
Ponatinib Inhibits Proliferation and Induces Apoptosis of Liver Cancer Cells, but Its Efficacy Is Compromised by Its Activation on PDK1/Akt/mTOR Signaling

Abstract
:1. Introduction
2. Results
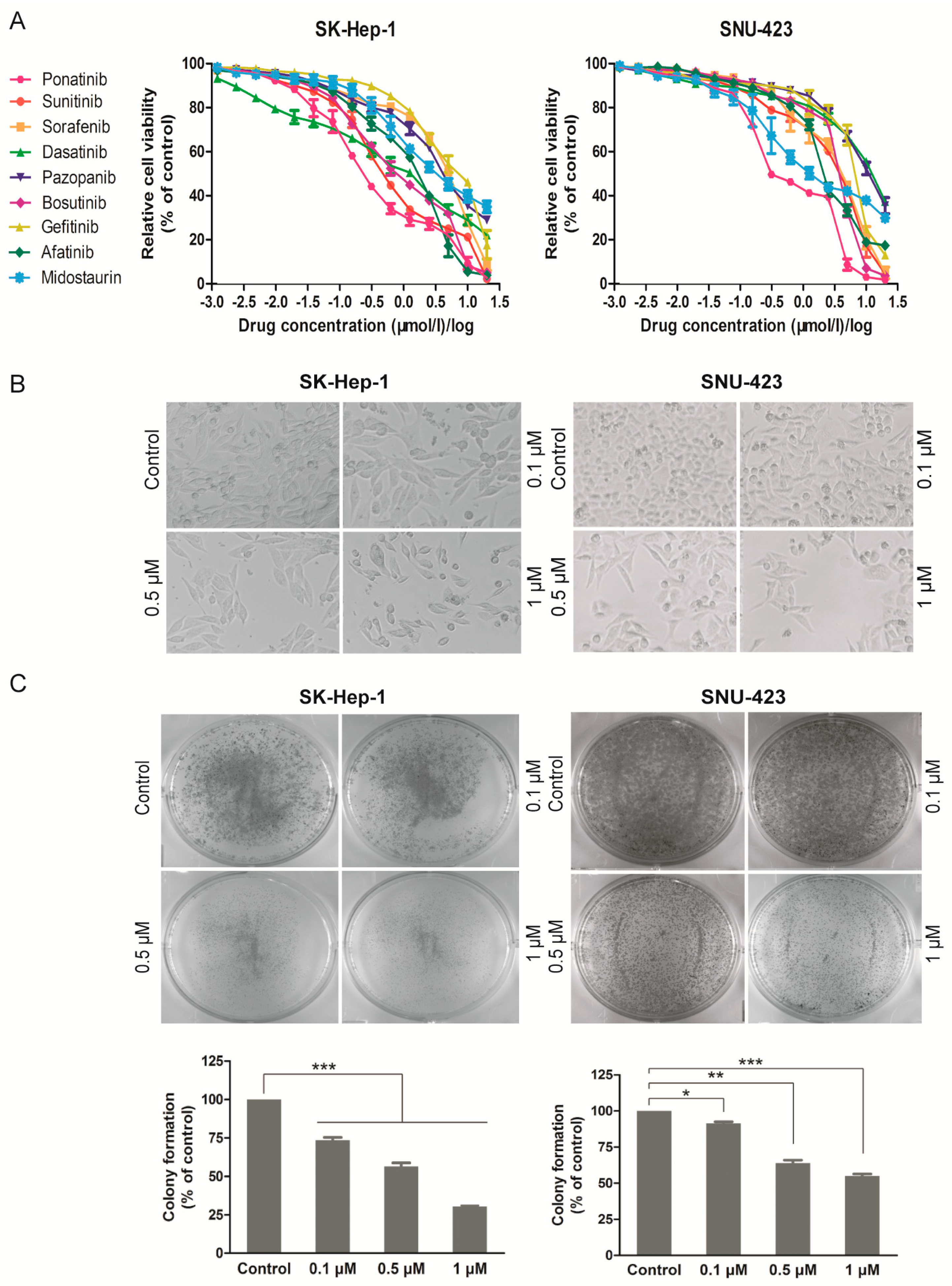
2.1. Ponatinib Potently Inhibits Viability and Proliferation of HCC Cell Lines
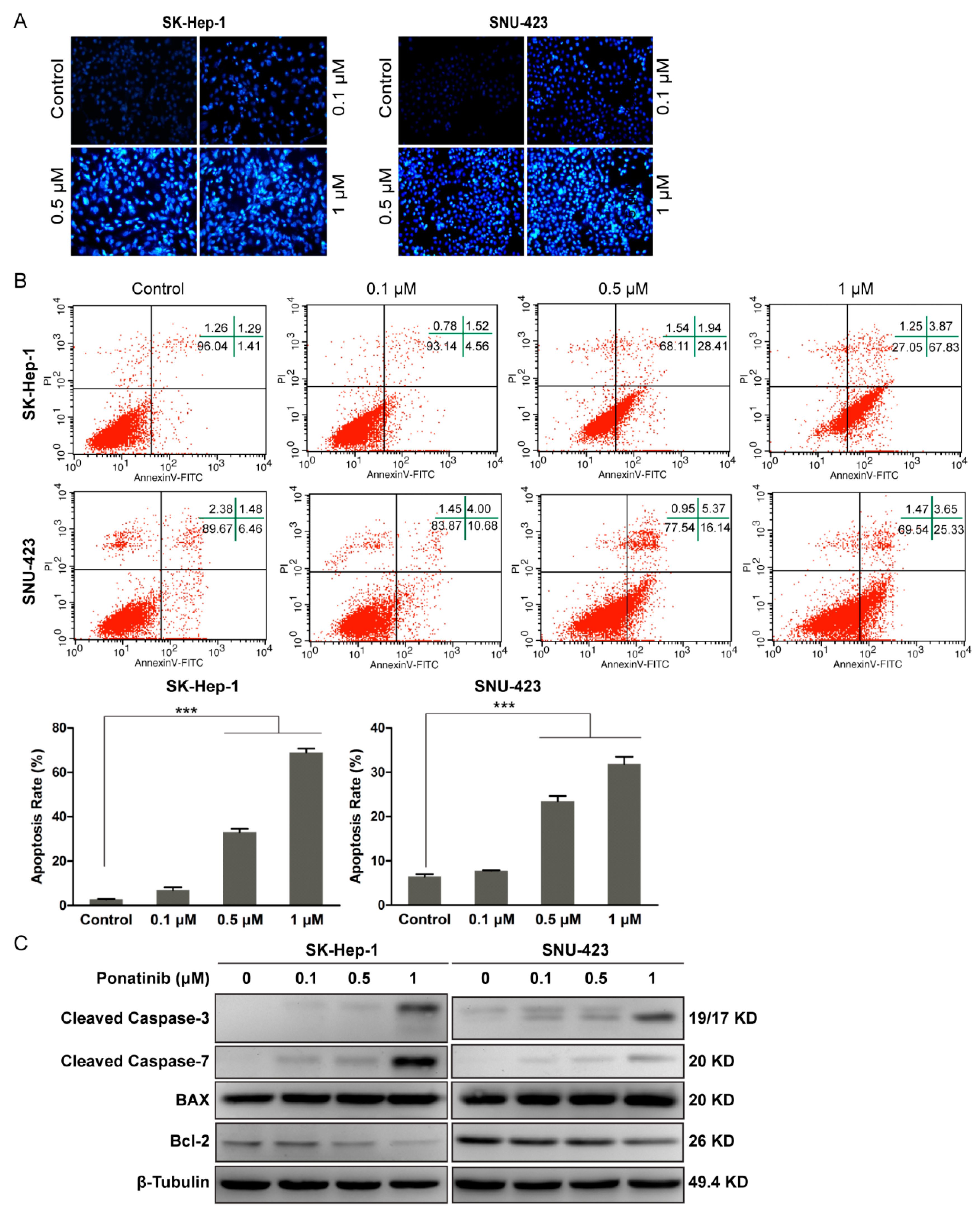
2.2. Ponatinib Induces Apoptosis in HCC Cells
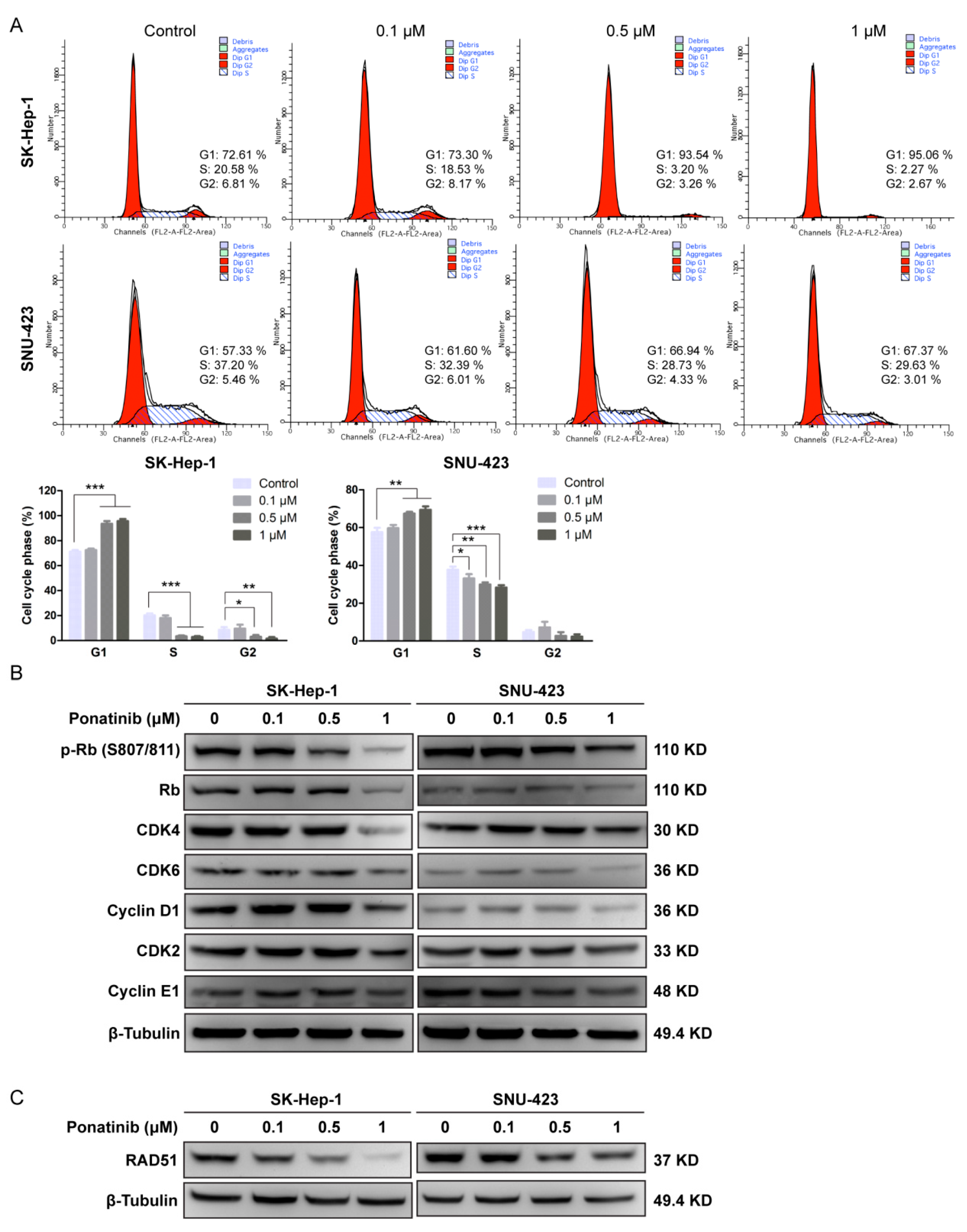
2.3. Ponatinib Causes Cell Cycle G1 Phase Arrest in HCC Cells
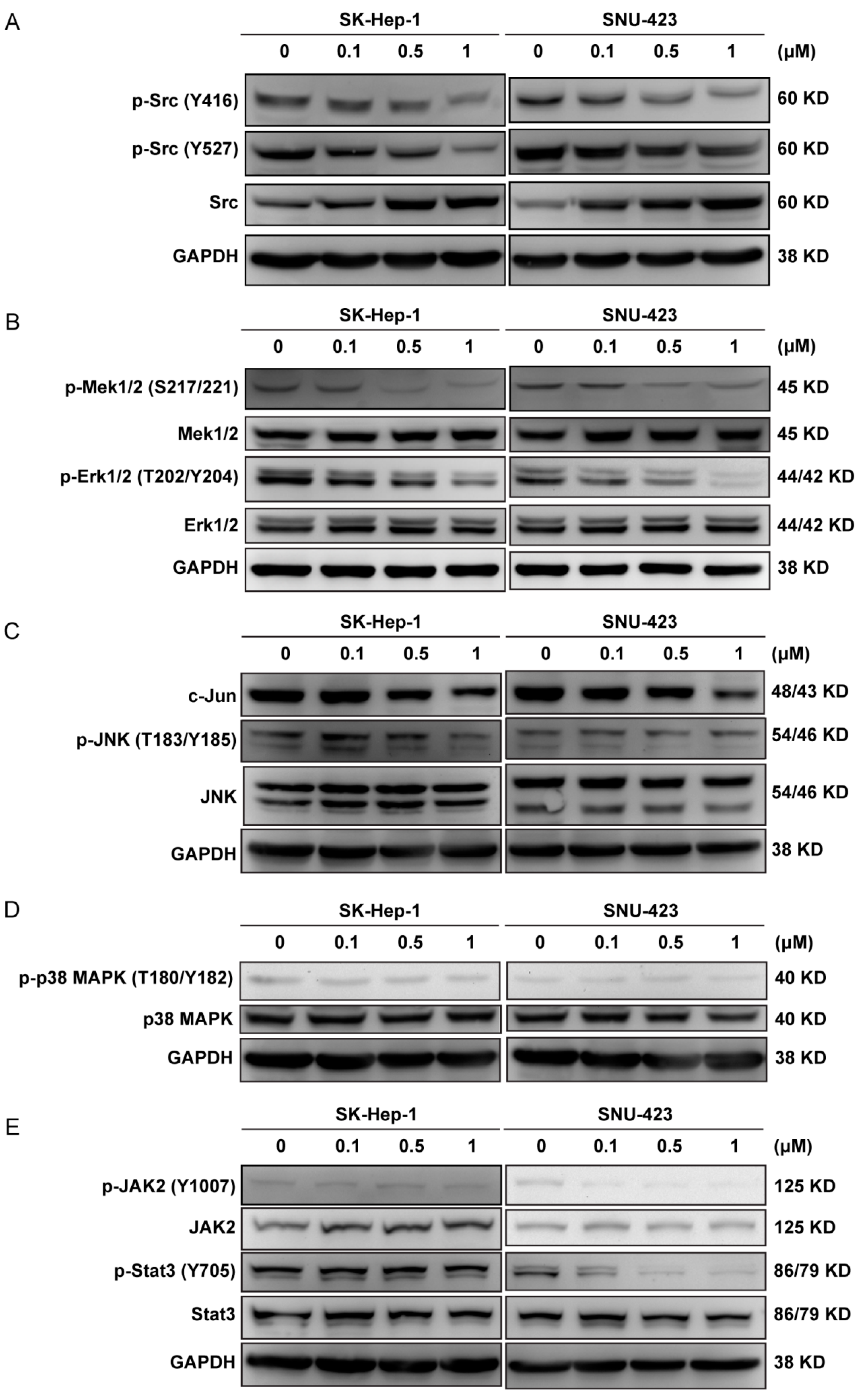
2.4. Ponatinib Interferes with Multiple Cell Signaling Pathways in the HCC Cells
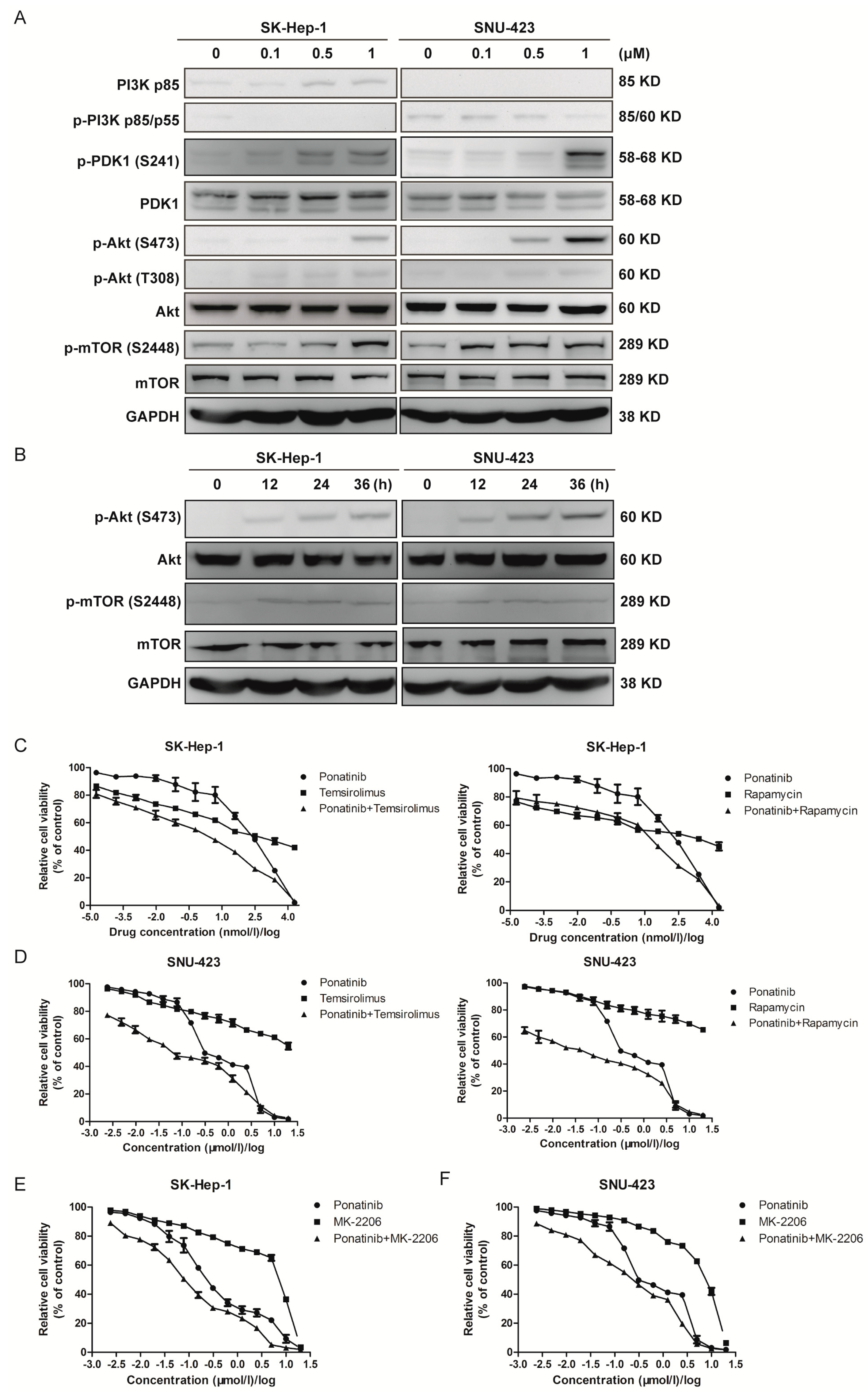
2.5. Ponatinib Activates the Akt/mTOR Pathway, and Blocking Akt/mTOR Signaling Sensitizes HCC Cells to Ponatinib Inhibition
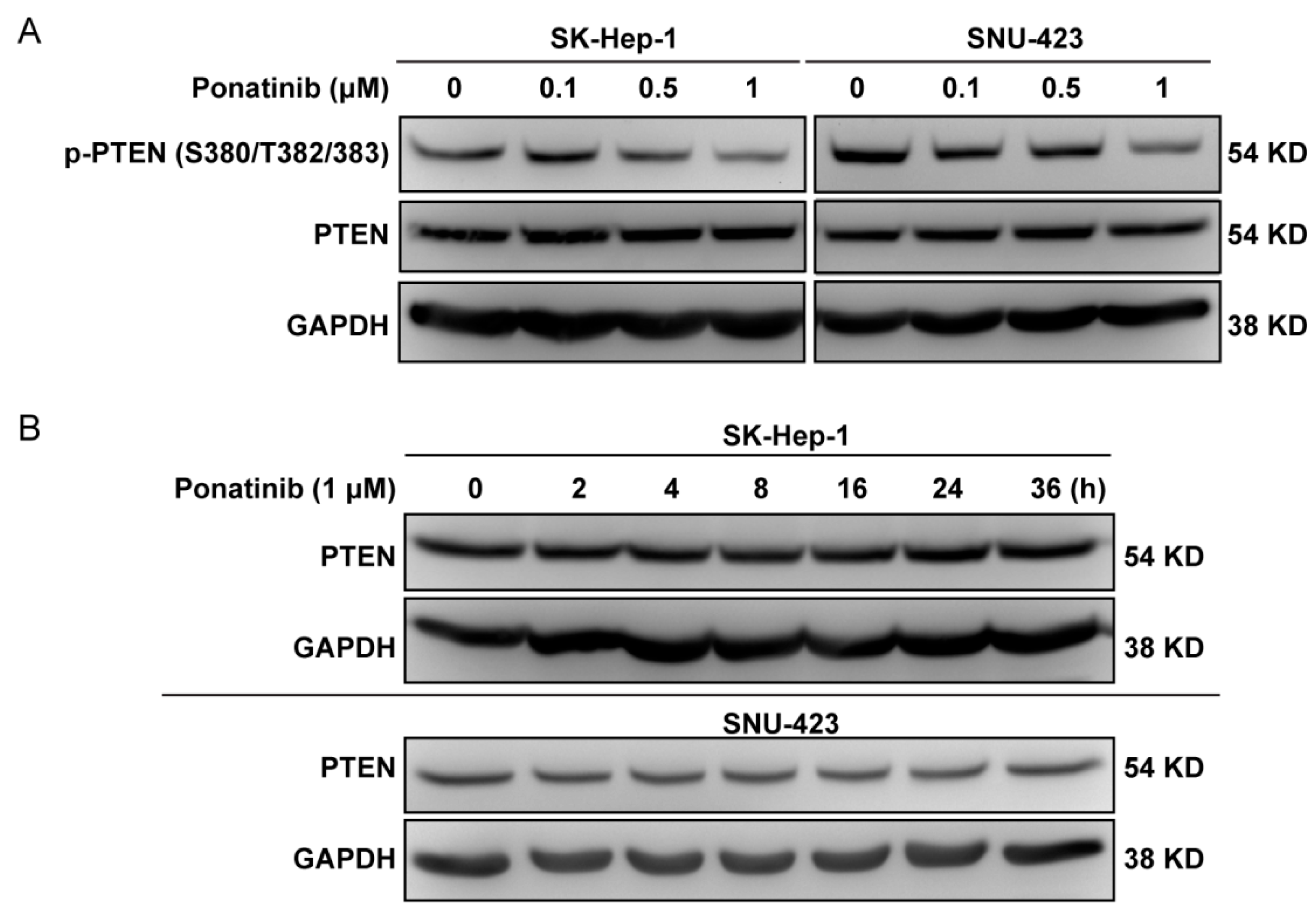
2.6. Ponatinib Did Not Affect PTEN Expression Level
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Materials and Antibodies
4.3. Cell Proliferation Assay
4.4. Colony Formation Assay
4.5. In-Vitro Drug Combination Study
4.6. Apoptosis Assay
4.7. Cell Cycle Analysis
4.8. Morphological Analysis by Hoechst Staining
4.9. Western Blot Analysis
4.10. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Fitzmaurice, C.; Allen, C.; Barber, R.M.; Barregard, L.; Bhutta, Z.A.; Brenner, H.; Dicker, D.J.; Chimed-Orchir, O.; Dandona, R.; Dandona, L.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-years for 32 Cancer Groups, 1990 to 2015. JAMA Oncol. 2017, 3, 524. [Google Scholar]
- Thillai, K.; Ross, P.; Sarker, D. Molecularly targeted therapy for advanced hepatocellular carcinoma—A drug development crisis? World J. Gastrointest. Oncol. 2016, 8, 173–185. [Google Scholar] [CrossRef]
- Bodzin, A.S. Hepatocellular carcinoma: Advances in diagnosis, management, and long term outcome. World J. Hepatol. 2015, 7, 1157. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Kimura, T.; Kato, Y.; Ozawa, Y.; Kodama, K.; Ito, J.; Ichikawa, K.; Yamada, K.; Hori, Y.; Tabata, K.; Takase, K.; et al. Immunomodulatory activity of lenvatinib contributes to antitumor activity in the Hepa1-6 hepatocellular carcinoma model. Cancer Sci. 2018, 109, 3993–4002. