The Concept of an Ideal Antibiotic: Implications for Drug Design


Abstract
:1. Introduction
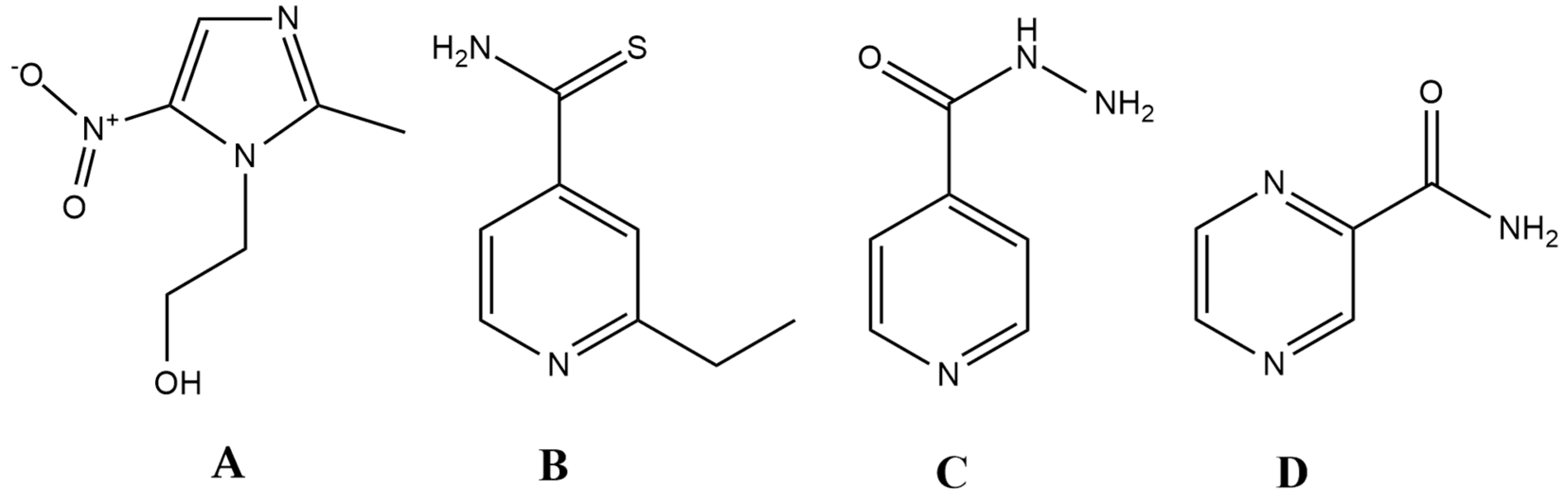
2. The Ideal Antibiotic (Prodrug) Model
3. Prodrug Antibiotics in Clinical Use
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACP | acyl carrier protein |
| AMP | antimicrobial peptide |
| BCS | Biopharmaceutical Classification System |
| CC | combinatorial chemistry |
| CDC | Centers for Disease Control and Prevention |
| CNS | central nervous system |
| ECDC | European Center for Disease Prevention and Control |
| EMA | European Medicines Agency |
| EPI: | efflux pump inhibitor |
| ETH | ethionamide |
| FDA | Food and Drug Administration |
| HTS | high-throughput screening |
| IDSA | Infectious Diseases Society of America |
| IM | inner membrane |
| IMI | Innovative Medicines Initiative |
| INH | isoniazid |
| IV | intravenous therapy |
| MDR | multidrug resistant |
| MRSA | methicillin-resistant Staphylococcus aureus |
| NAD | nicotinamide adenine dinucleotide |
| ND4BB | New Drugs 4 Bad Bugs |
| OM | outer membrane |
| PO | oral therapy |
| POA | pyrazinoic acid |
| PYR | pyrazinamide |
| PDR | pandrug resistant |
| RDD | rational drug design |
| ROS | reactive oxygen species |
| R&D | research and development |
| RO5 | Lipinsky’s Rule of Five |
| TB | tuberculosis |
| XDR | extensively drug resistant |
| WHO | World Health Organization |
References
- Gaynes, R. The Discovery of Penicillin—New Insights after More Than 75 Years of Clinical Use. Emerg. Infect. Dis. 2017, 23, 849–853. [Google Scholar] [CrossRef]
- McNulty, C.A.M.; Cookson, B.D.; Lewis, M.A.O. Education of healthcare professionals and the public. J. Antimicrob. Chemother. 2012, 67 (Suppl. 1), i11–i18. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-R.; Cho, I.H.; Jeong, B.C.; Lee, S.H. Strategies to Minimize Antibiotic Resistance. Int. J. Environ. Res. Public Health 2013, 10, 4274–4305. [Google Scholar] [CrossRef] [PubMed]
- Dyar, O.J.; Huttner, B.; Schouten, J.; Pulcini, C. What is antimicrobial stewardship? Clin. Microbiol. Infect. 2017, 23, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Gajdács, M.; Paulik, E.; Szabó, A. [The opinions of community pharmacists related to antibiotic use and resistance] (article in Hungarian). Acta Pharm. Hung. 2018, 88, 249–252. [Google Scholar]
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.M.; Wertheim, H.F.L.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic resistance-the need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef]
- Shallcross, L.J.; Howard, S.J.; Fowler, T.; Davies, S.C. Tackling the threat of antimicrobial resistance: From policy to sustainable action. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140082. [Google Scholar] [CrossRef] [PubMed]
- Ali, J.; Rafiq, Q.A.; Ratcliffe, E. Antimicrobial resistance mechanisms and potential synthetic treatments. Future Sci. OA 2018, 4, FSO290. [Google Scholar] [CrossRef] [PubMed]
- Munita, J.M.; Arias, C.A. Mechanisms of Antibiotic Resistance. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Chang, H.-H.; Cohen, T.; Grad, Y.H.; Hanage, W.P.; O’Brien, T.F.; Lipsitch, M. Origin and Proliferation of Multiple-Drug Resistance in Bacterial Pathogens. Microbiol. Mol. Biol. Rev. 2015, 79, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Multidrug resistance in bacteria. Annu. Rev. Biochem. 2009, 78, 119–146. [Google Scholar] [CrossRef] [PubMed]
- Magiorakos, A.-P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, R.; Cantón, R.; Brown, D.F.J.; Giske, C.G.; Heisig, P.; MacGowan, A.P.; Mouton, J.W.; Nordmann, P.; Rodloff, A.C.; Rossolini, G.M.; et al. EUCAST expert rules in antimicrobial susceptibility testing. Clin. Microbiol. Infect. 2013, 19, 141–160. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, J. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations. Available online: https://amr-review.org/sites/default/files/AMRReviewPaper-Tacklingacrisisforthehealthandwealthofnations_1.pdf (accessed on 23 January 2019).
- World Health Organisation. Antimicrobial Resistance: Global Report on Surveillance. 2014, pp. 1–256. Available online: http://apps.who.int/iris/bitstream/10665/112642/1/9789241564748_eng.pdf?ua=1 (accessed on 23 January 2019).
