Regulation of the Nrf2 Pathway by Glycogen Synthase Kinase-3β in MPP+-Induced Cell Damage

 ,
,  , ,
, , 
Abstract
:
1. Introduction
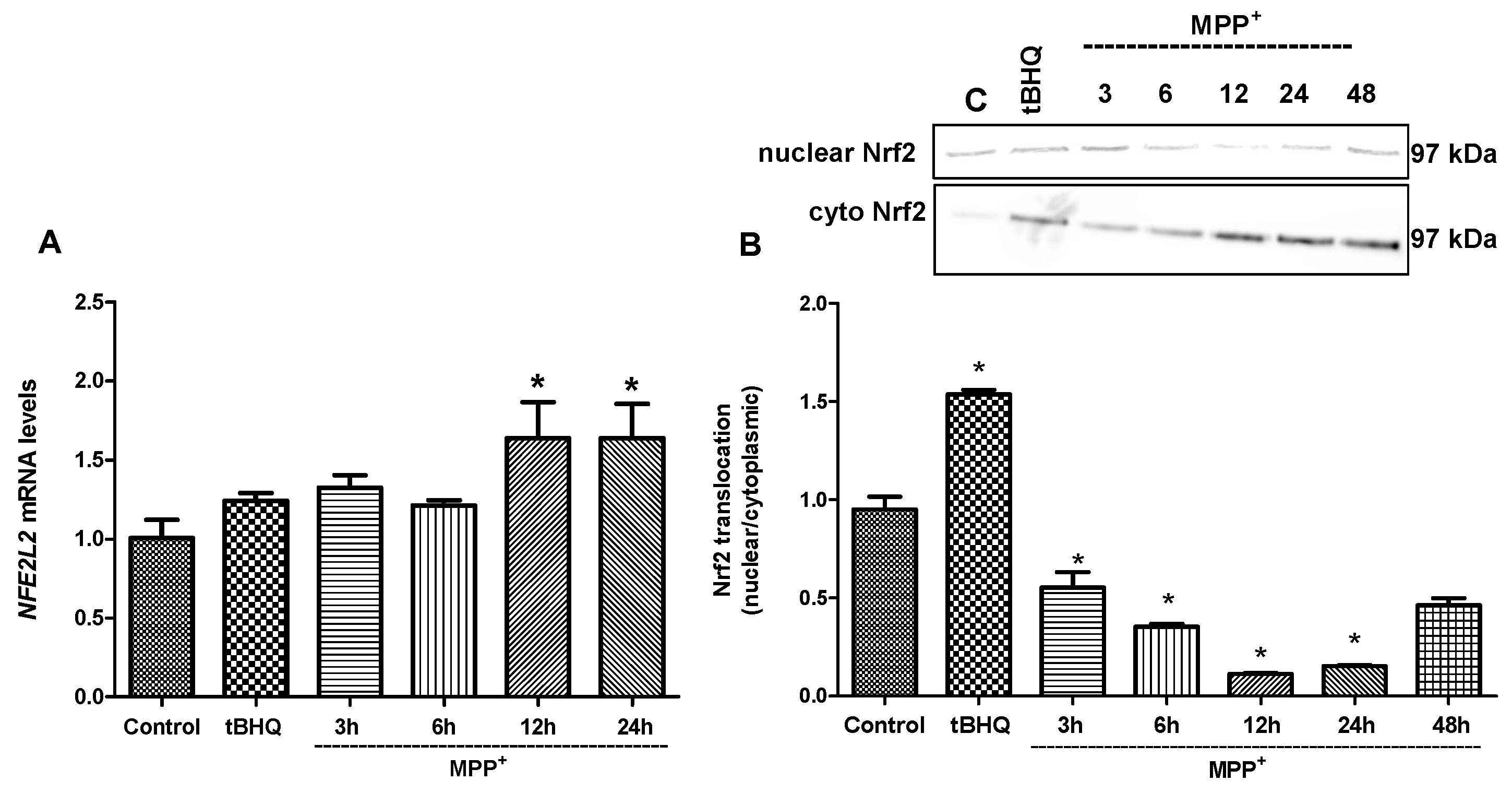
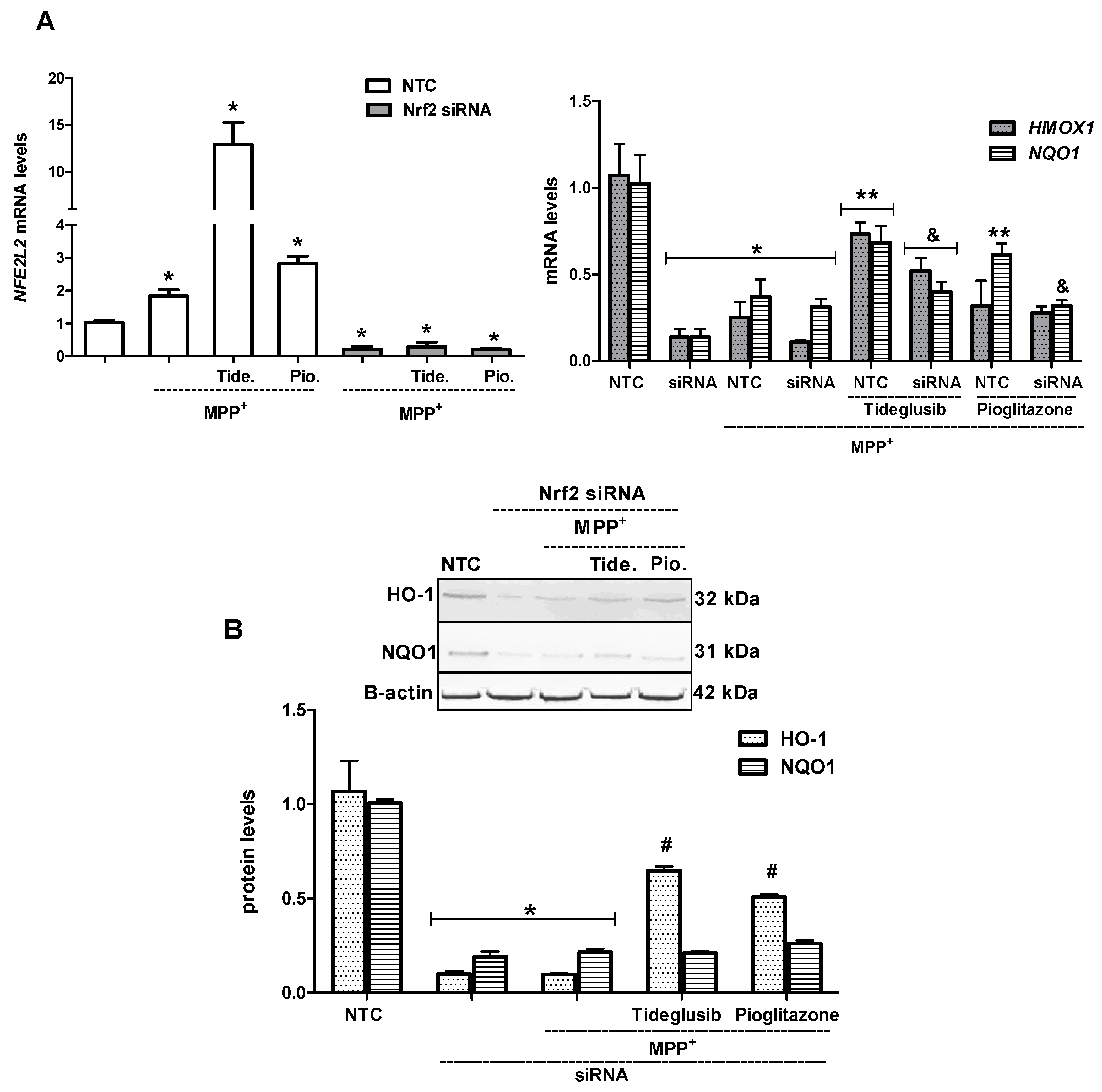
2. Results
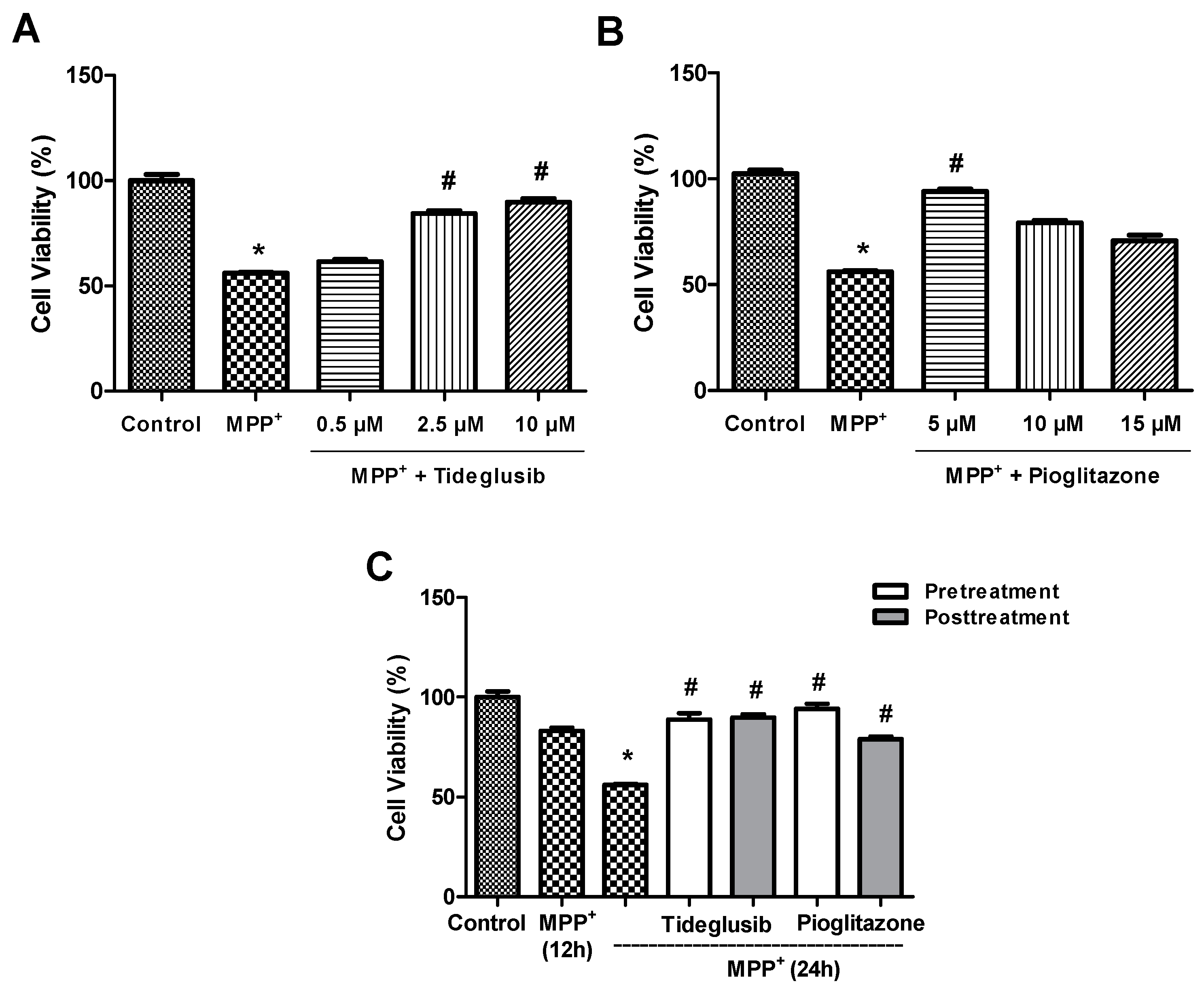
2.1. Tideglusib and Pioglitazone Significantly Increased Cell Viability in MPP+-Treated Cells.
2.2. ROS Production and MMP Were Altered by Tideglusib or Pioglitazone Treatment in MPP+-Treated Cells
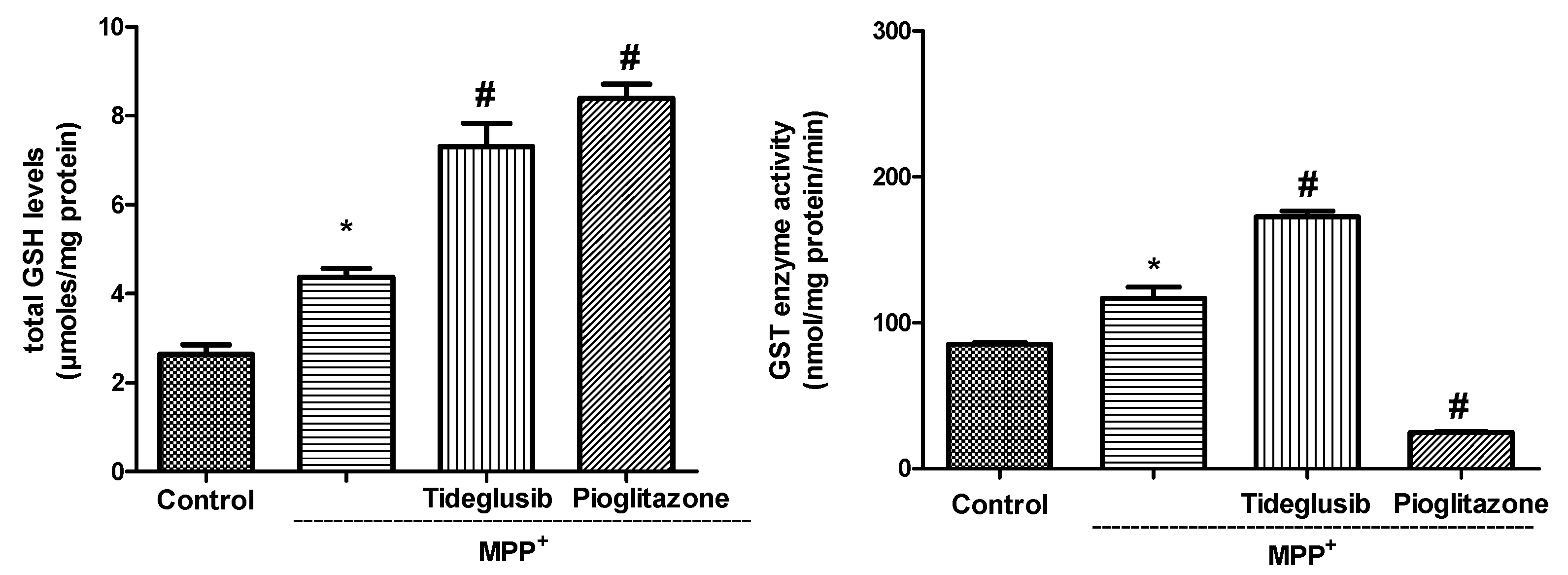
2.3. Total GSH Levels and GST Enzyme Activity Were Regulated by Tideglusib in MPP+-Treated Cells
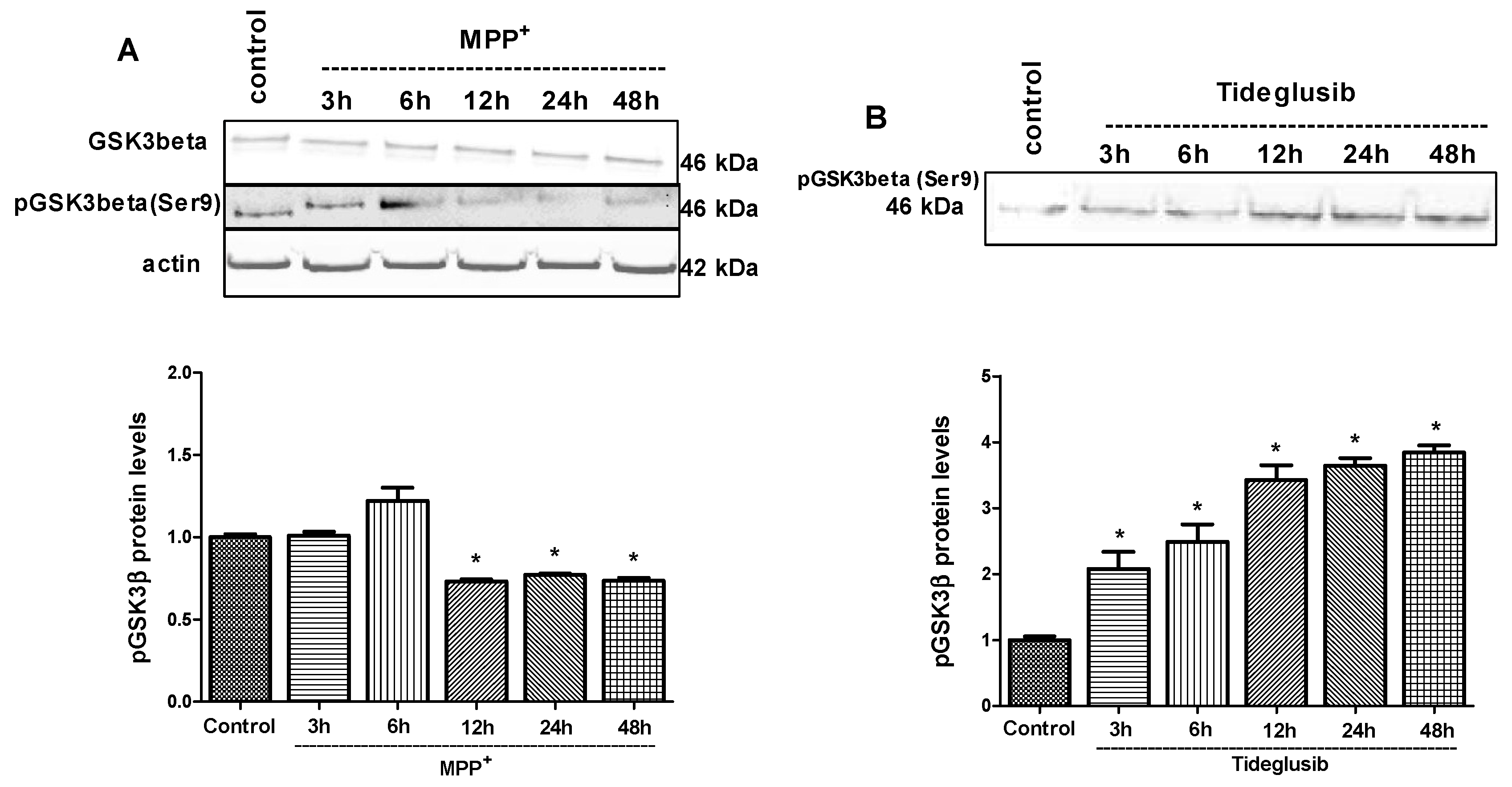
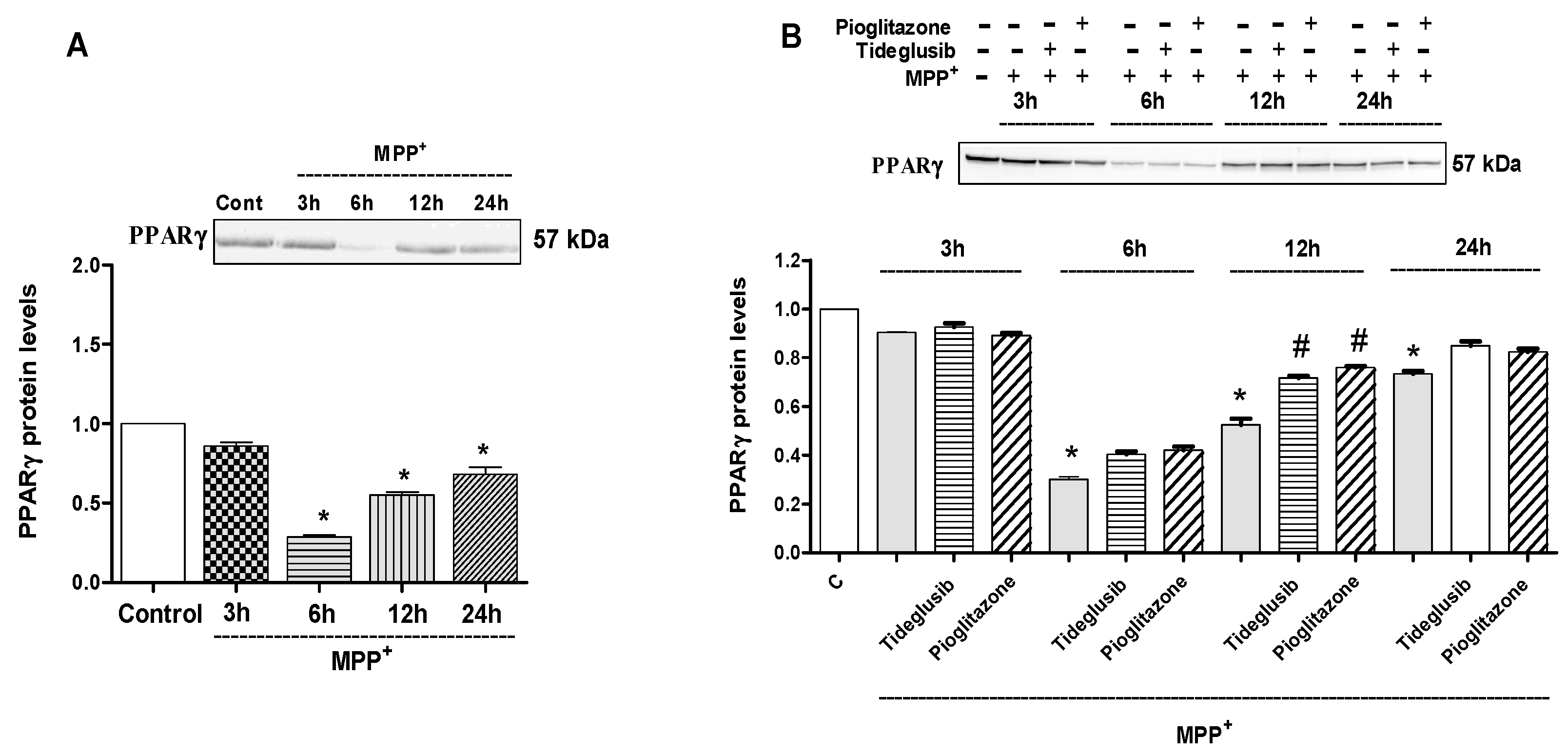
2.4. GSK-3β and PPARγ Levels Were Regulated by Tideglusib in MPP+-Treated Cells
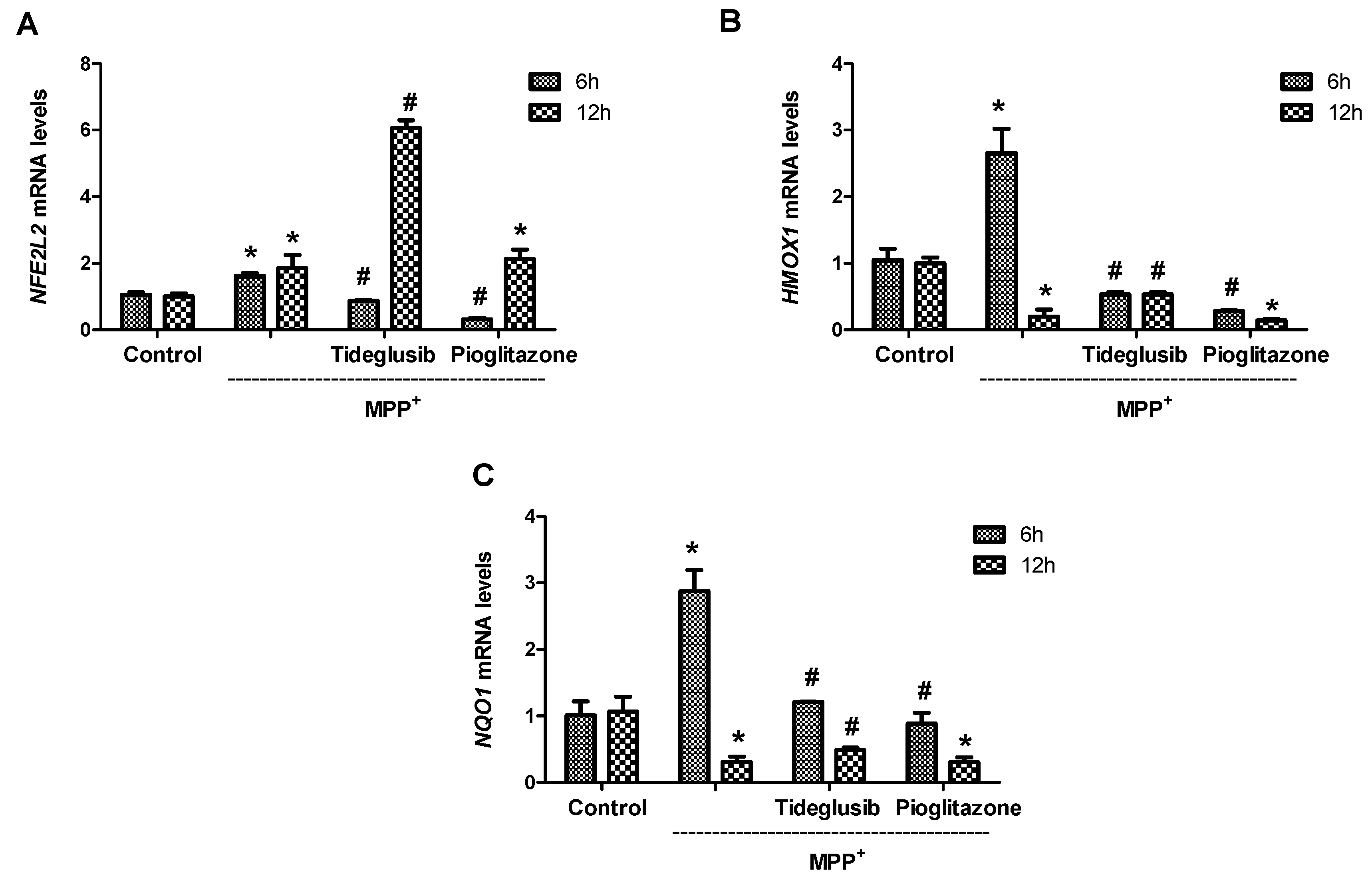
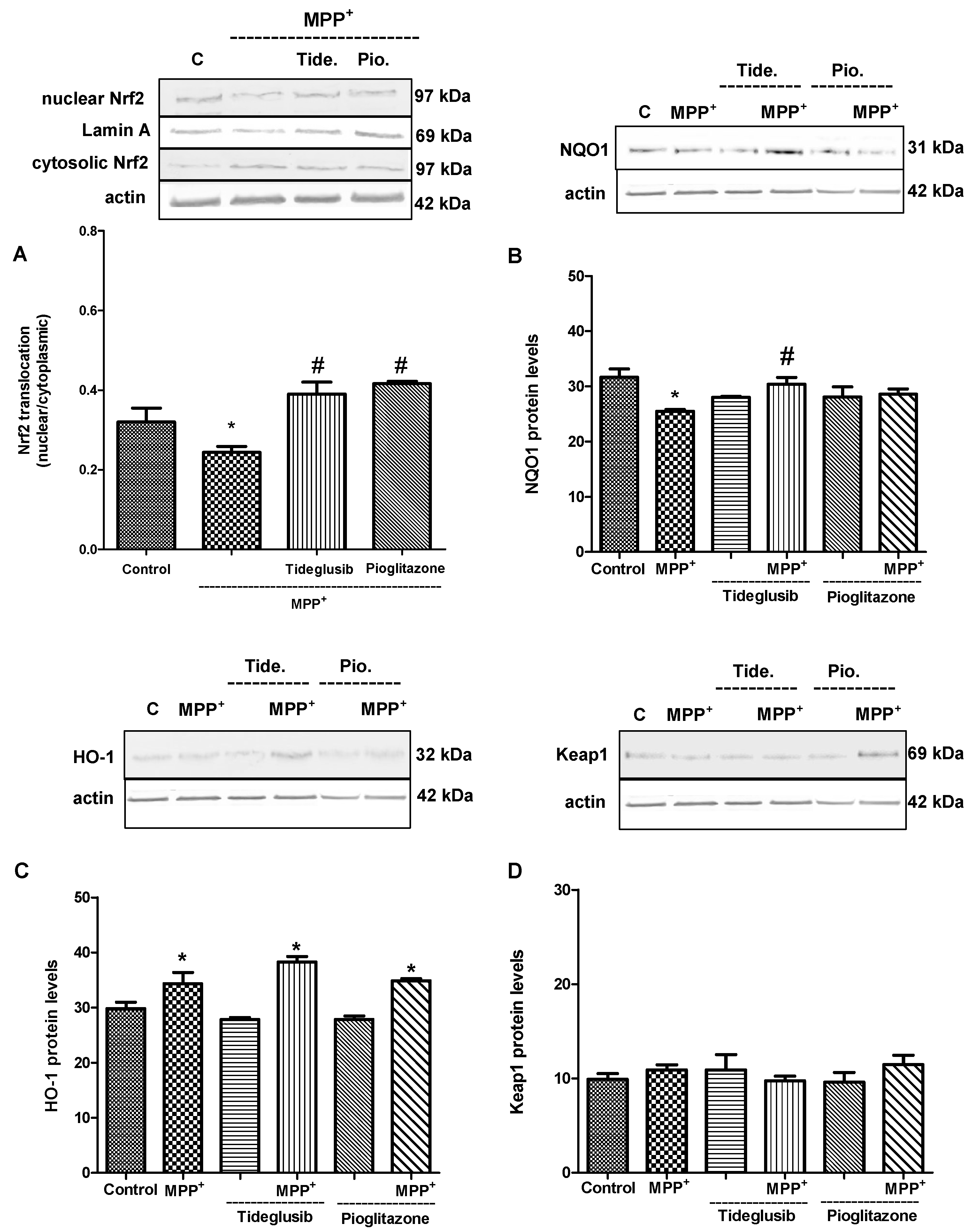
2.5. Nrf2 and Nrf2-Related Proteins Were Regulated at Transcriptional and Translational Levels by Tideglusib
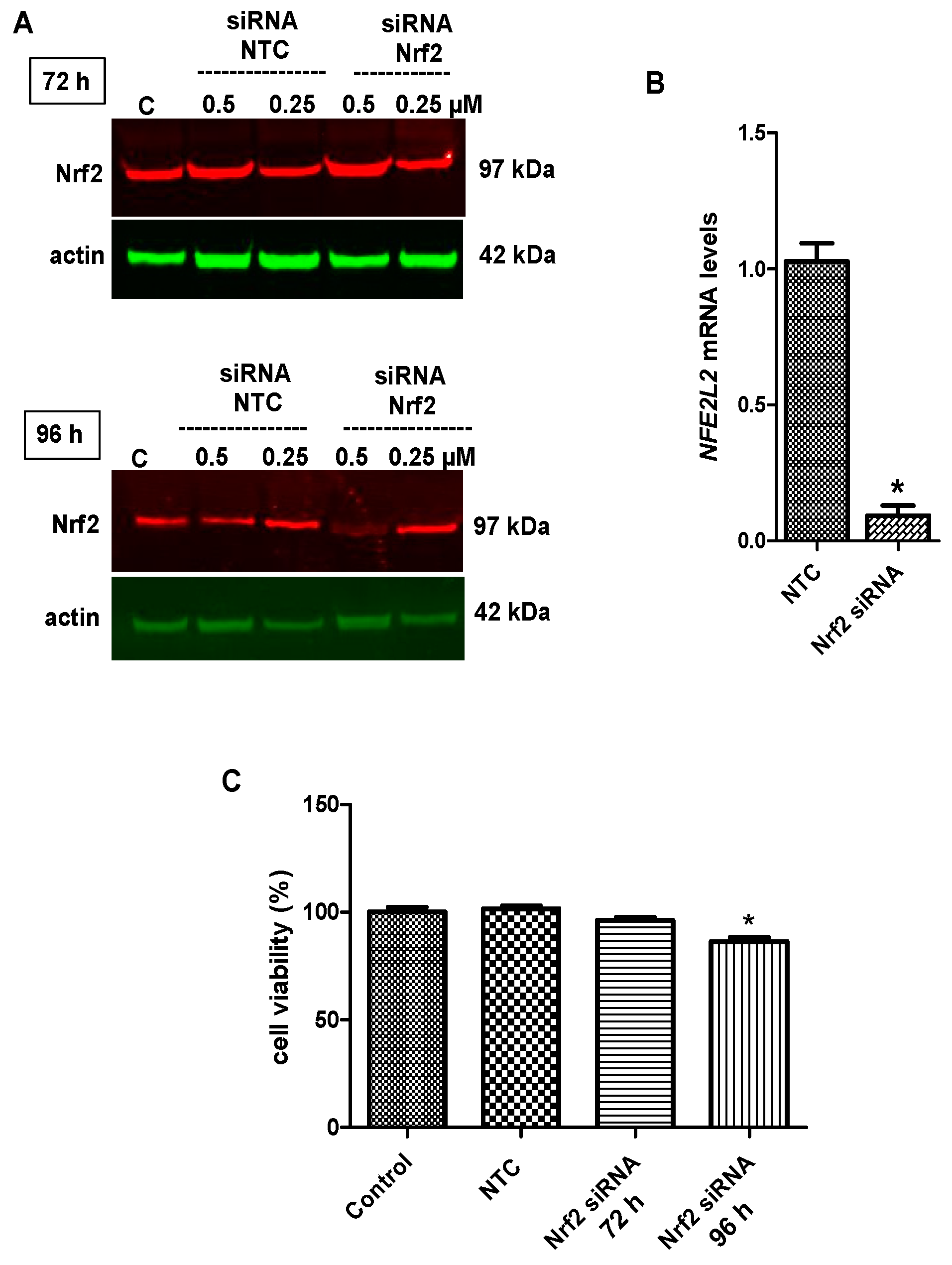
2.6. Nrf2 Silencing Reversed Tideglusib-Mediated Neuroprotection
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Cell Viability Assessment
4.3. Measurement of ROS Production
4.4. Analysis of MMP
4.5. Total Glutathione Levels
4.6. GST Enzyme Activity
4.7. Total RNA Isolation, Reverse Transcription PCR, and Real-Time PCR
4.8. Nrf2 siRNA Transfection
4.9. Isolation of Cytoplasmic and Nuclear Fraction
4.10. Western Blotting
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Drechsel, D.A.; Patel, M. Role of reactive oxygen species in the neurotoxicity of environmental agents implicated in Parkinson’s disease. Free Radic. Biol. Med. 2008, 44, 1873–1886. [Google Scholar] [CrossRef] [PubMed]