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Tak, W.Y.; Gasbarrini, A.; Santoro, A.; Colombo, M.; Lim, H.Y.; Mazzaferro, V.; Wiest, R.; Reig, M.; Wagner, A.; et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: Multicentre, open-label, phase II safety study. Eur. J. Cancer 2013, 49, 3412–3419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, A.; Hernandez-Gea, V.; Llovet, J.M. Medical therapies for hepatocellular carcinoma: A critical view of the evidence. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Levitzki, A.; Klein, S. Signal transduction therapy of cancer. Mol. Aspects Med. 2010, 31, 287–329. [Google Scholar] [CrossRef]
- Colombino, M.; Sperlongano, P.; Izzo, F.; Tatangelo, F.; Botti, G.; Lombardi, A.; Accardo, M.; Tarantino, L.; Sordelli, I.; Agresti, M.; et al. BRAF and PIK3CA genes are somatically mutated in hepatocellular carcinoma among patients from South Italy. Cell Death Dis. 2012, 3, e259. [Google Scholar] [CrossRef]
- Bromberg, J. Stat proteins and oncogenesis. J. Clin. Investig. 2002, 109, 1139–1142. [Google Scholar] [CrossRef] [Green Version]
- Pilati, C.; Amessou, M.; Bihl, M.P.; Balabaud, C.; Nhieu, J.T.; Paradis, V.; Nault, J.C.; Izard, T.; Bioulac-Sage, P.; Couchy, G.; et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J. Exp. Med. 2011, 208, 1359–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greten, T.F.; Korangy, F.; Manns, M.P.; Malek, N.P. Molecular therapy for the treatment of hepatocellular carcinoma. Br. J. Cancer 2009, 100, 19–23. [Google Scholar] [CrossRef]
- Lee, Y.H.; Seo, D.; Choi, K.J.; Andersen, J.B.; Won, M.A.; Kitade, M.; Gomez-Quiroz, L.E.; Judge, A.D.; Marquardt, J.U.; Raggi, C.; et al. Antitumor effects in hepatocarcinoma of isoform-selective inhibition of HDAC2. Cancer Res. 2014, 74, 4752–4761. [Google Scholar] [CrossRef]
- Chuma, M.; Terashita, K.; Sakamoto, N. New molecularly targeted therapies against advanced hepatocellular carcinoma: From molecular pathogenesis to clinical trials and future directions. Hepatol. Res. 2015, 45, E1–E11. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Shah, A.N.; Gallick, G.E. Src continues aging: Current and future clinical directions. Clin. Cancer Res. 2007, 13, 7232–7236. