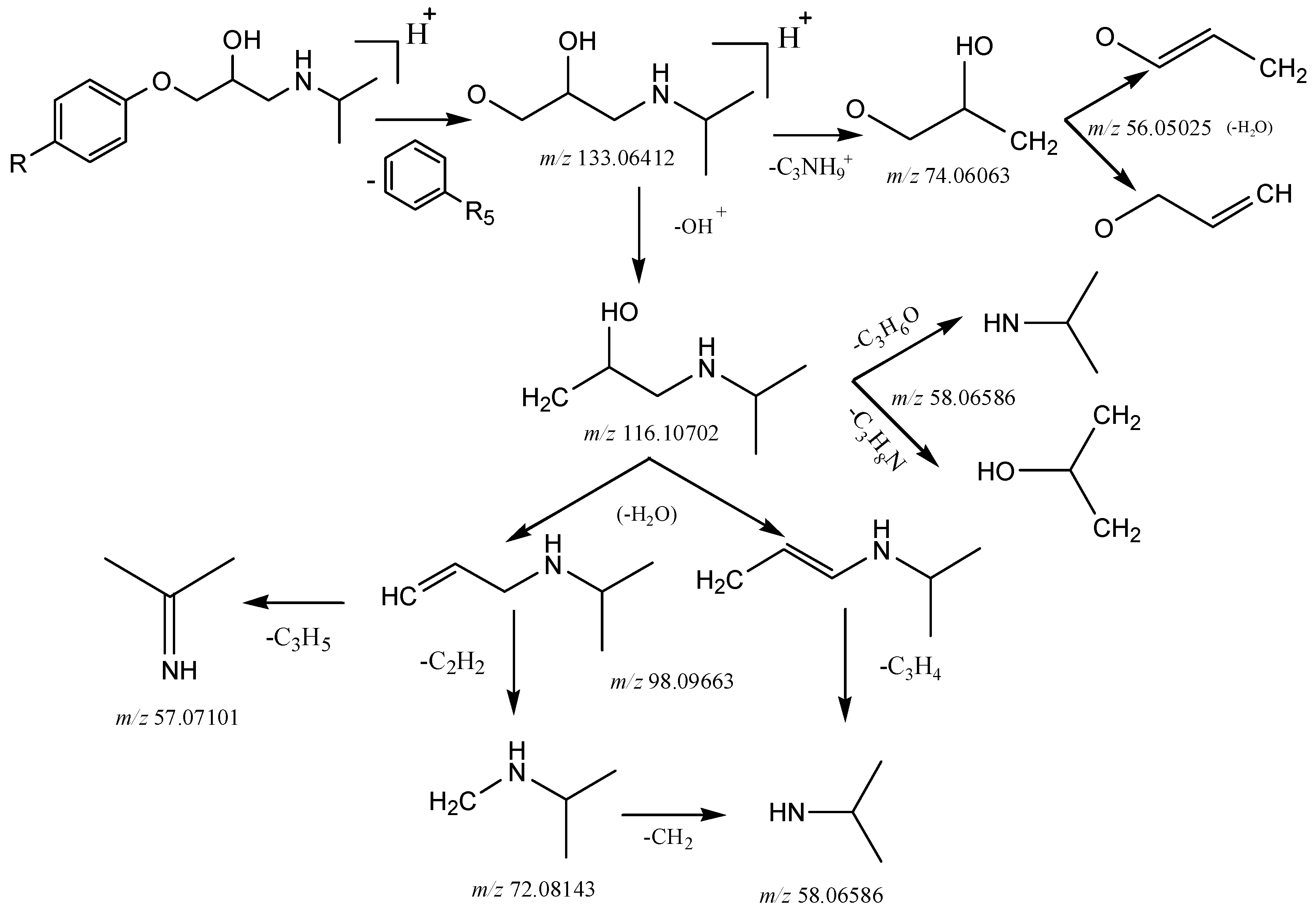
2.3. The Proposed Fragmentation Pathways for 27 β-Blockers
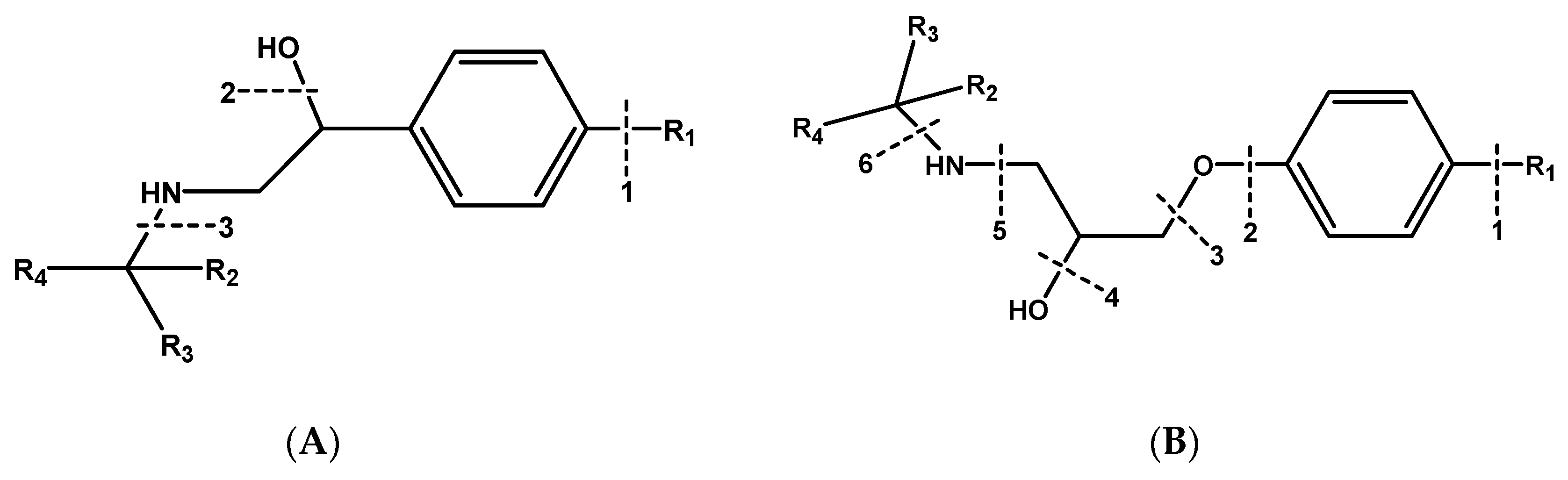
2.3.1. Phenylethanolamines Structure
2.3.2. Aryloxypropanolamines Structure
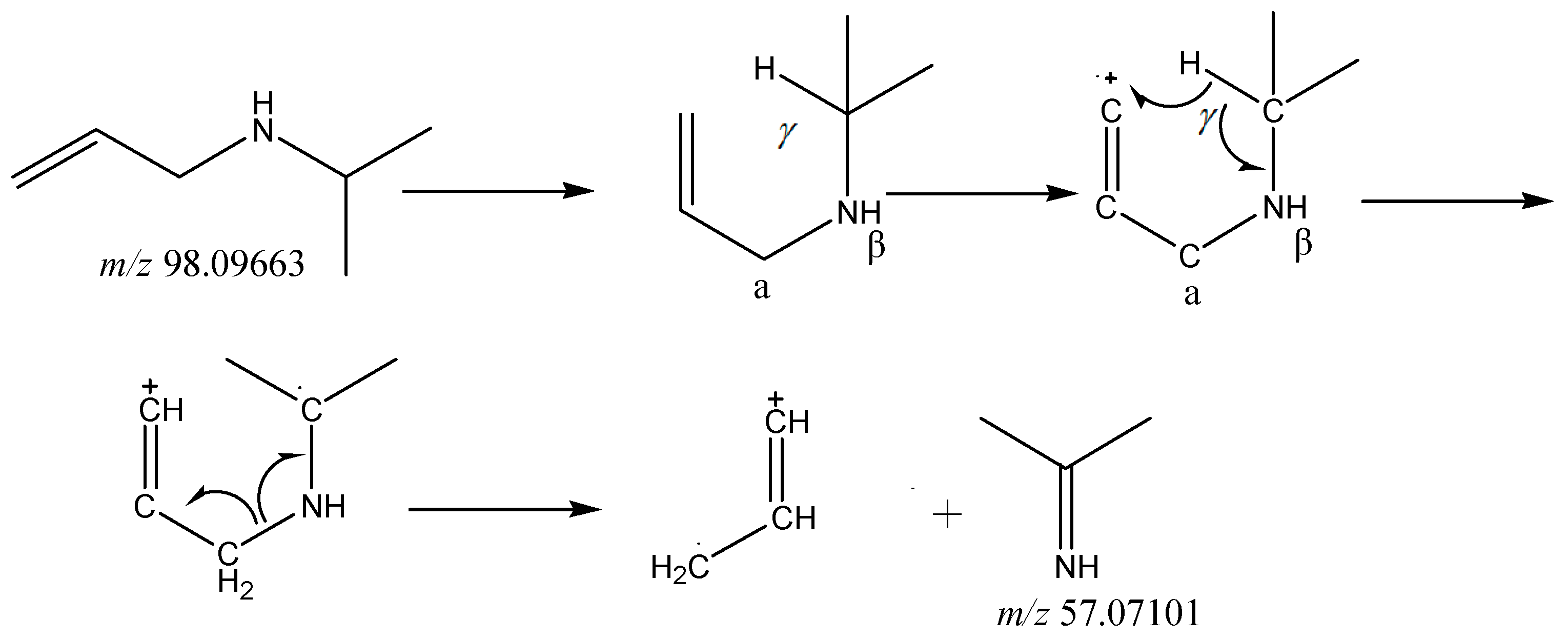
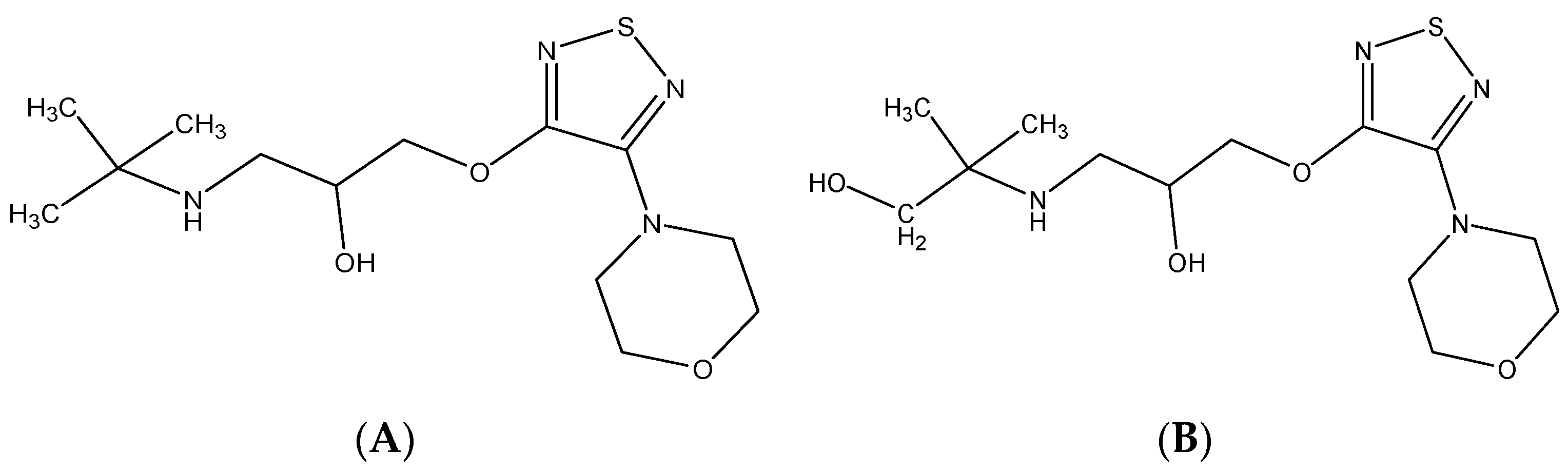
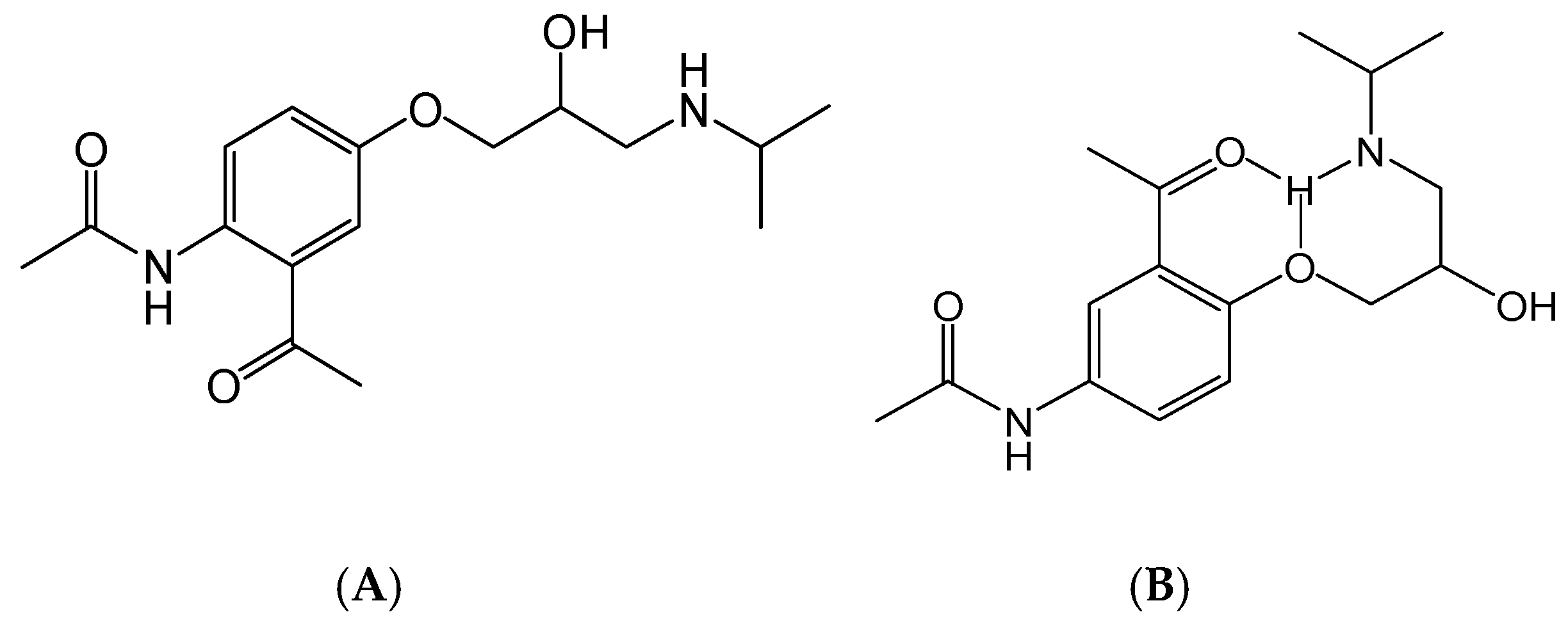
2.3.3. Special Structures
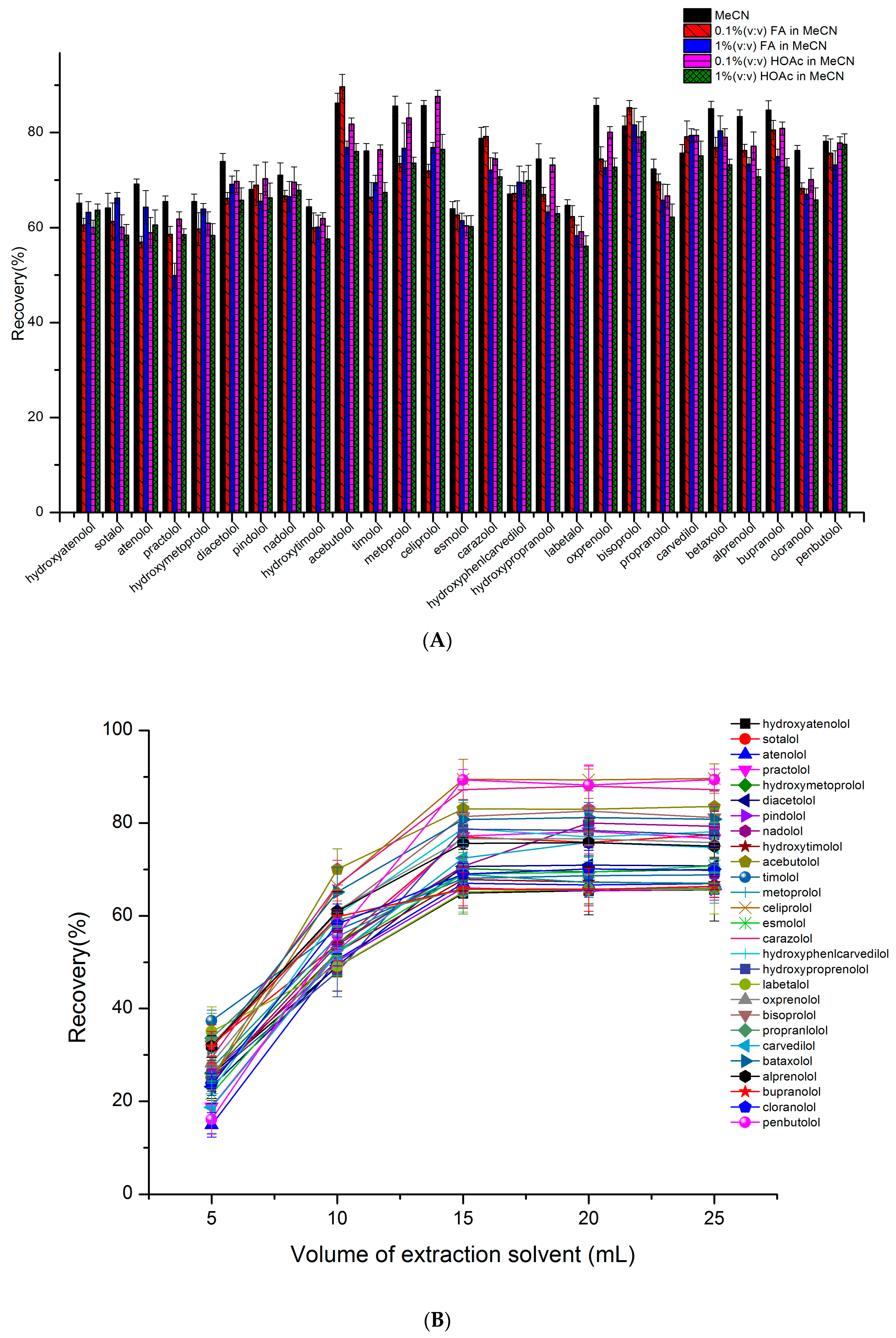
2.4. The Optimization of the Sample Preparation Procedure
2.5. Validation of the Proposed Method
2.5.1. Linearity and Sensitivity
2.5.2. Matrix effect
2.5.3. Trueness and Precision
2.6. Real Samples Analysis
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Instrument and Analytical Conditions
3.3. Sample Preparation
3.4. Method Validation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Xu, H.; Zhang, H.W.; Wang, F.M. Determination of 9 β-blockers residues in dairy products by liquid chromatography-tandem mass spectrometry. J. Food Safety Qual. 2014, 12, 3884–3890. [Google Scholar]
- Zhang, J.; Shao, B.; Yin, J. Simultaneous detection of residues of β-adrenergic receptor blockers and sedatives in animal tissues by high-performance liquid chromatography/tandem mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 1915–1922. [Google Scholar] [CrossRef] [PubMed]
- Mitrowska, K.; Posyniak, A.; Zmudzki, J. Rapid method for the determination of tranquilizers and a β-blocker in porcine and bovine kidney by liquid chromatography with tandem mass spectrometry. Anal. Chim. Acta 2009, 637, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Jiang, J.; Shao, R.T.; Ding, X.Y.; Shi, N.; Lu, Y. Determination of Eleven β-Blocker residues in Animal Derived Foods by Ultra High Performance Liquid Chromatography-Tandem Mass Spectrometry with Molecularly Imprinted Solid Phase Extraction. J. Instrum. Anal. 2016, 10, 1278–1282. [Google Scholar]
- Gehr, T.W.B.; Tenero, D.M.; Boyle, D.A.; Qian, Y.; Sica, D.A.; Shusterman, N.H. The pharmacokinetics of carvedilol and its metabolites after single and multiple dose oral administration in patients with hypertension and renal insufficiency. Eur. J. Clin. Pharmaco. 1999, 55, 269–277. [Google Scholar] [CrossRef]