- Todorovic, M.; Wood, S.A.; Mellick, G.D. Nrf2: A modulator of Parkinson’s disease? J. Neural Transm. 2016, 123, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Hao, D.L.; Wu, B.N.; Mao, L.L.; Zhang, J. Uric acid demonstrates neuroprotective effect on Parkinson’s disease mice through Nrf2-ARE signaling pathway. Biochem. Biophys. Res. Commun. 2017, 493, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Izumi, Y.; Kataoka, H.; Inose, Y. Neuroprotective effect of an Nrf2-ARE activator identified from a chemical library on dopaminergic neurons. Eur. J. Pharm. 2018, 5, 470–479. [Google Scholar] [CrossRef]
- Salazar, M.; Rojo, A.I.; Velasco, D. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef]
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133. [Google Scholar] [CrossRef]
- Jain, A.K.; Jaiswal, A.K. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J. Biol. Chem. 2007, 282, 16502–16510. [Google Scholar] [CrossRef]
- Facci, L.; Stevens, D.A.; Skaper, S.D. Glycogen synthase kinase-3 inhibitors protect central neurons against excitotoxicity. Neuroreport 2003, 14, 1467–1470. [Google Scholar]
- Goodenough, S.; Conrad, S.; Skutella, T. Inactivation of glycogen synthase kinase-3beta protects against kainic acid-induced neurotoxicity in vivo. Brain Res. 2004, 1026, 116–125. [Google Scholar] [CrossRef]