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Kawakatsu, H.; Takeda, T.; Sakon, M.; Nagano, H.; Sakai, T.; Miyoshi, E.; Noda, K.; Tsujimoto, M.; Wakasa, K.; et al. Activation of c-Src gene product in hepatocellular carcinoma is highly correlated with the indices of early stage phenotype. J. Hepatol. 2001, 35, 68–73. [Google Scholar] [CrossRef]
- O’Hare, T.; Shakespeare, W.C.; Zhu, X.; Eide, C.A.; Rivera, V.M.; Wang, F.A.; Adrian, L.T.; Zhou, T.; Huang, W.S.; Xu, Q.; et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell 2009, 16, 401–412. [Google Scholar] [CrossRef]
- Jin, B.; Ding, K.; Pan, J. Ponatinib induces apoptosis in imatinib-resistant human mast cells by dephosphorylating mutant D816V KIT and silencing beta-catenin signaling. Mol. Cancer Ther. 2014, 13, 1217–1230. [Google Scholar] [CrossRef]
- Gozgit, J.M.; Wong, M.J.; Wardwell, S.; Tyner, J.W.; Loriaux, M.M.; Mohemmad, Q.K.; Narasimhan, N.I.; Shakespeare, W.C.; Wang, F.; Druker, B.J. Potent Activity of Ponatinib (AP24534) in Models of FLT3-Driven Acute Myeloid Leukemia and Other Hematologic Malignancies. Mol. Cancer Ther. 2011, 10, 1028–1035. [Google Scholar] [CrossRef] [Green Version]
- Petrilli, A.M.; Garcia, J.; Bott, M.; Klingeman Plati, S.; Dinh, C.T.; Bracho, O.R.; Yan, D.; Zou, B.; Mittal, R.; Telischi, F.F.; et al. Ponatinib promotes a G1 cell-cycle arrest of merlin/NF2-deficient human schwann cells. Oncotarget 2017, 8, 31666–31681. [Google Scholar] [CrossRef] [Green Version]
- Lierman, E.; Smits, S.; Cools, J.; Dewaele, B.; Debiec-Rychter, M.; Vandenberghe, P. Ponatinib is active against imatinib-resistant mutants of FIP1L1-PDGFRA and KIT and against FGFR1-derived fusion kinases. Leukemia 2012, 26, 1693–1695. [Google Scholar] [CrossRef]
- Gozgit, J.M.; Wong, M.J.; Moran, L.; Wardwell, S.; Mohemmad, Q.K.; Narasimhan, N.I.; Shakespeare, W.C.; Wang, F.; Clackson, T.; Rivera, V.M. Ponatinib (AP24534), a multitargeted pan-FGFR inhibitor with activity in multiple FGFR-amplified or mutated cancer models. Mol. Cancer Ther. 2012, 11, 690–699. [Google Scholar] [CrossRef]
- Shao, W.; Li, S.; Li, L.; Lin, K.; Liu, X.; Wang, H.; Wang, H.; Wang, D. Chemical genomics reveals inhibition of breast cancer lung metastasis by Ponatinib via c-Jun. Protein Cell 2018. [Google Scholar] [CrossRef]