- The Council of the European Communities. Council Regulation 2377/90/EEC. Off. J. Eur. Commun. 1990, 224, 1. [Google Scholar]
- Cooper, J.; Delahaut, P.; Fodey, T.L. Development of a rapid screening test for veterinary sedatives and the β-blocker carazolol in porcine kidney by ELISA. Analyst 2004, 129, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Delbeke, F.T.; Debackere, M.; Desme, T.N. Comparative study of extraction methods for the GC and GC-MS screening of urine for β-blocker abuse. J. Pharmaceu. Biomed. Anal. 1988, 6, 827–835. [Google Scholar] [CrossRef]
- Ternes, T.A.; Hirsch, R.; Mueller, J. Methods for the determination of neutral drugs as well as β-blockers and β2-sympathomimetics in aqueous matrices using GC/MS and LC/MS/MS. Anal. Bioanal. Chem. 1998, 362, 329–340. [Google Scholar]
- Magiera, S.; Uhlschmied, C.; Rainer, M. GC–MS method for the simultaneous determination of β-blockers, flavonoids, isoflavones and their metabolites in human urine. J. Pharmaceu. Biomed. Anal. 2011, 56, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Amendola, L.; Molaioni, F.; Botrè, F. Detection of β-blockers in human urine by GC-MS-MS-EI: Perspectives for the antidoping control. J. Pharmaceu. Biomed. Anal. 2000, 23, 211–221. [Google Scholar] [CrossRef]
- Delamoye, M.; Duverneuil, C.; Paraire, F. Simultaneous determination of thirteen β-blockers and one metabolite by gradient high-performance liquid chromatography with photodiode-array UV detection. Forensic Sci. Int. 2004, 141, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Zuppa, A.F.; Shi, H.; Adamson, P.C. Adamson, Liquid chromatography-electrospray mass spectrometry (LC-MS) method for determination of esmolol concentration in human plasma. J. Chromatogr. B 2003, 796, 293–301. [Google Scholar] [CrossRef]
- Delahaut, P.; Levaux, C.; Eloy, P. Validation of a method for detecting and quantifying tranquillisers and a β-blocker in pig tissues by liquid chromatography–tandem mass spectrometry. Anal. Chim. Acta 2003, 483, 335–340. [Google Scholar] [CrossRef]