- Szatmari, E.; Habas, A.; Yang, P. A positive feedback loop between glycogen synthase kinase 3beta and protein phosphatase 1 after stimulation of NR2B NMDA receptors in forebrain neurons. J. Biol. Chem. 2005, 280, 37526–37535. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, Y.; Ying, C. Inhibition of glycogen synthase kinase-3beta protects dopaminergic neurons from MPTP toxicity. Neuropharmacology 2007, 52, 1678–1684. [Google Scholar] [CrossRef]
- Armagan, G.; Keser, A.; Atalayın, Ç.; Dağcı, T. Tideglusib protects neural stem cells against NMDA receptor overactivation. Pharmacol. Rep. 2015, 67, 823–831. [Google Scholar] [CrossRef]
- Xu, C.; Kim, N.G.; Gumbiner, B.M. Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle 2009, 8, 4032–4039. [Google Scholar] [CrossRef]
- Martinez, A.; Alonso, M.; Castro, A.; Pérez, C.; Moreno, F.J. First non-ATP competitive glycogen synthase kinase 3 beta (GSK-3 beta) inhibitors: Thiadiazolidinones (TDZD) as potential drugs for the treatment of Alzheimer’s disease. J. Med. Chem. 2002, 45, 1292–1299. [Google Scholar] [CrossRef]
- Martinez, A.; Castro, A.; Dorronsoro, I.; Alonso, M. Glycogen synthase kinase 3 (GSK-3) inhibitors as new promising drugs for diabetes, neurodegeneration, cancer, and inflammation. Med. Res. Rev. 2002, 22, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Luna-Medina, R.; Cortes-Canteli, M.; Alonso, M.; Santos, A.; Martínez, A.; Perez-Castillo, A. Regulation of inflammatory response in neural cells in vitro by thiadiazolidinones derivatives through peroxisome proliferator-activated receptor gamma activation. J. Biol. Chem. 2005, 280, 21453–21462. [Google Scholar] [CrossRef]
