
Heterologous Machine Learning for the Identification of Antimicrobial Activity in Human-Targeted Drugs



Abstract
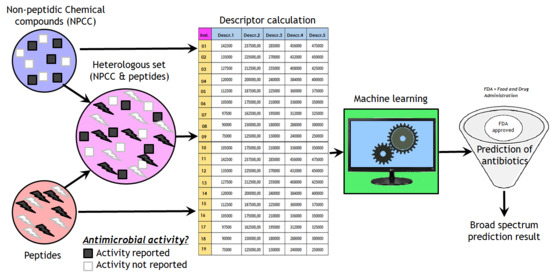
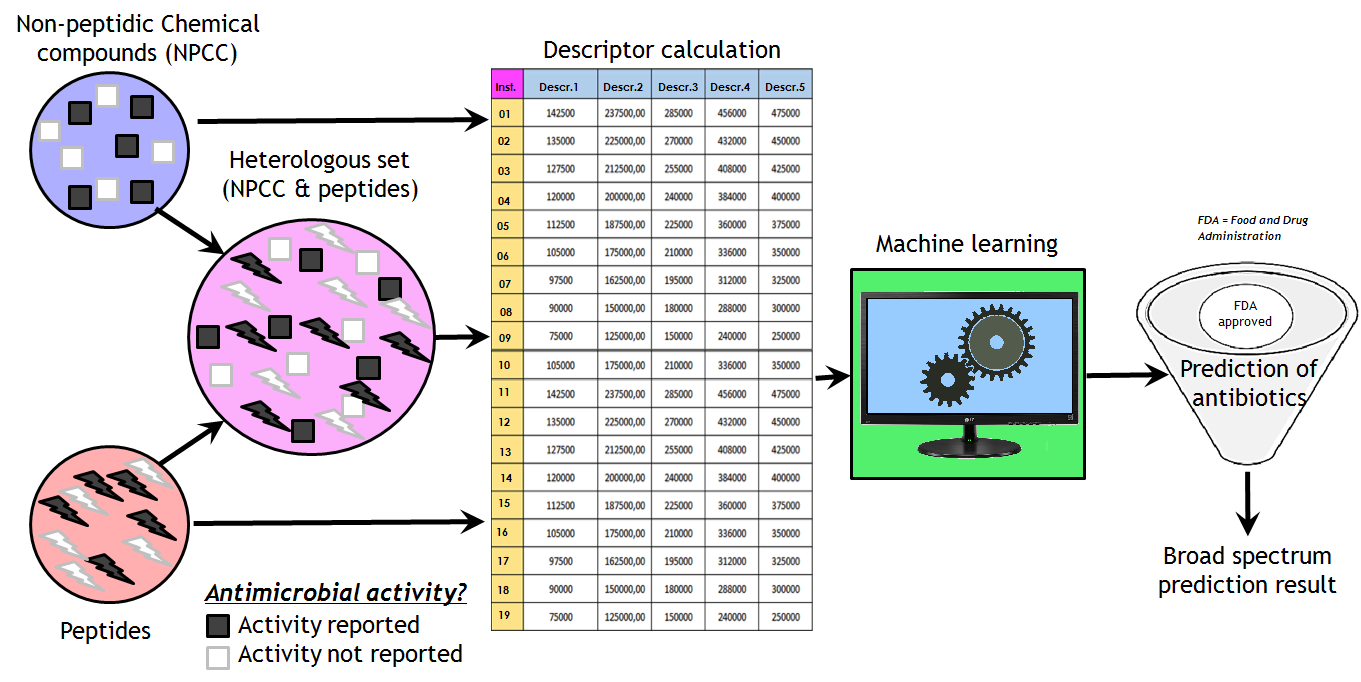
:
1. Introduction
2. Results
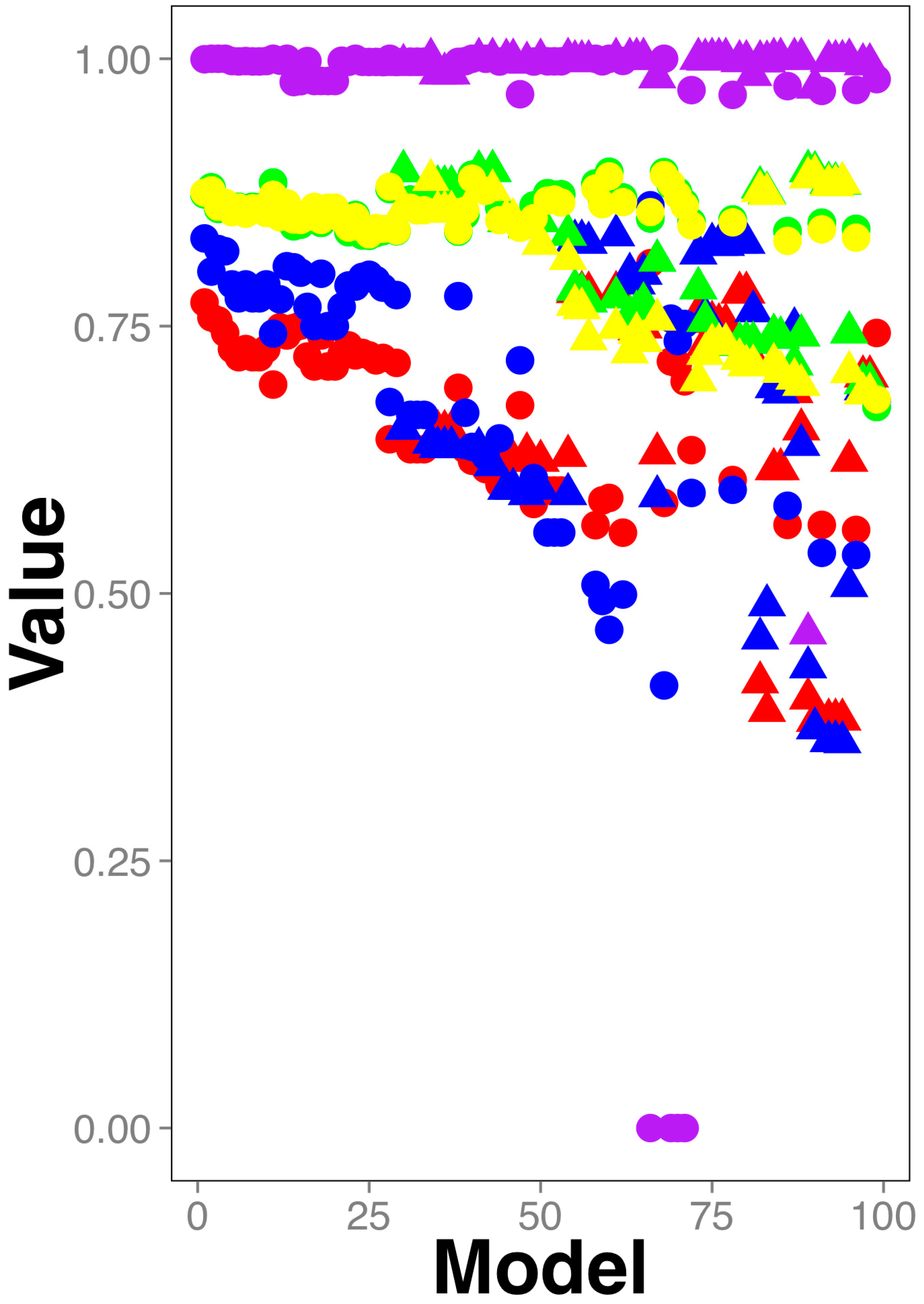
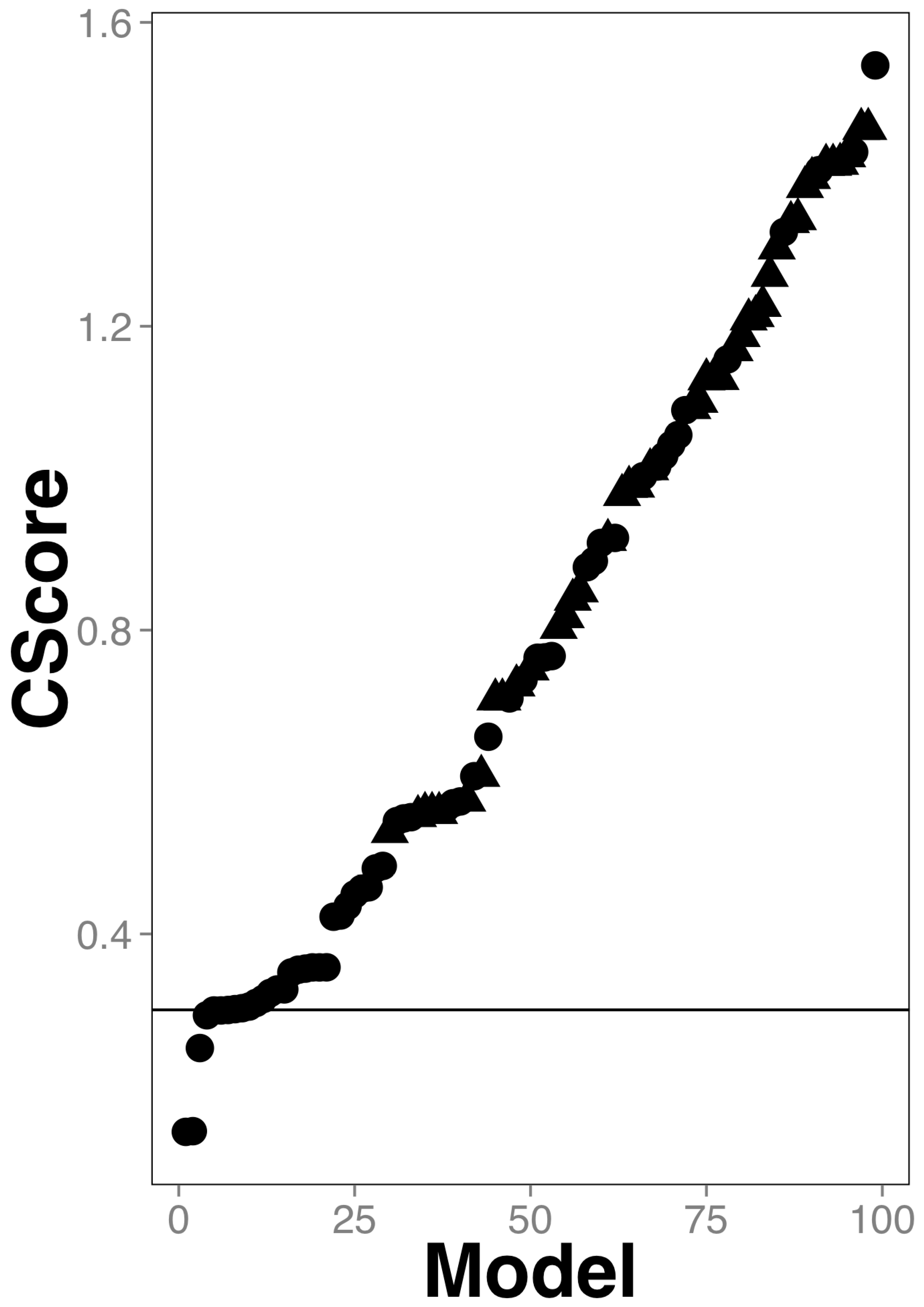
2.1. Training and Testing Gut Antimicrobial Classifiers
2.2. Identifying Broad-Spectrum Antibiotics among FDA-Approved Compounds
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Method
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Akova, M. Epidemiology of antimicrobial resistance in bloodstream infections. Virulence 2016, 7, 252–266. [Google Scholar] [CrossRef] [Green Version]
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; Salamat, M.K.F.; et al. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef]
- Roger, P.-M.; Montera, E.; Lesselingue, D.; Troadec, N.; Charlot, P.; Simand, A.; Rancezot, A.; Pantaloni, O.; Guichard, T.; Dautezac, V.; et al. Risk factors for unnecessary antibiotic therapy: A major role for clinical management. Clin Infect Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.A.; Goodrich, J.K.; Maxan, M.-E.; Freedberg, D.E.; Abrams, J.A.; Poole, A.C.; Sutter, J.L.; Welter, D.; Ley, R.E.; Bell, J.T.; et al. Proton pump inhibitors alter the composition of the gut microbiota. Gut 2016, 65, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.A.M.; Aronoff, D.M. The influence of non-steroidal anti-inflammatory drugs on the gut microbiome. Clin. Microbiol. Infect. 2016, 22, 178.e1–178.e9. [Google Scholar] [CrossRef]