- ECDC/EMEA Joint Technical Report (2009). The Bacterial Challenge: Time to React. Available online: http://ecdc.europa.eu/en/publications/Publications/0909_TER_The_Bacterial_Challenge_Time_to_React.pdf (accessed on 23 January 2019).
- CDC Antibiotic/Antimicrobial Resistance (AR/AMR). Available online: https://www.cdc.gov/drugresistance/biggest_threats.html (accessed on 23 January 2019).
- Santajit, S.; Indrawattana, N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. Biomed. Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Swierstra, J.; Wu, C.; Girard, G.; Choi, Y.H.; van Wamel, W.; Sandiford, S.K.; van Wezel, G.P. Eliciting antibiotics active against the ESKAPE pathogens in a collection of actinomycetes isolated from mountain soils. Microbiology 2014, 160, 1714–1725. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Jin, D.; Kim, H.B.; Stratton, C.W.; Wu, B.; Tang, Y.-W.; Sun, X. Update on Antimicrobial Resistance in Clostridium difficile: Resistance Mechanisms and Antimicrobial Susceptibility Testing. J. Clin. Microbiol. 2017, 55, 1998–2008. [Google Scholar] [CrossRef] [PubMed]
- Brooke, J.S. Stenotrophomonas maltophilia: An Emerging Global Opportunistic Pathogen. Clin. Microbiol. Rev. 2012, 25, 2–41. [Google Scholar] [CrossRef] [PubMed]
- Boucher, H.W.; Talbot, G.H.; Bradley, J.S.; Edwards, J.E.; Gilbert, D.; Rice, L.B.; Scheld, M.; Spellberg, B.; Bartlett, J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Elemam, A.; Rahimian, J.; Mandell, W. Infection with panresistant Klebsiella pneumoniae: A report of 2 cases and a brief review of the literature. Clin. Infect. Dis. 2009, 49, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Rafailidis, P.I.; Matthaiou, D.K.; Virtzili, S.; Nikita, D.; Michalopoulos, A. Pandrug-resistant Klebsiella pneumoniae, Pseudomonas aeruginosa and Acinetobacter baumannii infections: Characteristics and outcome in a series of 28 patients. Int. J. Antimicrob. Agents 2008, 32, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Medina, E.; Pieper, D.H. Tackling Threats and Future Problems of Multidrug-Resistant Bacteria. Curr. Top. Microbiol. Immunol. 2016, 398, 3–33. [Google Scholar] [PubMed]
- Lyddiard, D.; Jones, G.L.; Greatrex, B.W. Keeping it simple: Lessons from the golden era of antibiotic discovery. FEMS Microbiol. Lett. 2016, 363, fnw084. [Google Scholar] [CrossRef] [PubMed]
- Darrow, J.J.; Kesselheim, A.S. Drug development and FDA approval, 1938–2013. N. Engl. J. Med. 2014, 370, e39. [Google Scholar] [CrossRef] [PubMed]
- Finland, M.; Kirby, W.M.; Chabbert, Y.A.; Chain, E.B.; Dowling, H.F.; Garrod, L.P.; Pettinga, C.W.; Todd, A.C. Round table: Are new antibiotics needed? Antimicrob. Agents Chemother. 1965, 5, 1107–1114. [Google Scholar] [PubMed]
- Bozdogan, B.; Appelbaum, P.C. Oxazolidinones: Activity, mode of action, and mechanism of resistance. Int. J. Antimicrob. Agents 2004, 23, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Steenbergen, J.N.; Alder, J.; Thorne, G.M.; Tally, F.P. Daptomycin: A lipopeptide antibiotic for the treatment of serious Gram-positive infections. J. Antimicrob. Chemother. 2005, 55, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Bergen, P.J.; Landersdorfer, C.B.; Zhang, J.; Zhao, M.; Lee, H.J.; Nation, R.L.; Li, J. Pharmacokinetics and pharmacodynamics of “old” polymyxins: What is new? Diagn. Microbiol. Infect. Dis. 2012, 74, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Govil, D.; Kakar, P.N.; Prakash, O.; Arora, D.; Das, S.; Govil, P.; Malhotra, A. Colistin and polymyxin B: A re-emergence. Indian J. Crit. Care Med. 2009, 13, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Nation, R.L.; Li, J. Colistin in the 21st Century. Curr. Opin. Infect. Dis. 2009, 22, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Chen, J.; Liu, Y.; Hu, Y.; Liu, Y.; Lu, J.; Zhang, S.; Yu, Y.; Huang, X.; Yang, Q.; et al. In Vitro Activities of Ceftaroline/Avibactam, Ceftazidime/Avibactam, and Other Comparators against Pathogens from Various Complicated Infections in China. Clin. Infect. Dis. 2018, 67, S206–S216. [Google Scholar] [CrossRef] [PubMed]