- Höglinger, G.U.; Huppertz, H.J.; Wagenpfeil, S.; Andrés, M.V.; Belloch, V.; León, T.; Del Ser, T.; TAUROS MRI Investigators. Tideglusib reduces progression of brain atrophy in progressive supranuclear palsy in a randomized trial. Mov. Dis. 2014, 29, 479–487. [Google Scholar] [CrossRef]
- Serenó, L.; Coma, M.; Rodríguez, M.; Sánchez-Ferrer, P.; Sánchez, M.B.; Gich, I.; Agulló, J.M.; Pérez, M.; Avila, J.; Guardia-Laguarta, C.; et al. A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo. Neurobiol. Dis. 2009, 35, 359–367. [Google Scholar] [CrossRef]
- Jouha, J.; Loubidi, M.; Bouali, J.; Hamri, S.; Hafid, A.; Suzenet, F.; Guillaumet, G.; Dagcı, T.; Khouili, M.; Aydın, F.; et al. Synthesis of new heterocyclic compounds based on pyrazolopyridine scaffold and evaluation of their neuroprotective potential in MPP+-induced neurodegeneration. Eur. J. Med. Chem. 2017, 129, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Qaisiya, M.; Coda Zabetta, C.D.; Bellarosa, C.; Tiribelli, C. Bilirubin mediated oxidative stress involves antioxidant response activation via Nrf2 pathway. Cell. Signal. 2014, 26, 512–520. [Google Scholar] [CrossRef]
- Bayele, H.K.; Balesaria, S.; Srai, S.K. Phytoestrogens modulate hepcidin expression by Nrf2: Implications for dietary control of iron absorption. Free Radic. Biol. Med. 2015, 89, 1192–1202. [Google Scholar] [CrossRef]
- Tolosa, E.; Litvan, I.; Höglinger, G.U.; Burn, D.; Lees, A.; Andrés, M.V.; Gómez-Carrillo, B.; León, T.; Del Ser, T.; TAUROS Investigators. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov. Disorders 2014, 29, 470–478. [Google Scholar] [CrossRef]