- Lee, H.B.; Sarafin, K.; Peart, T.E. Determination of β-blockers and β2-agonists in sewage by solid-phase extraction and liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2007, 1148, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Umezawa, H.; Lee, X.P.; Arima, Y. Simultaneous determination of β-blockers in human plasma using liquid chromatography–tandem mass spectrometry. Biomed. Chromatogr. 2008, 22, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Farré, M.; Gros, M.; Hernández, B. Analysis of biologically active compounds in water by ultra-performance liquid chromatography quadrupole time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2008, 22, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Nie, X.M.; Wu, H.Q. A high-throughput screening method of bisphenols, bisphenols digycidyl ethers and their derivatives in dairy products by ultra-high performance liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta 2017, 950, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Lamshöft, M.; Zühlke, S. Determination of sedatives and adrenergic blockers in blood meal using accelerated solvent extraction and Orbitrap mass spectrometry. J. Chromatogr. A 2012, 1260, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shen, B.; Jiang, Z.; Huang, Y.; Zhuo, X. Rapid screening of drugs of abuse in human urine by high-performance liquid chromatography coupled with high resolution and high mass accuracy hybrid linear ion trap-Orbitrap mass spectrometry. J. Chromatogr. A 2013, 1302, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Shi, F.; Gong, L. Ultra-trace analysis of 12 β-agonists in pork, beef, mutton and chicken by ultrahigh-performance liquid-chromatography-quadrupole-orbitrap tandem mass spectrometry. J. Pharmaceu. Biomed. Anal. 2015, 107, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.H.; Xu, X.L.; Li, W.Q. A high-accuracy screening method of 44 cephalosporins in meat using liquid chromatography quadrupole-orbitrap hybrid mass spectrometry. Anal. Methods 2017, 10. [Google Scholar] [CrossRef]