- Karaiskos, I.; Galani, I.; Souli, M.; Giamarellou, H. Novel β-lactam-β-lactamase inhibitor combinations: Expectations for the treatment of carbapenem-resistant Gram-negative pathogens. Expert Opin. Drug Metab. Toxicol. 2019, 15, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B. The future of antibiotics. Crit. Care 2014, 18, 228. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B.; Talbot, G.H.; Brass, E.P.; Bradley, J.S.; Boucher, H.W.; Gilbert, D.N. Infectious Diseases Society of America Position paper: Recommended design features of future clinical trials of antibacterial agents for community-acquired pneumonia. Clin. Infect. Dis. 2008, 47 (Suppl. 3), S249–S265. [Google Scholar]
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Projan, S.J. Why is big Pharma getting out of antibacterial drug discovery? Curr. Opin. Microbiol. 2003, 6, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Boggs, A.F.; Miller, G.H. Antibacterial drug discovery: Is small pharma the solution? Clin. Microbiol. Infect. 2004, 10 (Suppl. 4), 32–36. [Google Scholar] [CrossRef] [PubMed]
- Rex, J.H. ND4BB: Addressing the antimicrobial resistance crisis. Nat. Rev. Microbiol. 2014, 12, 231–232. [Google Scholar] [CrossRef]
- Infectious Diseases Society of America. The 10 × ’20 Initiative: Pursuing a global commitment to develop 10 new antibacterial drugs by 2020. Clin. Infect. Dis. 2010, 50, 1081–1083. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.; Karlén, A. Discovery and preclinical development of new antibiotics. Ups. J. Med. Sci. 2014, 119, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Coates, A.R.; Halls, G.; Hu, Y. Novel classes of antibiotics or more of the same? Br. J. Pharmacol. 2011, 163, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. New approaches to antimicrobial discovery. Biochem. Pharmacol. 2017, 134, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Kealey, C.; Creaven, C.A.; Murphy, C.D.; Brady, C.B. New approaches to antibiotic discovery. Biotechnol. Lett. 2017, 39, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Leung, M.C.K.; Williams, P.L.; Benedetto, A.; Au, C.; Helmcke, K.J.; Aschner, M.; Meyer, J.N. Caenorhabditis elegans: An Emerging Model in Biomedical and Environmental Toxicology. Toxicol. Sci. 2008, 106, 5–28. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, E.J.; Conery, A.L.; Moy, T.I. Whole-animal high-throughput screens: The C. elegans model. Methods Mol. Biol. 2009, 486, 57–75. [Google Scholar] [PubMed]
- Domagk, G. Ein Beitrag zur Chemotherapie der bakteriellen Infektionen. Dtsch. Med. Wochenschr. 1935, 61, 250–253. [Google Scholar] [CrossRef]
- Vézina, C.; Kudelski, A.; Sehgal, S.N. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J. Antibiot. 1975, 28, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Waksman, S.A.; Woodruff, H.B. The Soil as a Source of Microorganisms Antagonistic to Disease-Producing Bacteria. J. Bacteriol. 1940, 40, 581–600. [Google Scholar] [PubMed]
- Zazopoulos, E.; Huang, K.; Staffa, A.; Liu, W.; Bachmann, B.O.; Nonaka, K.; Ahlert, J.; Thorson, J.S.; Shen, B.; Farnet, C.M. A genomics-guided approach for discovering and expressing cryptic metabolic pathways. Nat. Biotechnol. 2003, 21, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.; Cahoon, N.; Trakhtenberg, E.M.; Pham, L.; Mehta, A.; Belanger, A.; Kanigan, T.; Lewis, K.; Epstein, S.S. Use of Ichip for High-Throughput In Situ Cultivation of “Uncultivable” Microbial Species. Appl. Environ. Microbiol. 2010, 76, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Shannon, E.; Abu-Ghannam, N. Antibacterial Derivatives of Marine Algae: An Overview of Pharmacological Mechanisms and Applications. Mar. Drugs 2016, 14, 81. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.; Ausubel, F.M. Prospects for plant-derived antibacterials. Nat. Biotechnol. 2006, 24, 1504–1507. [Google Scholar] [CrossRef] [PubMed]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.H.; Neefs, J.-M.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, L.; Relman, D.A. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4554–4561. [Google Scholar] [CrossRef]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007, 6, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Nuti, R.; Goud, N.S.; Saraswati, A.P.; Alvala, R.; Alvala, M. Antimicrobial Peptides: A Promising Therapeutic Strategy in Tackling Antimicrobial Resistance. Curr. Med. Chem. 2017, 24, 4303–4314. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Ribeiro, A.M.; de Melo Carrasco, L.D. Novel Formulations for Antimicrobial Peptides. Int. J. Mol. Sci. 2014, 15, 18040–18083. [Google Scholar] [CrossRef] [PubMed]