- Chen, G.; Bower, K.A.; Ma, C.; Fang, S.; Thiele, C.J.; Luo, J. Glycogen synthase kinase 3beta (GSK3beta) mediates 6-hydroxydopamine-induced neuronal death. Faseb J. 2004, 18, 1162–1164. [Google Scholar] [CrossRef]
- Lai, J.S.; Zhao, C.; Warsh, J.J.; Li, P.P. Cytoprotection by lithium and valproate varies between cell types and cellular stresses. Eur. J. Pharmacol. 2006, 539, 18–26. [Google Scholar] [CrossRef]
- Petit-Paitel, A.; Brau, F.; Cazareth, J.; Chabry, J. Involvement of cytosolic and mitochondrial GSK-3 beta in mitochondrial dysfunction and neuronal cell death of MPTP/MPP+-treated neurons. PLoS ONE 2009, 4, e5491. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, W.; Wang, Y.; Zhao, S.; Chang, N. miR-135b plays a neuroprotective role by targeting GSK3β in MPP-intoxicated SH-SY5Y cells. Dis. Markers 2017, 2017, 5806146. [Google Scholar] [CrossRef]
- Luna-Medina, R.; Cortes-Canteli, M.; Sanchez-Galiano, S.; Morales-Garcia, J.A.; Martinez, A.; Santos, A.; Perez-Castillo, A. NP031112, a thiadiazolidinone compound, prevents inflammation and neurodegeneration under excitotoxic conditions: Potential therapeutic role in brain disorders. J. Neurosci. 2007, 27, 5766–5776. [Google Scholar] [CrossRef]
- Wei, W.Y.; Ma, Z.G.; Xu, S.C.; Zhang, N.; Tang, Q.Z. Pioglitazone protected against cardiac hypertrophy via inhibiting AKT/GSK3β and MAPK signaling pathways. PPAR Res. 2016, 2016, 9174190. [Google Scholar] [CrossRef]
- Swanson, C.R.; Joers, V.; Bondarenko, V.; Brunner, K.; Simmons, H.A.; Ziegler, T.E.; Kemnitz, J.W.; Johnson, J.A.; Emborg, M.E. The PPAR-γ agonist pioglitazone modulates inflammation and induces neuroprotection in parkinsonian monkeys. J. Neuroinflammation 2011, 8, 91. [Google Scholar] [CrossRef]