- Sidarovich, V.; De Mariano, M.; Aveic, S.; Pancher, M.; Adami, V.; Gatto, P.; Pizzini, S.; Pasini, L.; Croce, M.; Parodi, F.; et al. A High-Content Screening of Anticancer Compounds Suggests the Multiple Tyrosine Kinase Inhibitor Ponatinib for Repurposing in Neuroblastoma Therapy. Mol. Cancer Ther. 2018, 17, 1405–1415. [Google Scholar] [CrossRef]
- De Falco, V.; Buonocore, P.; Muthu, M.; Torregrossa, L.; Basolo, F.; Billaud, M.; Gozgit, J.M.; Carlomagno, F.; Santoro, M. Ponatinib (AP24534) is a novel potent inhibitor of oncogenic RET mutants associated with thyroid cancer. J. Clin. Endocrinol. Metab. 2013, 98, E811–E819. [Google Scholar] [CrossRef]
- Chase, A.; Bryant, C.; Score, J.; Cross, N.C. Ponatinib as targeted therapy for FGFR1 fusions associated with the 8p11 myeloproliferative syndrome. Haematologica 2013, 98, 103–106. [Google Scholar] [CrossRef]
- Mingard, C.; Paech, F.; Bouitbir, J.; Krahenbuhl, S. Mechanisms of toxicity associated with six tyrosine kinase inhibitors in human hepatocyte cell lines. J. Appl. Toxicol. 2018, 38, 418–431. [Google Scholar] [CrossRef]
- Kuribayashi, K.; Mayes, P.A.; El-Deiry, W.S. What are caspases 3 and 7 doing upstream of the mitochondria? Cancer Biol. Ther. 2006, 5, 763–765. [Google Scholar] [CrossRef] [Green Version]
- Lakhani, S.A.; Masud, A.; Kuida, K.; Porter, G.A.; Booth, C.J.; Mehal, W.Z.; Inayat, I.; Flavell, R.A. Caspases 3 and 7: Key mediators of mitochondrial events of apoptosis. Science 2006, 311, 847–851. [Google Scholar] [CrossRef]
- Shao, J.; Xu, Z.; Peng, X.; Chen, M.; Zhu, Y.; Xu, L.; Zhu, H.; Yang, B.; Luo, P.; He, Q. Gefitinib Synergizes with Irinotecan to Suppress Hepatocellular Carcinoma via Antagonizing Rad51-Mediated DNA-Repair. PLoS ONE 2016, 11, e0146968. [Google Scholar] [CrossRef]
- Ji, J.; Chen, W.; Lian, W.; Chen, R.; Yang, J.; Zhang, Q.; Weng, Q.; Khan, Z.; Hu, J.; Chen, X.; et al. (S)-crizotinib reduces gastric cancer growth through oxidative DNA damage and triggers pro-survival akt signal. Cell Death Dis. 2018, 9, 660. [Google Scholar] [CrossRef]