- Wang, X.J.; Zhang, F.; Li, W.Q. Simultaneous determination of 12 β-agonists in feeds by ultra-high-performance liquid chromatography-quadrupole-time-of-flight mass spectrometry. J. Chromatogr. A 2013, 1278, 82–88. [Google Scholar]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC–MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- Cappiello, A.; Famiglini, G.; Palma, P.; Pierini, E.; Termopoli, V.; Trufelli, Y. Overcoming matrix effects in liquid chromatography–mass spectrometry. Anal. Chem. 2008, 80, 9343–9348. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Formula | Theoretical Mass [m/z] | Measured Mass [m/z] | Accuracy α [ppm] | MS2 | Structure Type |
|---|---|---|---|---|---|---|
| Carazolol | C18H22N2O2 | 299.17540 | 299.17484 | 1.87 | 222.09090 116.10712 98.09675 72.08148 56.05032 | Type I |
| Oxprenolol | C15H23NO3 | 266.17507 | 266.17496 | 0.41 | 133.06451 116.10696 98.09662 72.08138 56.05027 | Type I |
| Propranolol | C16H21NO2 | 260.16451 | 260.16373 | 3.00 | 183.07979 116.10689 98.09659 72.08135 58.06586 | Type I |
| Alprenolol | C15H23NO2 | 250.18016 | 250.17953 | 2.52 | 173.09550 116.10687 98.09660 72.08134 56.05024 | Type I |
| Bisoprolol | C18H31NO4 | 326.23258 | 326.23169 | 3.00 | 133.06441 116.10689 98.09659 74.06059 56.05026 | Type I |
| Betaxolol | C18H29NO3 | 308.22202 | 308.22174 | 0.91 | 133.06445 116.10691 98.09661 72.08135 56.05004 | Type I |
| Sotalol | C12H20N2O3S | 273.12674 | 273.12680 | 0.46 | 255.11484 213.06822 176.12991 133.07552 198.05713 | phenylethanolamine |
| Pindolol | C14H20N2O2 | 249.15975 | 249.15961 | 0.56 | 172.07532 116.10711 98.09679 72.08147 58.06597 | Type I |
| Nadolol | C17H27NO4 | 310.20128 | 310.20084 | 1.42 | 354.13795 236.12750 201.09059 74.06068 56.05030 | Type II |
| Timolol | C13H24N4O3S | 317.16419 | 317.16367 | 1.64 | 261.10089 244.07440 188.04840 74.06068 57.07074 | Special structure |
| Acebutolol | C18H28N2O4 | 337.21218 | 337.21310 | 2.73 | 218.11726 116.10712 98.09680 72.08150 56.05036 | Type I |
| Celiprolol | C20H33N3O4 | 380.25438 | 380.25299 | 3.67 | 324.19070 307.16397 251.10155 74.06061 56.05026 | Type II |
| Labetalol | C19H24N2O3 | 329.18597 | 329.18613 | 0.49 | 311.17395 294.14755 207.11201 179.08063 162.05423 | phenylethanolamine |
| Cloranolol | C13H19Cl2NO2 | 292.08656 | 292.08658 | 0.07 | 236.02318 218.01273 174.97054 74.06063 56.05020 | Type II |
| Penbutolol | C18H29NO2 | 292.22711 | 292.22672 | 1.33 | 236.16374 201.12683 133.06451 74.06063 57.07070 | Type II |