- Flowers, S.A.; Evans, S.J.; Ward, K.M.; McInnis, M.G.; Ellingrod, V.L. Interaction between Atypical Antipsychotics and the Gut Microbiome in a Bipolar Disease Cohort. Pharmacother. J. Hum. Pharmacol. Drug Ther. 2017, 37, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 2018, 555, 623–628. [Google Scholar] [CrossRef] [PubMed]
- González-Díaz, H.; Prado-Prado, F.J.; Santana, L.; Uriarte, E. Unify QSAR approach to antimicrobials. Part 1: Predicting antifungal activity against different species. Bioorg. Med. Chem. 2006, 14, 5973–5980. [Google Scholar] [CrossRef]
- Rath, E.C.; Gill, H.; Bai, Y. Identification of potential antimicrobials against Salmonella typhimurium and Listeria monocytogenes using Quantitative Structure-Activity Relation modeling. PLoS ONE 2017, 12, e0189580. [Google Scholar] [CrossRef]
- Murcia-Soler, M.; Pérez-Giménez, F.; García-March, F.J.; Salabert-Salvador, M.T.; Díaz-Villanueva, W.; Castro-Bleda, M.J.; Villanueva-Pareja, A. Artificial Neural Networks and Linear Discriminant Analysis: A Valuable Combination in the Selection of New Antibacterial Compounds. J. Chem. Inf. Comput. Sci. 2004, 44, 1031–1041. [Google Scholar] [CrossRef]