- Haaf, T.; Golub, E.I.; Reddy, G.; Radding, C.M.; Ward, D.C. Nuclear foci of mammalian Rad51 recombination protein in somatic cells after DNA damage and its localization in synaptonemal complexes. Proc. Natl. Acad. Sci. USA 1995, 92, 2298–2302. [Google Scholar] [CrossRef]
- Tomicic, M.T.; Kaina, B. Topoisomerase degradation, DSB repair, p53 and IAPs in cancer cell resistance to camptothecin-like topoisomerase I inhibitors. Biochim. Biophys. Acta 2013, 1835, 11–27. [Google Scholar] [CrossRef]
- Sun, G.; Sharma, A.K.; Budde, R.J. Autophosphorylation of Src and Yes blocks their inactivation by Csk phosphorylation. Oncogene 1998, 17, 1587–1595. [Google Scholar] [CrossRef] [Green Version]
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harbor Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef]
- Coppo, P.; Flamant, S.; De Mas, V.; Jarrier, P.; Guillier, M.; Bonnet, M.L.; Lacout, C.; Guilhot, F.; Vainchenker, W.; Turhan, A.G. BCR-ABL activates STAT3 via JAK and MEK pathways in human cells. Br. J. Haematol. 2006, 134, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Mu, C.F.; Xiong, Y.; Bai, X.; Sheng, Y.J.; Cui, J. Codelivery of Ponatinib and SAR302503 by Active Bone-Targeted Polymeric Micelles for the Treatment of Therapy-Resistant Chronic Myeloid Leukemia. Mol. Pharm. 2017, 14, 274–283. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Wang, C.; Ho, C.; Ladu, S.; Lee, S.A.; Mattu, S.; Destefanis, G.; Delogu, S.; Zimmermann, A.; Ericsson, J.; et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011, 140, 1071–1083. [Google Scholar] [CrossRef]
- Jiang, S.; Wang, Q.; Feng, M.; Li, J.; Guan, Z.; An, D.; Dong, M.; Peng, Y.; Kuerban, K.; Ye, L. C2-ceramide enhances sorafenib-induced caspase-dependent apoptosis via PI3K/AKT/mTOR and Erk signaling pathways in HCC cells. Appl. Microbiol. Biotechnol. 2017, 101, 1535–1546. [Google Scholar] [CrossRef]
- Rodrik-Outmezguine, V.S.; Chandarlapaty, S.; Pagano, N.C.; Poulikakos, P.I.; Scaltriti, M.; Moskatel, E.; Baselga, J.; Guichard, S.; Rosen, N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011, 1, 248–259. [Google Scholar] [CrossRef] [Green Version]