| Practolol | C14H22N2O3 | 267.17032 | 267.16965 | 2.51 | 190.08589 116.10711 98.09682 72.08146 56.05036 | Type I |
| Carvedilol | C24H26N2O4 | 407.19653 | 407.19565 | 2.16 | 283.14340 224.12755 100.07599 74.06063 56.05036 | Type III |
| Bupranolol | C14H22ClNO2 | 272.14118 | 272.14020 | 3.60 | 216.07790 198.06741 181.04089 74.06061 56.05027 | Type II |
| Atenolol | C14H22N2O3 | 267.17032 | 267.16983 | 1.83 | 133.06412 116.10690 98.09663 74.06060 56.05026 | Type I |
| Esmolol | C16H25NO4 | 296.18563 | 296.18558 | 0.17 | 133.06467 116.10737 98.09705 72.08168 56.05050 | Type I |
| Metoprolol | C15H25NO3 | 268.19072 | 268.19028 | 1.64 | 133.06435 116.10693 98.09660 74.06057 56.05026 | Type I |
| Diacetolol | C16H24N2O4 | 308.18088 | 308.18088 | 0.00 | 291.16943 116.10702 98.09670 72.08143 56.05031 | Type I |
| α-hydroxymetoprolol | C15H25NO4 | 284.18563 | 284.18472 | 3.20 | 133.06435 116.10691 98.09663 74.06059 56.05026 | Type I |
| α-hydroxyatenolol | C14H22N2O4 | 283.16523 | 283.16507 | 0.57 | 133.08632 116.10760 89.06059 74.06103 57.07010 | Type I |
| (S)-Hydroxytimolol | C13H24N4O4S | 333.15910 | 333.15823 | 2.61 | 261.10059 188.04814 146.11705 74.06059 56.05025 | Special structure |
| 7-Hydroxyproprenolol | C16H21NO3 | 276.15942 | 276.15930 | 0.43 | 199.07463 116.1067 98.09663 74.06057 58.06586 | Type I |
| 4-Hydroxyphenylcarvedilol | C24H26N2O5 | 423.19145 | 423.19141 | 0.09 | 283.14267 240.12180 100.07578 74.06049 56.05022 | Type III |
| No. | m/z | The Molecular Formula | The Possible Structure |
|---|---|---|---|
| 1 | 56.05025 | C3H4O |  or or  |
| 2 | 57.07101 | C3H7N |  |
| 3 | 58.06586 | C3H6O or C3H8N |  or or  |
| 4 | 72.08143 | C4H10N |  |
| 5 | 74.06063 | C3H6O2 |  |
| 6 | 98.09663 | C6H12N |  or or  |
| 7 | 116.10702 | C6H14NO |  |
| 8 | 133.06412 | C6H15NO2+ |  |
| NO. | Analyte | Matrix Effect C (%) | QC Concentration (μg kg−1) | Average Recovery (%) | Intra-Day Precision (%) (n = 5) | Inter-Day Precision (%) (n = 5) | |
|---|---|---|---|---|---|---|---|
| PRiME HLB | Centrifugation | ||||||
| 1 | Atenolol | 115.7 | 118.5 | 2 | 72.5 | 2 | 3.1 |
| 4 | 76.3 | 3.1 | 2.2 | ||||