- Nguyen, M.; Long, S.W.; McDermott, P.F.; Olsen, R.J.; Olson, R.; Stevens, R.L.; Tyson, G.H.; Zhao, S.; Davis, J.J. Using machine learning to predict antimicrobial minimum inhibitory concentrations and associated genomic features for nontyphoidal Salmonella. J. Clin. Microbiol. 2018. Available online: http://www.ncbi.nlm.nih.gov/pubmed/30333126 (accessed on 19 January 2019). [CrossRef] [PubMed]
- Wang, Z.; Wang, G. APD: The Antimicrobial Peptide Database. Nucleic Acids Res. 2004, 32, D590–D592. [Google Scholar] [CrossRef] [PubMed]
- Pirtskhalava, M.; Gabrielian, A.; Cruz, P.; Griggs, H.L.; Squires, R.B.; Hurt, D.E.; Grigolava, M.; Chubinidze, M.; Gogoladze, G.; Vishnepolsky, B.; et al. DBAASP v.2: An enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res. 2016, 44, D1104–D1112. [Google Scholar] [CrossRef] [PubMed]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMP R3: A database on sequences, structures and signatures of antimicrobial peptides: Table 1. Nucleic Acids Res. 2016, 44, D1094–D1097. [Google Scholar] [CrossRef] [PubMed]
- Del Rio, G.; Castro-Obregon, S.; Rao, R.; Ellerby, H.M.; Bredesen, D.E. APAP, a sequence-pattern recognition approach identifies substance P as a potential apoptotic peptide. FEBS Lett. 2001, 494, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Toropova, M.A.; Veselinović, A.M.; Veselinović, J.B.; Stojanović, D.B.; Toropov, A.A. QSAR modeling of the antimicrobial activity of peptides as a mathematical function of a sequence of amino acids. Comput. Biol. Chem. 2015, 59, 126–130. [Google Scholar] [CrossRef]