- Drawz, S.M.; Bonomo, R.A. Three Decades of β-Lactamase Inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef] [PubMed]
- Spengler, G.; Kincses, A.; Gajdacs, M.; Amaral, L. New Roads Leading to Old Destinations: Efflux Pumps as Targets to Reverse Multidrug Resistance in Bacteria. Molecules 2017, 22, 468. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Warren, M.S.; Lee, A.; Galazzo, J.; Fronko, R.; Lee, M.; Blais, J.; Cho, D.; Chamberland, S.; Renau, T.; et al. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: Novel agents for combination therapy. Antimicrob. Agents Chemother. 2001, 45, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Mühlen, S.; Dersch, P. Anti-virulence Strategies to Target Bacterial Infections. In How to Overcome the Antibiotic Crisis: Facts, Challenges, Technologies and Future Perspectives; Stadler, M., Dersch, P., Eds.; Current Topics in Microbiology and Immunology; Springer International Publishing: Cham, Switzerland, 2016; pp. 147–183. ISBN 978-3-319-49284-1. [Google Scholar]
- Cegelski, L.; Marshall, G.R.; Eldridge, G.R.; Hultgren, S.J. The biology and future prospects of antivirulence therapies. Nat. Rev. Microbiol. 2008, 6, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Ahmad, F.; Dar, S.A.; Jawed, A.; Mandal, R.K.; Wahid, M.; Lohani, M.; Khan, S.; Singh, V.; Akhter, N. Developments in strategies for Quorum Sensing virulence factor inhibition to combat bacterial drug resistance. Microb. Pathog. 2018, 121, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Lowy, I.; Molrine, D.C.; Leav, B.A.; Blair, B.M.; Baxter, R.; Gerding, D.N.; Nichol, G.; Thomas, W.D.; Leney, M.; Sloan, S.; et al. Treatment with Monoclonal Antibodies against Clostridium difficile Toxins. N. Engl. J. Med. 2010, 362, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Love, M.J.; Bhandari, D.; Dobson, R.C.J.; Billington, C. Potential for Bacteriophage Endolysins to Supplement or Replace Antibiotics in Food Production and Clinical Care. Antibiotics (Basel) 2018, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Schmelcher, M.; Donovan, D.M.; Loessner, M.J. Bacteriophage endolysins as novel antimicrobials. Future Microbiol. 2012, 7, 1147–1171. [Google Scholar] [CrossRef] [PubMed]
- Briers, Y.; Walmagh, M.; Puyenbroeck, V.V.; Cornelissen, A.; Cenens, W.; Aertsen, A.; Oliviera, H.; Azeredo, J.; Verween, G.; Pirnay, J.P.; et al. Engineered Endolysin-Based “Artilysins” To Combat Multidrug-Resistant Gram-Negative Pathogens. mBio 2014, 5, e01379-14. [Google Scholar] [CrossRef] [PubMed]
- Kashani, H.H.; Schmelcher, M.; Sabzalipoor, H.; Hosseini, E.S.; Moniri, R. Recombinant Endolysins as Potential Therapeutics against Antibiotic-Resistant Staphylococcus aureus: Current Status of Research and Novel Delivery Strategies. Clin. Microbiol. Rev. 2018, 31, 1–26. [Google Scholar]
- Singh, S.B.; Young, K.; Silver, L.L. What is an “ideal” antibiotic? Discovery challenges and path forward. Biochem. Pharmacol. 2017, 133, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Pankey, G.A.; Sabath, L.D. Clinical Relevance of Bacteriostatic versus Bactericidal Mechanisms of Action in the Treatment of Gram-Positive Bacterial Infections. Clin. Infect. Dis. 2004, 38, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, J.; Oesch, G.; Kuster, S.P. Bacteriostatic versus bactericidal antibiotics for patients with serious bacterial infections: Systematic review and meta-analysis. J. Antimicrob. Chemother. 2015, 70, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Wald-Dickler, N.; Holtom, P.; Spellberg, B. Busting the Myth of “Static vs Cidal”: A Systemic Literature Review. Clin. Infect. Dis. 2018, 66, 1470–1474. [Google Scholar] [CrossRef] [PubMed]