| 8 | 74.8 | 4.1 | 5 | ||||
| 2 | Sotalol | 85.6 | 86.8 | 2 | 83.6 | 3.2 | 3.2 |
| 4 | 87 | 5.6 | 1.9 | ||||
| 8 | 81.1 | 2.7 | 7.9 | ||||
| 3 | Pindolol | 101.7 | 108.2 | 1 | 89.2 | 1.6 | 5.2 |
| 2 | 100.4 | 4.8 | 2.7 | ||||
| 4 | 83.7 | 3.6 | 6.4 | ||||
| 4 | Nadolol | 102.2 | 112.8 | 0.5 | 83.6 | 7.9 | 5.5 |
| 1 | 93.2 | 2.1 | 3.1 | ||||
| 2 | 78.5 | 4.4 | 5.3 | ||||
| 5 | Metoprolol | 120.9 | 140 | 1 | 80.4 | 3.9 | 5 |
| 2 | 90 | 6.2 | 3.5 | ||||
| 4 | 84.2 | 2.1 | 7.7 | ||||
| 6 | Timolol | 116.9 | 133.7 | 1 | 76.5 | 2.1 | 7.4 |
| 2 | 83.8 | 5.2 | 3.9 | ||||
| 4 | 78.8 | 3.8 | 3.2 | ||||
| 7 | Acebutolol | 129.2 | 155.3 | 0.5 | 95.6 | 2.2 | 3.2 |
| 1 | 89.3 | 5.7 | 5 | ||||
| 2 | 92.1 | 2.4 | 7.5 | ||||
| 8 | Oxprenolol | 109.9 | 123.3 | 1 | 69.6 | 1.9 | 4.4 |
| 2 | 89.7 | 6.7 | 7.5 | ||||
| 4 | 84.8 | 3.3 | 3.9 | ||||
| 9 | Celiprolol | 165.1 | 181.5 | 1.5 | 98.5 | 7.3 | 5.6 |
| 3 | 87.4 | 3.5 | 5.4 | ||||
| 6 | 93.3 | 2.1 | 7.7 | ||||
| 10 | Bisoprolol | 134.6 | 156.3 | 0.5 | 93.8 | 7.1 | 7.2 |
| 1 | 90.4 | 4.4 | 3.6 | ||||
| 2 | 84.6 | 2.5 | 5.6 | ||||
| 11 | Labetalol | 91.2 | 102.1 | 0.5 | 91.7 | 2.1 | 2.8 |
| 1 | 86.6 | 7.5 | 6.4 | ||||
| 2 | 83.4 | 3.3 | 6.5 | ||||
| 12 | Alprenolol | 102.1 | 117 | 0.5 | 74.8 | 5.5 | 4.2 |
| 1 | 81.3 | 7.2 | 8.9 | ||||
| 2 | 81.1 | 4.1 | 5.4 | ||||
| 13 | Propranolol | 98.5 | 120.7 | 0.5 | 80.2 | 8.7 | 2.3 |
| 1 | 83.5 | 3.5 | 1.8 | ||||
| 2 | 80.4 | 6.4 | 5.6 | ||||
| 14 | Betaxolol | 117.6 | 146.5 | 2 | 79.8 | 7.1 | 4.5 |
| 4 | 91.5 | 2.7 | 3.7 | ||||
| 8 | 85.2 | 5.6 | 2.5 | ||||
| 15 | Cloranolol | 109.2 | 126.1 | 2 | 72.1 | 8.1 | 3.5 |
| 4 | 75.5 | 3.5 | 5.4 | ||||
| 8 | 76.6 | 3.1 | 7.2 | ||||
| 16 | Penbutolol | 109.4 | 134.3 | 1 | 85.5 | 3.4 | 6.5 |
| 2 | 97.6 | 2.6 | 4.4 | ||||
| 4 | 76.4 | 1.7 | 2.5 | ||||
| 17 | Practolol | 115.7 | 120.6 | 0.5 | 71.9 | 4.4 | 2.6 |
| 1 | 73.6 | 3.4 | 7.5 | ||||
| 2 | 75.2 | 5 | 5.8 | ||||
| 18 | Carazolol | 80.1 | 96.1 | 0.5 | 99.3 | 3.1 | 2.8 |
| 1 | 85.3 | 4 | 5.7 | ||||
| 2 | 85.9 | 7.9 | 4.9 | ||||
| 19 | Carvedilol | 82.7 | 105.6 | 2 | 79.5 | 5.7 | 2.5 |
| 4 | 78 | 2.5 | 7.5 | ||||
| 8 | 84.6 | 3.4 | 5.6 | ||||
| 20 | Esmolol | 101.8 | 106.9 | 3 | 72.5 | 5.1 | 3.6 |