- Sahu, N.; Chan, E.; Chu, F.; Pham, T.; Koeppen, H.; Forrest, W.; Merchant, M.; Settleman, J. Cotargeting of MEK and PDGFR/STAT3 Pathways to Treat Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2017, 16, 1729–1738. [Google Scholar] [CrossRef]
- Bostner, J.; Karlsson, E.; Pandiyan, M.J.; Westman, H.; Skoog, L.; Fornander, T.; Nordenskjold, B.; Stal, O. Activation of Akt, mTOR, and the estrogen receptor as a signature to predict tamoxifen treatment benefit. Breast Cancer Res. Treat. 2013, 137, 397–406. [Google Scholar] [CrossRef]
- Yim, E.-K.; Peng, G.; Dai, H.; Hu, R.; Li, K.; Lu, Y.; Mills, G.B.; Meric-Bernstam, F.; Hennessy, B.T.; Craven, R.J.; et al. Rak Functions as a Tumor Suppressor by Regulating PTEN Protein Stability and Function. Cancer Cell 2009, 15, 304–314. [Google Scholar] [CrossRef]
- Kim, D.H.; Kwak, Y.; Kim, N.D.; Sim, T. Antitumor effects and molecular mechanisms of ponatinib on endometrial cancer cells harboring activating FGFR2 mutations. Cancer Biol. Ther. 2016, 17, 65–78. [Google Scholar] [CrossRef]
- Zirm, E.; Spies-Weisshart, B.; Heidel, F.; Schnetzke, U.; Bohmer, F.D.; Hochhaus, A.; Fischer, T.; Scholl, S. Ponatinib may overcome resistance of FLT3-ITD harbouring additional point mutations, notably the previously refractory F691I mutation. Br. J. Haematol. 2012, 157, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Baro, M.; de Llobet, L.I.; Figueras, A.; Skvortsova, I.; Mesia, R.; Balart, J. Dasatinib worsens the effect of cetuximab in combination with fractionated radiotherapy in FaDu- and A431-derived xenografted tumours. Br. J. Cancer 2014, 111, 1310–1318. [Google Scholar] [CrossRef] [Green Version]
- Rhim, J.H.; Luo, X.; Gao, D.; Xu, X.; Zhou, T.; Li, F.; Wang, P.; Wong, S.T.; Xia, X. Cell type-dependent Erk-Akt pathway crosstalk regulates the proliferation of fetal neural progenitor cells. Sci. Rep. 2016, 6, 26547. [Google Scholar] [CrossRef] [Green Version]
- Paech, F.; Mingard, C.; Grunig, D.; Abegg, V.F.; Bouitbir, J.; Krahenbuhl, S. Mechanisms of mitochondrial toxicity of the kinase inhibitors ponatinib, regorafenib and sorafenib in human hepatic HepG2 cells. Toxicology 2018, 395, 34–44. [Google Scholar] [CrossRef]
- Hasinoff, B.B.; Patel, D.; Wu, X. The Myocyte-Damaging Effects of the BCR-ABL1-Targeted Tyrosine Kinase Inhibitors Increase with Potency and Decrease with Specificity. Cardiovasc. Toxicol. 2017, 17, 297–306. [Google Scholar] [CrossRef]