- Laloux, C.; Petrault, M.; Lecointe, C.; Devos, D.; Bordet, R. Differential susceptibility to the PPAR-γ agonist pioglitazone in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 6-hydroxydopamine rodent models of Parkinson’s disease. Pharmacol. Res. 2012, 65, 514–522. [Google Scholar] [CrossRef]
- Bonato, J.M.; Bassani, T.B.; Milani, H.; Vital, M.A.B.F.; de Oliveira, R.M.W. Pioglitazone reduces mortality, prevents depressive-like behavior, and impacts hippocampal neurogenesis in the 6-OHDA model of Parkinson’s disease in rats. Exp. Neurol. 2017, 300, 188–200. [Google Scholar] [CrossRef]
- Wild, A.C.; Moinova, H.R.; Mulcahy, R.T. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J. Biol. Chem. 1999, 274, 33627–33636. [Google Scholar] [CrossRef]
- Hara, H.; Ohta, M.; Ohta, K.; Kuno, S.; Adachi, T. Increase of antioxidative potential by tert-butylhydroquinone protects against cell death associated with 6hydroxydopamine-induced oxidative stress in neuroblastoma SH-SY5Y cells. Brain Res. Mol. Brain Res. 2003, 119, 125–131. [Google Scholar] [CrossRef]
- Kraft, A.D.; Johnson, D.A.; Johnson, J.A. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J. Neurosci. 2004, 24, 1101–1112. [Google Scholar] [CrossRef]
- Jakel, R.J.; Kern, J.T.; Johnson, D.A.; Johnson, J.A. Induction of the protective antioxidant response element pathway by 6-hydroxydopamine in vivo and in vitro. Toxicol. Sci. 2005, 87, 176–186. [Google Scholar] [CrossRef]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef]