| 6 | 83.2 | 8.1 | 2.3 | ||||
| 12 | 73.4 | 4.3 | 7.5 | ||||
| 21 | Bupranolol | 112.6 | 122.9 | 0.5 | 73.4 | 3.2 | 5.9 |
| 1 | 79.4 | 5.2 | 3.5 | ||||
| 2 | 80 | 1.7 | 5.4 | ||||
| 22 | Diacetolol | 134 | 152.2 | 1 | 81.7 | 3.8 | 3.5 |
| 2 | 87.7 | 2.3 | 5.9 | ||||
| 4 | 82.9 | 6.9 | 2.5 | ||||
| 23 | α-Hydroxymetoprolol | 107.5 | 113.8 | 1 | 85.8 | 2.4 | 7.1 |
| 2 | 89.8 | 4.7 | 4.6 | ||||
| 4 | 84.5 | 5.6 | 5.6 | ||||
| 24 | α-Hydroxyatenolol | 80.1 | 72.8 | 5 | 67.7 | 4.3 | 3.2 |
| 10 | 66.1 | 3.4 | 5.4 | ||||
| 20 | 68.6 | 7.7 | 6.9 | ||||
| 25 | (s)-Hydroxytimolol | 93.4 | 103.6 | 1 | 78.5 | 2.2 | 5.8 |
| 2 | 91.3 | 5.5 | 5.7 | ||||
| 4 | 85.7 | 6.3 | 4.6 | ||||
| 26 | 7-Hydroxypropranolol | 84.7 | 99.9 | 1 | 69.8 | 5.6 | 3.5 |
| 2 | 73.6 | 2.2 | 5.3 | ||||
| 4 | 78.8 | 3.2 | 7.2 | ||||
| 27 | 4-Hydroxyphenlcarvedilol | 85.6 | 103.1 | 2 | 73.5 | 3.7 | 1.8 |
| 4 | 66 | 7 | 4.5 | ||||
| 8 | 67.4 | 6.3 | 6.9 | ||||
| Analytes | Linear Equation | Linear Range (μg kg−1) | Correlation Coefficient (r2) | LOD (μg kg−1) | LOQ (μg kg−1) |
|---|---|---|---|---|---|
| Atenolol | Y = −0.0196094 + 0.0399798X | 2–200 | 0.9994 | 0.6 | 2 |
| Sotalol | Y = −0.0302291 + 0.0364729X | 2–200 | 0.9995 | 0.6 | 2 |
| Pindolol | Y = 0.374297 + 0.109209X | 1–200 | 0.9967 | 0.3 | 1 |
| Nadolol | Y = 0.00696148 + 0.0330124X | 0.5–50 | 0.9987 | 0.2 | 0.5 |
| Metoprolol | Y = 0.250919 + 0.0925317X | 0.5–50 | 0.9975 | 0.3 | 1 |
| Timolol | Y = −0.0667935 + 0.0862132X | 1–100 | 0.9997 | 0.3 | 1 |
| Acebutolol | Y = 0.0484461 + 0.0478361X | 0.5–50 | 0.9977 | 0.2 | 0.5 |
| Oxprenolol | Y = −0.157711 + 0.0272438X | 1–100 | 0.9990 | 0.3 | 1 |
| Celiprolol | Y = −0.00691133 + 0.068192X | 2–200 | 0.9966 | 0.5 | 1.5 |
| Bisoprolol | Y = 0.130461 + 0.111734X | 0.5–50 | 0.9969 | 0.2 | 0.5 |
| Labetalol | Y = −0.0396352 + 0.0389041X | 0.5–50 | 0.9997 | 0.2 | 0.5 |
| Alprenolol | Y = −0.164222 + 0.588251X | 0.5–50 | 0.9997 | 0.2 | 0.5 |
| Propranolol | Y = 0.0870785 + 0.148381X | 0.5–50 | 0.9996 | 0.2 | 0.5 |
| Betaxolol | Y = 0.132601 + 0.130952X | 2–200 | 0.9987 | 0.6 | 2 |
| Cloranolol | Y = 0.214191 + 0.103506X | 2–200 | 0.9991 | 0.6 | 2 |