- Redelinghuys, M.J.; Ehlers, M.M.; Dreyer, A.W.; Lombaard, H.A.; Kock, M.M. Antimicrobial susceptibility patterns of Ureaplasma species and Mycoplasma hominis in pregnant women. BMC Infect. Dis. 2014, 14, 171. [Google Scholar] [CrossRef] [PubMed]
- Briers, Y.; Staubli, T.; Schmid, M.C.; Wagner, M.; Schuppler, M.; Loessner, M.J. Intracellular Vesicles as Reproduction Elements in Cell Wall-Deficient L-Form Bacteria. PLoS ONE 2012, 7, e38514. [Google Scholar] [CrossRef] [PubMed]
- Errington, J. L-form bacteria, cell walls and the origins of life. Open Biol. 2013, 3, 120143. [Google Scholar] [CrossRef] [PubMed]
- Errington, J.; Mickiewicz, K.; Kawai, Y.; Wu, L.J. L-form bacteria, chronic diseases and the origins of life. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150494. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.K.; Knabel, S.J.; Kwan, B.W. Bacterial Persister Cell Formation and Dormancy. Appl. Environ. Microbiol. 2013, 79, 7116–7121. [Google Scholar] [CrossRef] [PubMed]
- Van den Bergh, B.; Fauvart, M.; Michiels, J. Formation, physiology, ecology, evolution and clinical importance of bacterial persisters. FEMS Microbiol. Rev. 2017, 41, 219–251. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 2017, 15, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Moser, C.; Wang, H.-Z.; Høiby, N.; Song, Z.-J. Strategies for combating bacterial biofilm infections. Int. J. Oral Sci. 2015, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Limoli, D.H.; Jones, C.J.; Wozniak, D.J. Bacterial Extracellular Polysaccharides in Biofilm Formation and Function. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Costerton, J.W. Cystic fibrosis pathogenesis and the role of biofilms in persistent infection. Trends Microbiol. 2001, 9, 50–52. [Google Scholar] [CrossRef]
- Belfield, K.; Bayston, R.; Hajduk, N.; Levell, G.; Birchall, J.P.; Daniel, M. Evaluation of combinations of putative anti-biofilm agents and antibiotics to eradicate biofilms of Staphylococcus aureus and Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2017, 72, 2531–2538. [Google Scholar] [CrossRef] [PubMed]
- Chung, P.Y.; Toh, Y.S. Anti-biofilm agents: Recent breakthrough against multi-drug resistant Staphylococcus aureus. Pathog. Dis. 2014, 70, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Zgurskaya, H.I.; Löpez, C.A.; Gnanakaran, S. Permeability Barrier of Gram-Negative Cell Envelopes and Approaches to Bypass It. ACS Infect. Dis. 2015, 1, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Silver, L.L. Are natural products still the best source for antibacterial discovery? The bacterial entry factor. Expert Opin. Drug. Discov. 2008, 3, 487–500. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, R.; Moser, H.E. Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery. J. Med. Chem. 2008, 51, 2871–2878. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.I. Antibiotic Resistance and Regulation of the Gram-Negative Bacterial Outer Membrane Barrier by Host Innate Immune Molecules. mBio 2016, 7, e01541-16. [Google Scholar] [CrossRef] [PubMed]
- Tiz, D.B.; Kikelj, D.; Zidar, N. Overcoming problems of poor drug penetration into bacteria: Challenges and strategies for medicinal chemists. Expert Opin. Drug Discov. 2018, 13, 497–507. [Google Scholar]
- Fernández, L.; Hancock, R.E.W. Adaptive and Mutational Resistance: Role of Porins and Efflux Pumps in Drug Resistance. Clin. Microbiol. Rev. 2012, 25, 661–681. [Google Scholar] [CrossRef] [PubMed]
- Delcour, A.H. Outer Membrane Permeability and Antibiotic Resistance. Biochim. Biophys. Acta 2009, 1794, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.A.; Piddock, L.J.V. The importance of efflux pumps in bacterial antibiotic resistance. J. Antimicrob. Chemother. 2003, 51, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Amaral, L.; Martins, A.; Spengler, G.; Molnar, J. Efflux pumps of Gram-negative bacteria: What they do, how they do it, with what and how to deal with them. Front. Pharmacol. 2014, 4, 168. [Google Scholar] [CrossRef] [PubMed]
- Tegos, G.P.; Haynes, M.; Strouse, J.J.; Khan, M.M.T.; Bologa, C.G.; Oprea, T.I.; Sklar, L.A. Microbial Efflux Pump Inhibition: Tactics and Strategies. Curr. Pharm. Des. 2011, 17, 1291–1302. [Google Scholar] [CrossRef] [PubMed]