- Durrant, J.D.; Amaro, R.E. Machine-Learning Techniques Applied to Antibacterial Drug Discovery. Chem. Biol. Drug Des. 2015, 85, 14–21. [Google Scholar] [CrossRef]
- Battisti, A.; Zamuner, S.; Sarti, E.; Laio, A. Toward a unified scoring function for native state discrimination and drug-binding pocket recognition. Phys. Chem. Chem. Phys. 2018, 20, 17148–17155. [Google Scholar] [CrossRef]
- Del Rio, G.; Koschützki, D.; Coello, G. How to identify essential genes from molecular networks? BMC Syst. Biol. 2009, 3, 102. [Google Scholar] [CrossRef] [PubMed]
- Calahorra, M.; Sánchez, N.S.; Peña, A. Influence of phenothiazines, phenazines and phenoxazine on cation transport in Candida albicans. J. Appl. Microbiol. 2018, 125, 1728–1738. [Google Scholar] [CrossRef] [PubMed]
- Acar, J. Broad- and narrow-spectrum antibiotics: An unhelpful categorization. Clin. Microbiol. Infect. 1997, 3, 395–396. [Google Scholar] [CrossRef]
- Sarpong, E.M.; Miller, G.E. Narrow- and Broad-Spectrum Antibiotic Use among U.S. Children. Health Serv. Res. 2015, 50, 830–846. [Google Scholar] [CrossRef] [PubMed]
- Beltran, J.A.; Aguilera-Mendoza, L.; Brizuela, C.A. Optimal selection of molecular descriptors for antimicrobial peptides classification: An evolutionary feature weighting approach. BMC Genomics 2018, 19, 672. [Google Scholar] [CrossRef]
- NIH DailyMed. 26/November 2018. Available online: https://dailymed.nlm.nih.gov/dailymed/index.cfm (accessed on 19 January 2019).