- Jazwa, A.; Rojo, A.I.; Innamorato, N.G.; Hesse, M.; Fernández-Ruiz, J.; Cuadrado, A. Pharmacological targeting of the transcription factor nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid. Redox Signal. 2011, 14, 2347–2360. [Google Scholar] [CrossRef]
- Schipper, H.M.; Liberman, A.; Stopa, E.G. Neural heme oxygenase-1 expression in idiopathic Parkinson’s disease. Exp. Neurol. 1998, 150, 60–68. [Google Scholar] [CrossRef]
- Yoo, M.S.; Chun, H.S.; Son, J.J.; DeGiorgio, L.A.; Kim, D.J.; Peng, C.; Son, J.H. Oxidative stress regulated genes in nigral dopaminergic neuronal cells: Correlation with the known pathology in Parkinson’s disease. Brain Res. Mol. Brain Res. 2003, 110, 76–84. [Google Scholar] [CrossRef]
- Schipper, H.M. Heme oxygenase-1: Role in brain aging and neurodegeneration. Exp. Gerontol. 2000, 35, 821–830. [Google Scholar] [CrossRef]
- Xiao, H.; Lv, F.; Xu, W.; Zhang, L.; Jing, P.; Cao, X. Deprenyl prevents MPP+-induced oxidative damage in PC12 cells by the upregulation of Nrf2-mediated NQO1 expression through the activation of PI3K/Akt and Erk. Toxicology 2011, 290, 286–294. [Google Scholar] [CrossRef]
- Hung, S.Y.; Liou, H.C.; Kang, K.H.; Wu, R.M.; Wen, C.C.; Fu, W.M. Overexpression of heme oxygenase-1 protects dopaminergic neurons against 1-methyl-4-phenylpyridinium-induced neurotoxicity. Mol. Pharmacol. 2008, 74, 1564–1575. [Google Scholar] [CrossRef]
- Choi, H.J.; Kim, S.W.; Lee, S.Y.; Hwang, O. Dopamine-dependent cytotoxicity of tetrahydrobiopterin: A possible mechanism for selective neurodegeneration in Parkinson’s disease. J. Neurochem. 2003, 86, 143–152. [Google Scholar] [CrossRef]