- Drlica, K. The mutant selection window and antimicrobial resistance. J. Antimicrob. Chemother. 2003, 52, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Hefti, F.F. Requirements for a lead compound to become a clinical candidate. BMC Neurosci. 2008, 9, S7. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, R.R.; Graziani, A.L. Oral administration of antibiotics: A rational alternative to the parenteral route. Clin. Infect. Dis. 1997, 24, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Li, H.K.; Agweyu, A.; English, M.; Bejon, P. An unsupported preference for intravenous antibiotics. PLoS Med. 2015, 12, e1001825. [Google Scholar] [CrossRef] [PubMed]
- Brunton, L.; Chabner, B.A.; Knollman, B. Goodman & Gillman’s The Pharmacological Basis of Therapeutics, 12th ed.; McGraw-Hill: New York, NY, USA, 2011. [Google Scholar]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Gajdács, M.; Handzlik, J.; Sanmartín, C.; Domínguez-Álvarez, E.; Spengler, G. [Prediction of ADME properties for selenocompounds with anticancer and efflux pump inhibitory activity using preliminary computational methods] (article in Hungarian). Acta Pharm. Hung. 2018, 88, 67–74. [Google Scholar]
- Takagi, T.; Ramachandran, C.; Bermejo, M.; Yamashita, S.; Yu, L.X.; Amidon, G.L. A Provisional Biopharmaceutical Classification of the Top 200 Oral Drug Products in the United States, Great Britain, Spain, and Japan. Mol. Pharm. 2006, 3, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B.; Lipsky, B.A. Systemic Antibiotic Therapy for Chronic Osteomyelitis in Adults. Clin. Infect. Dis. 2012, 54, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.; Sauermann, R.; Joukhadar, C. Principles of antibiotic penetration into abscess fluid. Pharmacology 2006, 78, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Carryn, S.; Chanteux, H.; Seral, C.; Mingeot-Leclercq, M.-P.; Van Bambeke, F.; Tulkens, P.M. Intracellular pharmacodynamics of antibiotics. Infect. Dis. Clin. N. Am. 2003, 17, 615–634. [Google Scholar] [CrossRef]
- Van Bambeke, F.; Barcia-Macay, M.; Lemaire, S.; Tulkens, P.M. Cellular pharmacodynamics and pharmacokinetics of antibiotics: Current views and perspectives. Curr. Opin. Drug Discov. Dev. 2006, 9, 218–230. [Google Scholar]
- McClure, E.E.; Oliva Chávez, A.S.; Shaw, D.K.; Carlyon, J.A.; Ganta, R.R.; Noh, S.M.; Wood, D.O.; Bavoil, P.M.; Brayton, K.A.; Martinez, J.J.; et al. Engineering of obligate intracellular bacteria: Progress, challenges and paradigms. Nat. Rev. Microbiol. 2017, 15, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Candel, F.J.; Peñuelas, M. Delafloxacin: Design, development and potential place in therapy. Drug Des. Dev. Ther. 2017, 11, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, S.; Tulkens, P.M.; Bambeke, F.V. Contrasting Effects of Acidic pH on the Extracellular and Intracellular Activities of the Anti-Gram-Positive Fluoroquinolones Moxifloxacin and Delafloxacin against Staphylococcus aureus. Antimicrob. Agents Chemother. 2011, 55, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Blais, J.; Beauchamp, D.; Chamberland, S. Azithromycin uptake and intracellular accumulation by Toxoplasma gondii-infected macrophages. J. Antimicrob. Chemother. 1994, 34, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Bosnar, M.; Kelnerić, Ž.; Munić, V.; Eraković, V.; Parnham, M.J. Cellular Uptake and Efflux of Azithromycin, Erythromycin, Clarithromycin, Telithromycin, and Cethromycin. Antimicrob. Agents Chemother. 2005, 49, 2372–2377. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, I. Antibiotic chemotherapy during pregnancy and lactation period: Aspects for consideration. Arch. Gynecol. Obstet. 2011, 283, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Nahum, G.G.; Uhl, K.; Kennedy, D.L. Antibiotic use in pregnancy and lactation: What is and is not known about teratogenic and toxic risks. Obstet. Gynecol. 2006, 107, 1120–1138. [Google Scholar] [CrossRef] [PubMed]
- Youngster, I.; Avorn, J.; Belleudi, V.; Cantarutti, A.; Díez-Domingo, J.; Kirchmayer, U.; Park, B.-J.; Peiró, S.; Sanfélix-Gimeno, G.; Schröder, H.; et al. Antibiotic Use in Children—A Cross-National Analysis of 6 Countries. J. Pediatr. 2017, 182, 239–244.e1. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Ross, J.S.; Kapczynski, A. Pediatric Exclusivity and Regulatory Authority: Implications of Amgen v HHS. JAMA 2018, 319, 21–22. [Google Scholar] [CrossRef] [PubMed]