- File, T.M.; Wilcox, M.H.; Stein, G.E. Summary of Ceftaroline Fosamil Clinical Trial Studies and Clinical Safety. Clin. Infect. Dis. 2012, 55, S173–S180. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef]
- Hammami, R.; Zouhir, A.; Le Lay, C.; Ben Hamida, J.; Fliss, I. BACTIBASE second release: A database and tool platform for bacteriocin characterization. BMC Microbiol. 2010, 10, 22. [Google Scholar] [CrossRef]
- De Jong, A.; van Heel, A.J.; Kok, J.; Kuipers, O.P. BAGEL2: Mining for bacteriocins in genomic data. Nucleic Acids Res. 2010, 38, W647–W651. [Google Scholar] [CrossRef]
- Novković, M.; Simunić, J.; Bojović, V.; Tossi, A.; Juretić, D. DADP: The database of anuran defense peptides. Bioinformatics 2012, 28, 1406–1407. [Google Scholar] [CrossRef] [PubMed]
- Seshadri Sundararajan, V.; Gabere, M.N.; Pretorius, A.; Adam, S.; Christoffels, A.; Lehväslaiho, M.; Archer, J.A.C.; Bajic, V.B. DAMPD: A manually curated antimicrobial peptide database. Nucleic Acids Res. 2012, 40, D1108–D1112. [Google Scholar] [CrossRef]
- Seebah, S.; Suresh, A.; Zhuo, S.; Choong, Y.H.; Chua, H.; Chuon, D.; Beuerman, R.; Verma, C. Defensins knowledgebase: A manually curated database and information source focused on the defensins family of antimicrobial peptides. Nucleic Acids Res. 2007, 35, D265–D268. [Google Scholar] [CrossRef]
- Qureshi, A.; Thakur, N.; Kumar, M. HIPdb: A Database of Experimentally Validated HIV Inhibiting Peptides. PLoS ONE 2013, 8, e54908. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wu, H.; Lu, H.; Li, G.; Huang, Q. LAMP: A Database Linking Antimicrobial Peptides. PLoS ONE 2013, 8, e66557. [Google Scholar] [CrossRef] [PubMed]
- Théolier, J.; Fliss, I.; Jean, J.; Hammami, R. MilkAMP: A comprehensive database of antimicrobial peptides of dairy origin. Dairy Sci. Technol. 2014, 94, 181–193. [Google Scholar] [CrossRef]
- Hammami, R.; Ben Hamida, J.; Vergoten, G.; Fliss, I. PhytAMP: A database dedicated to antimicrobial plant peptides. Nucleic Acids Res. 2009, 37, D963–D968. [Google Scholar] [CrossRef] [PubMed]
- Gueguen, Y.; Garnier, J.; Robert, L.; Lefranc, M.; Mougenot, I.; Lorgeril, J.; Janech, M.; Gross, P.S.; Warr, G.W.; Cuthbertson, B.; et al. PenBase, the shrimp antimicrobial peptide penaeidin database: Sequence-based classification and recommended nomenclature. Dev. Comp. Immunol. 2006, 30, 283–288. [Google Scholar] [CrossRef]