- Moslehi, J.J.; Deininger, M. Tyrosine Kinase Inhibitor-Associated Cardiovascular Toxicity in Chronic Myeloid Leukemia. J. Clin. Oncol. 2015, 33, 4210–4218. [Google Scholar] [CrossRef]
- Spraggs, C.F.; Xu, C.F.; Hunt, C.M. Genetic characterization to improve interpretation and clinical management of hepatotoxicity caused by tyrosine kinase inhibitors. Pharmacogenomics 2013, 14, 541–554. [Google Scholar] [CrossRef]
- Cortes, J.E.; Apperley, J.F.; DeAngelo, D.J.; Deininger, M.W.; Kota, V.K.; Rousselot, P.; Gambacorti-Passerini, C. Management of adverse events associated with bosutinib treatment of chronic-phase chronic myeloid leukemia: Expert panel review. J. Hematol. Oncol. 2018, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J. Hematol. Oncol. 2018, 11, 84. [Google Scholar] [CrossRef]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds used in this study are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drugs | Molecular Weight (g/mol) | Target PTKs |
|---|---|---|
| Ponatinib | 532.56 | BCR-ABL, VEGFR, PDGFR, FGFR, EpH kinase, SRC-family protein-tyrosine kinases, and Kit, RET, TIE2, Flt-3 kinase inhibitor |
| Sunitinib | 398.47 | PDGFRα, PDGFRβ, VEGFR1, VEGFR2, VEGFR3, c-KIT, Flt-3, CSF-1R, RET tyrosine kinase inhibitor |
| Sorafenib | 464.83 | KIT, Flt-3, VEGFR-2, VEGFR-3, PDGFR-β tyrosine kinase, C-Raf, B-Raf and mutant B-Raf kinase inhibitor. Unique in targeting the Raf/Mek/Erk pathway. |
| Dasatinib | 488.01 | SRC family (SRC, LCK, YES, FYN) tyrosine kinase, BCR-ABL, c-Kit, EpHA2, PDGFRβ kinase inhibitor |
| Pazopanib | 437.52 | VEGFR1, VEGFR2, VEGFR3, PDGFRα, PDGFRβ, c-kit tyrosine kinase inhibitor |
| Bosutinib | 530.45 | Src-family (Src, Lyn, Hck) tyrosine kinases, BCR-ABL kinase inhibitor |
| Gefitinib | 446.90 | EGFR/Her1/ErbB-1 tyrosine kinase inhibitor |
| Afatinib | 485.94 | EGFR (ErbB1)/HER2 (ErbB2)/ HER4 (ErbB4) tyrosine kinase inhibitor |
| Midostaurin | 570.64 | PKCα/β/γ, Syk, Flk-1, Akt, PKA, c-Kit, c-Fgr, c-Src, Flt-3, PDFRβ and VEGFR1/2 tyrosine kinase inhibitor |
| SK-Hep-1 | SNU-423 | |
|---|---|---|