| Penbutolol | Y = −0.0422939 + 0.063028X | 1–100 | 0.9989 | 0.3 | 1 |
| Practolol | Y = −0.0153134 + 0.114317X | 0.5–50 | 0.9990 | 0.2 | 0.5 |
| Carazolol | Y = −0.015998 + 0.0807354X | 0.5–50 | 0.9998 | 0.2 | 0.5 |
| Carvedilol | Y = −0.00662109 + 0.0671112X | 2–200 | 0.9998 | 0.6 | 2 |
| Esmolol | Y = −0.142646 + 0.159519X | 5–500 | 0.9994 | 1 | 3 |
| Bupranolol | Y = −0.126666 + 0.325598X | 0.5–50 | 0.9995 | 0.3 | 0.5 |
| Diacetolol | Y = 0.204209 + 0.0797893X | 1–100 | 0.9973 | 0.3 | 1 |
| α-Hydroxymetoprolol | Y = 0.0134667 + 0.105121X | 1–100 | 0.9992 | 0.3 | 1 |
| α-Hydroxyatenolol | Y = −0.0738747 + 0.0396782X | 5–500 | 0.9993 | 1.5 | 5 |
| (S)-Hydroxytimolol | Y = 0.168441 + 0.119248X | 1–100 | 0.9989 | 0.3 | 1 |
| 7-Hydroxypropranolol | Y = 0.0104 + 0.167864X | 1–100 | 0.9999 | 0.3 | 1 |
| 4-Hydroxyphenlcarvedilol | Y = 0.0993695 + 0.159881X | 2–200 | 0.9991 | 0.6 | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, J.-Q.; Liu, T.; Nie, X.-M.; Chen, F.-M.; Wang, C.-S.; Zhang, F. Analysis of 27 β-Blockers and Metabolites in Milk Powder by High Performance Liquid Chromatography Coupled to Quadrupole Orbitrap High-Resolution Mass Spectrometry. Molecules 2019, 24, 820. https://doi.org/10.3390/molecules24040820
Cheng J-Q, Liu T, Nie X-M, Chen F-M, Wang C-S, Zhang F. Analysis of 27 β-Blockers and Metabolites in Milk Powder by High Performance Liquid Chromatography Coupled to Quadrupole Orbitrap High-Resolution Mass Spectrometry. Molecules. 2019; 24(4):820. https://doi.org/10.3390/molecules24040820
Chicago/Turabian StyleCheng, Jian-Qiao, Tong Liu, Xue-Mei Nie, Feng-Ming Chen, Chuan-Sheng Wang, and Feng Zhang. 2019. "Analysis of 27 β-Blockers and Metabolites in Milk Powder by High Performance Liquid Chromatography Coupled to Quadrupole Orbitrap High-Resolution Mass Spectrometry" Molecules 24, no. 4: 820. https://doi.org/10.3390/molecules24040820
APA StyleCheng, J. -Q., Liu, T., Nie, X. -M., Chen, F. -M., Wang, C. -S., & Zhang, F. (2019). Analysis of 27 β-Blockers and Metabolites in Milk Powder by High Performance Liquid Chromatography Coupled to Quadrupole Orbitrap High-Resolution Mass Spectrometry. Molecules, 24(4), 820. https://doi.org/10.3390/molecules24040820