- Whitmore, L.; Wallace, B.A. The Peptaibol Database: A database for sequences and structures of naturally occurring peptaibols. Nucleic Acids Res. 2004, 32, D593–D594. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Z. RAPD: A database of recombinantly-produced antimicrobial peptides. FEMS Microbiol. Lett. 2008, 289, 126–129. [Google Scholar] [CrossRef]
- Fjell, C.D.; Hancock, R.E.W.; Cherkasov, A. AMPer: A database and an automated discovery tool for antimicrobial peptides. Bioinformatics 2007, 23, 1148–1155. Available online: http://www.ncbi.nlm.nih.gov/pubmed/17341497 (accessed on 23 January 2019). [CrossRef]
- UniProt Consortium T. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar]
- Piotto, S.P.; Sessa, L.; Concilio, S.; Iannelli, P. YADAMP: Yet another database of antimicrobial peptides. Int. J. Antimicrob. Agents 2012, 39, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Tossi, A.; Sandri, L. Molecular diversity in gene-encoded, cationic antimicrobial polypeptides. Curr. Pharm. Des. 2002, 8, 743–761. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef]
- Qureshi, A.; Thakur, N.; Tandon, H.; Kumar, M. AVPdb: A database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res. 2014, 42, D1147–D1153. [Google Scholar] [CrossRef] [PubMed]
- Corral-Corral, R.; Beltrán, J.; Brizuela, C.; Del Rio, G. Systematic Identification of Machine-Learning Models Aimed to Classify Critical Residues for Protein Function from Protein Structure. Molecules 2017, 22, 1673. [Google Scholar] [CrossRef]
- Witten, I.H.; Ian, H.; Frank, E.; Hall, M.A.; Mark, A. Data Mining: Practical Machine Learning Tools and Techniques; Morgan Kaufmann: Burlington, MA, USA, 2011; 629p. [Google Scholar]
- Kotthoff, L.; Thornton, C.; Hoos, H.H.; Hutter, F.; Leyton-Brown, K. Auto-WEKA 2.0: Automatic model selection and hyperparameter optimization in WEKA. J. Mach. Learn. Res. 2017, 18, 1–5. [Google Scholar]
- Newland, J.G.; Stach, L.M.; De Lurgio, S.A.; Hedican, E.; Yu, D.; Prasad, P.A.; Jackson, M.A.; Myers, A.L.; Zaoutis, T.E. Impact of a Prospective-Audit-With-Feedback Antimicrobial Stewardship Program at a Children’s Hospital. J. Pediatric Infect. Dis. Soc. 2012, 1, 179–186. [Google Scholar] [CrossRef]
- Newman, R.E.; Hedican, E.B.; Herigon, J.C.; Williams, D.D.; Williams, A.R.; Jason, G. Newland. Impact of a Guideline on Management of Children Hospitalized With Community-Acquired Pneumonia. Pediatrics 2012, 129, e597–e604. [Google Scholar] [CrossRef]