| Ponatinib (μmol/L) | 0.288 ± 0.044 | 0.553 ± 0.041 |
| Sunitinib (μmol/L) | 0.568 ± 0.070 | 2.720 ± 0.036 |
| Sorafenib (μmol/L) | 3.896 ± 0.037 | 3.125 ± 0.034 |
| Dasatinib (μmol/L) | 0.902 ± 0.107 | 9.758 ± 0.044 |
| Pazopanib(μmol/L) | 3.951 ± 0.041 | 9.777 ± 0.030 |
| Bosutinib (μmol/L) | 0.840 ± 0.042 | 3.062 ± 0.041 |
| Gefitinib (μmol/L) | 5.557 ± 0.029 | 5.752 ± 0.048 |
| Afatinib (μmol/L) | 1.218 ± 0.031 | 2.400 ± 0.024 |
| Midostaurin (μmol/L) | 3.363 ± 0.053 | 1.640 ± 0.070 |
| Parameter | SK-Hep-1 | SNU-423 |
|---|---|---|
| IC50-ponatinib (μM) | 0.288 ± 0.044 | 0.553 ± 0.041 |
| IC50-temsirolimus (μM) | 0.341 ± 0.197 | 11.407 ± 0.104 |
| IC50-rapamycin (μM) | 1.718 ± 0.282 | 22.665 ± 0.097 |
| IC50-ponatinib + temsirolimus (μM) | 0.002 ± 0.001 | 0.066 ± 0.009 |
| IC50-ponatinib + rapamycin (μM) | 0.019 ± 0.009 | 0.042 ± 0.007 |
| CI50-ponatinib + temsirolimus | 0.013 | 0.122 |
| CI50-ponatinib + rapamycin | 0.077 | 0.078 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, C.; Mu, X.; Wang, X.; Zhang, C.; Zhang, L.; Yu, B.; Sun, G. Ponatinib Inhibits Proliferation and Induces Apoptosis of Liver Cancer Cells, but Its Efficacy Is Compromised by Its Activation on PDK1/Akt/mTOR Signaling. Molecules 2019, 24, 1363. https://doi.org/10.3390/molecules24071363
Liu C, Mu X, Wang X, Zhang C, Zhang L, Yu B, Sun G. Ponatinib Inhibits Proliferation and Induces Apoptosis of Liver Cancer Cells, but Its Efficacy Is Compromised by Its Activation on PDK1/Akt/mTOR Signaling. Molecules. 2019; 24(7):1363. https://doi.org/10.3390/molecules24071363
Chicago/Turabian StyleLiu, Chang, Xiuli Mu, Xuan Wang, Chan Zhang, Lina Zhang, Baofeng Yu, and Gongqin Sun. 2019. "Ponatinib Inhibits Proliferation and Induces Apoptosis of Liver Cancer Cells, but Its Efficacy Is Compromised by Its Activation on PDK1/Akt/mTOR Signaling" Molecules 24, no. 7: 1363. https://doi.org/10.3390/molecules24071363
APA StyleLiu, C., Mu, X., Wang, X., Zhang, C., Zhang, L., Yu, B., & Sun, G. (2019). Ponatinib Inhibits Proliferation and Induces Apoptosis of Liver Cancer Cells, but Its Efficacy Is Compromised by Its Activation on PDK1/Akt/mTOR Signaling. Molecules, 24(7), 1363. https://doi.org/10.3390/molecules24071363