- Manikandan, P.; Nagini, S. Cytochrome P450 Structure, Function and Clinical Significance: A Review. Curr. Drug Targets 2018, 19, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Nettleton, D.O.; Einolf, H.J. Assessment of cytochrome p450 enzyme inhibition and inactivation in drug discovery and development. Curr. Top. Med. Chem. 2011, 11, 382–403. [Google Scholar] [CrossRef] [PubMed]
- Fox, L.M.; Saravolatz, L.D. Nitazoxanide: A New Thiazolide Antiparasitic Agent. Clin. Infect. Dis. 2005, 40, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Löfmark, S.; Edlund, C.; Nord, C.E. Metronidazole is still the drug of choice for treatment of anaerobic infections. Clin. Infect. Dis. 2010, 50 (Suppl. 1), S16–S23. [Google Scholar] [CrossRef] [PubMed]
- Adachi, T.; Matsui, S.; Watanabe, T.; Okamoto, K.; Okamoto, A.; Kono, M.; Yamada, M.; Nagai, T.; Komeda, Y.; Minaga, K.; et al. Comparative Study of Clarithromycin- versus Metronidazole-Based Triple Therapy as First-Line Eradication for Helicobacter pylori. Oncology 2017, 93 (Suppl. 1), 15–19. [Google Scholar] [CrossRef]
- Butenko, T.; Jeverica, S.; Orel, R.; Homan, M. Antibacterial resistance and the success of tailored triple therapy in Helicobacter pylori strains isolated from Slovenian children. Helicobacter 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Ahn, H.J.; Lee, D.S. Helicobacter pylori in gastric carcinogenesis. World J. Gastrointest. Oncol. 2015, 7, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Sóki, J.; Gal, M.; Brazier, J.S.; Rotimi, V.O.; Urbán, E.; Nagy, E.; Duerden, B.I. Molecular investigation of genetic elements contributing to metronidazole resistance in Bacteroides strains. J. Antimicrob. Chemother. 2006, 57, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Gajdács, M.; Spengler, G.; Urbán, E. Identification and Antimicrobial Susceptibility Testing of Anaerobic Bacteria: Rubik’s Cube of Clinical Microbiology? Antibiotics (Basel) 2017, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Jeverica, S.; Kolenc, U.; Mueller-Premru, M.; Papst, L. Evaluation of the routine antimicrobial susceptibility testing results of clinically significant anaerobic bacteria in a Slovenian tertiary-care hospital in 2015. Anaerobe 2017, 47, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Shinn, D.L.S. Metronidazole in acute ulcerative gingivitis. Lancet 1962, 279, 1191. [Google Scholar] [CrossRef]
- Sotgiu, G.; Centis, R.; D’ambrosio, L.; Migliori, G.B. Tuberculosis Treatment and Drug Regimens. Cold Spring Harb. Perspect. Med. 2015, 5, a017822. [Google Scholar] [CrossRef] [PubMed]
- Pieters, J. Mycobacterium tuberculosis and the Macrophage: Maintaining a Balance. Cell Host Microbe 2008, 3, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, W.; Zhang, W.; Mitchison, D. Mechanisms of Pyrazinamide Action and Resistance. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Langley, R.; Gulten, G.; Dover, L.G.; Besra, G.S.; Jacobs, W.R.; Sacchettini, J.C. Mechanism of thioamide drug action against tuberculosis and leprosy. J. Exp. Med. 2007, 204, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Unissa, A.N.; Subbian, S.; Hanna, L.E.; Selvakumar, N. Overview on mechanisms of isoniazid action and resistance in Mycobacterium tuberculosis. Infect. Genet. Evol. 2016, 45, 474–492. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Dubnau, E.; Quemard, A.; Balasubramanian, V.; Um, K.S.; Wilson, T.; Collins, D.; de Lisle, G.; Jacobs, W.R. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 1994, 263, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Zhang, X.; Jiang, X.; Yuan, H.; Lee, J.S.; Barry, C.E.; Wang, H.; Zhang, W.; Zhang, Y. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 2011, 333, 1630–1632. [Google Scholar] [PubMed]
- The Selection and Use of Essential Medicines. World Health Organization. Available online: https://www.who.int/medicines/publications/essentialmedicines/EML_2017_ExecutiveSummary.pdf?ua=1 (accessed on 23 January 2019).