- Di Pentima, M.C.; Chan, S. Impact of Antimicrobial Stewardship Program on Vancomycin Use in a Pediatric Teaching Hospital. Pediatr. Infect. Dis. J. 2010, 29, 707–711. [Google Scholar] [CrossRef]
- Kreitmeyr, K.; von Both, U.; Pecar, A.; Borde, J.P.; Mikolajczyk, R.; Huebner, J. Pediatric antibiotic stewardship: Successful interventions to reduce broad-spectrum antibiotic use on general pediatric wards. Infection 2017, 45, 493–504. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: All data used in this study are available as supplemental data. |




{kind=link}
{kind=link}
{kind=link}
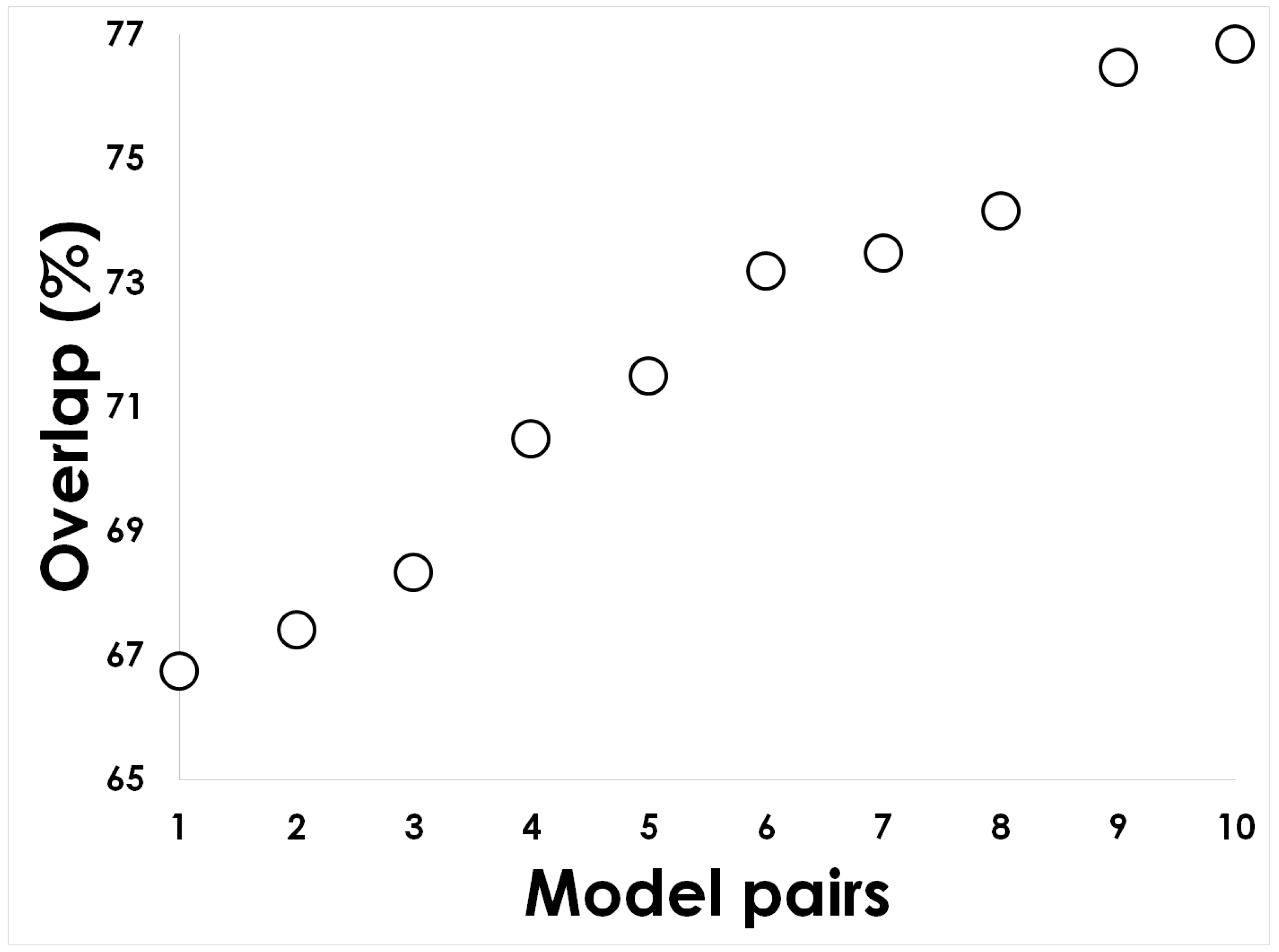{kind=link}
| Training Set | Entries | Description |
|---|---|---|
| TrOnlyPeptides | 11,546 | 8000 antimicrobial peptides, 3546 peptides with no known antimicrobial activity |
| TrNPCC1 | 431 | 164 antimicrobial non-peptides, 267 non-peptides with no known antimicrobial activity |
| TrNPCC2 | 430 | 164 antimicrobial non-peptides, 266 non-peptides with no known antimicrobial activity |
| TrNPCC3 | 430 | 164 antimicrobial non-peptides, 266 non-peptides with no known antimicrobial activity |
| TrNPCC4 | 431 | 164 antimicrobial non-peptides, 267 non-peptides with no known antimicrobial activity |
| TrHeterologous1 | 6204 | 4164 antimicrobial compounds (4000 peptides and 164 non-peptidic compounds), 2040 no antimicrobial compounds (1773 peptides and 267 non-peptidic compounds) |
| TrHeterologous2 | 6203 | 4164 antimicrobial compounds (4000 peptides and 164 non-peptidic compounds), 2039 no antimicrobial compounds (1773 peptides and 266 non-peptidic compounds) |
| TrHeterologous3 | 6203 | 4164 antimicrobial compounds (4000 peptides and 164 non-peptidic compounds), 2039 no antimicrobial compounds (1773 peptides and 266 non-peptidic compounds) |
| TrHeterologous4 | 6204 | 4164 antimicrobial compounds (4000 peptides and 164 non-peptidic compounds), 2040 no antimicrobial compounds (1773 peptides and 267 non-peptidic compounds) |
| Testing Set | Entries | Description |
|---|---|---|
| TeOnlyPeptides | 861 | 328 antimicrobial and 533 non-antimicrobial non-peptides |
| TeNPCC1 | 430 | 164 antimicrobial non-peptides, 266 non-peptides with no known antimicrobial activity. Same as TrNPCC2. |
| TeNPCC2 | 431 | 164 antimicrobial non-peptides, 267 non-peptides with no known antimicrobial activity. Same as TrNPCC1. |
| TeNPCC3 | 431 | 164 antimicrobial non-peptides, 267 non-peptides with no known antimicrobial activity. Same as TrNPCC4. |