- Almeida Da Silva, P.E.A.; Palomino, J.C. Molecular basis and mechanisms of drug resistance in Mycobacterium tuberculosis: Classical and new drugs. J. Antimicrob. Chemother. 2011, 66, 1417–1430. [Google Scholar] [CrossRef] [PubMed]
- Witek, K.; Nasim, M.J.; Bischoff, M.; Gaupp, R.; Arsenyan, P.; Vasiljeva, J.; Marć, M.A.; Olejarz, A.; Latacz, G.; Kieć-Kononowicz, K.; et al. Selenazolinium Salts as “Small Molecule Catalysts” with High Potency against ESKAPE Bacterial Pathogens. Molecules 2017, 22, 2174. [Google Scholar] [CrossRef] [PubMed]
- Acker, H.V.; Coenye, T. The Role of Reactive Oxygen Species in Antibiotic-Mediated Killing of Bacteria. Trends Microbiol. 2017, 25, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.C. Antimicrobial Actions of Reactive Oxygen Species. mBio 2011, 2, e00141-11. [Google Scholar] [CrossRef] [PubMed]
- Keren, I.; Wu, Y.; Inocencio, J.; Mulcahy, L.R.; Lewis, K. Killing by Bactericidal Antibiotics Does Not Depend on Reactive Oxygen Species. Science 2013, 339, 1213–1216. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Imlay, J.A. Cell Death from Antibiotics without the Involvement of Reactive Oxygen Species. Science 2013, 339, 1210–1213. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D. Antibiotic Adjuvants: Rescuing Antibiotics from Resistance. Trends Microbiol. 2016, 24, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Ha, D.R.; Haste, N.M.; Gluckstein, D.P. The Role of Antibiotic Stewardship in Promoting Appropriate Antibiotic Use. Am. J. Lifestyle Med. 2017. [Google Scholar] [CrossRef]
- Gajdács, M.; Paulik, E.; Szabó, A. [The attitude of community pharmacists towards their widening roles in the prevention and treatment of infectious diseases in the southeast region of Hungary] (article in Hungarian). Gyógyszerészet 2019, 63, 26–30. [Google Scholar]
- Infectious Diseases Society of America. An unmet medical need: Rapid molecular diagnostics tests for respiratory tract infections. Clin. Infect. Dis. 2011, 52 (Suppl. 4), S384–S395. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Platform | Brief Description of Pros and Cons | Compounds in Clinical Practice (Examples) |
|---|---|---|
| Domagk-platform/In situ screening-platform |
| Sulfonamides (sulfamidochrysoidine) |
| Waksmann-platform/Natural products-platform |
| Penicillin (First antibiotic discovered) Streptomycin (First drug active against tuberculosis (TB)) Daptomycin (MDR Gram-positives) Fidaxomicin (Clostridioides difficile) |
| Species-selective platform | Bedaquiline F1F0-ATPase-inhibitor in Mycobacterium tuberculosis complex Ethambutol Arabinosyl-transferase-inhibitor in Mycobacterium tuberculosis complex | |
| High-throughput screening (HTS) Combinatorial chemistry (CC) Rational drug design (RDD) |
| Oxazolidinones Inhibitors of protein synthesis by interfering with the ribosomal 50S subunit |
| Antimicrobial peptides (AMPs) |
| No AMP has been approved yet for clinical use |
| Resistance reversing compounds |
| Beta-lactamase inhibitors (clavulanic acid, sulbactam, tazobactam, avibactam etc.) No EPI has been approved yet for clinical use |
| Virulence modulation |
| No virulence modulator has been approved yet for clinical use |
| Drug-Specific | Pathogen-Specific |
|---|---|
| Available for oral administration | Broad-spectrum bactericidal activity (including Gram-positive and Gram-negative bacteria, Mycoplasma/Ureaplasma ssp. and intracellular pathogens) |
| Acts as a prodrug | Antibacterial activity against persisters and pathogens in biofilms |
| Class I in the Biopharmaceutical Classification System | Activity at very low (nanomolar) concentrations |
| Accumulation in macrophages | Useful in hard-to-reach infected sites, e.g., abscesses, central nervous system (CNS), bone tissue |
| No teratogenic effects (safe in pregnancy, lactation and childhood) | Acts on multiple, unrelated, essential bacterial targets |
| No drug–drug interactions | Forms irreversible covalent bonds inside bacterial cells (ruling out drug efflux) |
| The drug is excreted from the body unchanged |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gajdács, M. The Concept of an Ideal Antibiotic: Implications for Drug Design. Molecules 2019, 24, 892. https://doi.org/10.3390/molecules24050892
Gajdács M. The Concept of an Ideal Antibiotic: Implications for Drug Design. Molecules. 2019; 24(5):892. https://doi.org/10.3390/molecules24050892
Chicago/Turabian StyleGajdács, Márió. 2019. "The Concept of an Ideal Antibiotic: Implications for Drug Design" Molecules 24, no. 5: 892. https://doi.org/10.3390/molecules24050892
APA StyleGajdács, M. (2019). The Concept of an Ideal Antibiotic: Implications for Drug Design. Molecules, 24(5), 892. https://doi.org/10.3390/molecules24050892