| TeNPCC4 | 430 | 164 antimicrobial non-peptides, 266 non-peptides with no known antimicrobial activity. Same as TrNPCC3. |
| TeHeterologous1 | 430 | Same as TeNPCC1. |
| TeHeterologous2 | 431 | Same as TeNPCC2. |
| TeHeterologous3 | 431 | Same as TeNPCC3. |
| TeHeterologous4 | 430 | Same as TeNPCC4. |
| Predicted Gut Antimicrobial | Predicted No Antimicrobial | |
|---|---|---|
| Pathogenic antimicrobial | 72 | 61 |
| No antimicrobial | 140 | 556 |
| Compound Name | Annotation |
|---|---|
| Amoxicillin | Narrow spectrum |
| Phenoxymethylpenicillin | Narrow spectrum |
| Cephalexin | Narrow spectrum |
| Database | Focused on | Reference |
|---|---|---|
| BACTIBASE | Bacteriocins | [28] |
| Bagel | Bacteriocins | [29] |
| CAMP | General and Patented AMPs | [14] |
| DADP | Anuran AMPs | [30] |
| DAMPD | General AMPs * | [31] |
| DBAASP | General AMPs | [13] |
| Defensins | Defensins | [32] |
| HIPdb | Anti-HIV peptides | [33] |
| LAMP | General and Patented AMPs | [34] |
| MilkAMP | AMPs of dairy origin | [35] |
| PhytAMP | Plant AMPs | [36] |
| PenBase | Penaeidin AMPs | [37] |
| Peptaibol | Peptaibols | [38] |
| RAPD | Recombinant AMPs | [39] |
| AMPer | Eukaryotic AMPs | [40] |
| UniprotKb | General AMPs | [41] |
| YADAMP | General AMPs | [42] |
| AMSDb | Eukaryotic AMPs | [43] |
| APD | General AMPs | [44] |
| AVPdb | Antiviral peptides | [45] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nava Lara, R.A.; Aguilera-Mendoza, L.; Brizuela, C.A.; Peña, A.; Del Rio, G. Heterologous Machine Learning for the Identification of Antimicrobial Activity in Human-Targeted Drugs. Molecules 2019, 24, 1258. https://doi.org/10.3390/molecules24071258
Nava Lara RA, Aguilera-Mendoza L, Brizuela CA, Peña A, Del Rio G. Heterologous Machine Learning for the Identification of Antimicrobial Activity in Human-Targeted Drugs. Molecules. 2019; 24(7):1258. https://doi.org/10.3390/molecules24071258
Chicago/Turabian StyleNava Lara, Rodrigo A., Longendri Aguilera-Mendoza, Carlos A. Brizuela, Antonio Peña, and Gabriel Del Rio. 2019. "Heterologous Machine Learning for the Identification of Antimicrobial Activity in Human-Targeted Drugs" Molecules 24, no. 7: 1258. https://doi.org/10.3390/molecules24071258
APA StyleNava Lara, R. A., Aguilera-Mendoza, L., Brizuela, C. A., Peña, A., & Del Rio, G. (2019). Heterologous Machine Learning for the Identification of Antimicrobial Activity in Human-Targeted Drugs. Molecules, 24(7), 1258. https://doi.org/10.3390/molecules24071258