- Han, J.M.; Lee, Y.J.; Lee, S.Y.; Kim, E.M.; Moon, Y.; Kim, H.W.; Hwang, O. Protective effect of sulforaphane against dopaminergic cell death. J. Pharmacol. Exp. Ther. 2007, 321, 249–256. [Google Scholar] [CrossRef]
- De Vries, H.E.; Witte, M.; Hondius, D.; Rozemuller, A.J.; Drukarch, B.; Hoozemans, J.; van Horssen, J. Nrf2-induced antioxidant protection: A promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic Biol. Med. 2008, 45, 1375–1383. [Google Scholar] [CrossRef]
- Krönke, G.; Kadl, A.; Ikonomu, E.; Blüml, S.; Fürnkranz, A.; Sarembock, I.J.; Bochkov, V.N.; Exner, M.; Binder, B.R.; Leitinger, N. Expression of heme oxygenase-1 in human vascular cells is regulated by peroxisome proliferator-activated receptors. Arterioscler Thromb Vasc Biol. 2007, 27, 1276–1282. [Google Scholar] [CrossRef]
- Untergrasser, A.; Cutcutache, I.; Koressaar, T. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′→3′) | Reverse Primer (5′→3′) | Base Pair |
|---|---|---|---|
| NFE2L2 (NM_006164) | agc gac gga aag agt atg ag | tgg gca acc tgg gag tag | 192 bp |
| NQO1 (NM_000903) | cag ctc acc gag agc cta gt | gag tga gcc agt acg atc agt g | 122 bp |
| HMOX1 (X06985) | cca gcg ggc cag caa caa agt gc | aag cct tca gtg ccc acg gta agg | 265 bp |
| GAPDH (NM_002046) | agc cac atc gct cag aca c | gcc caa tac gac caa atc c | 65 bp |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armagan, G.; Sevgili, E.; Gürkan, F.T.; Köse, F.A.; Bilgiç, T.; Dagcı, T.; Saso, L. Regulation of the Nrf2 Pathway by Glycogen Synthase Kinase-3β in MPP+-Induced Cell Damage. Molecules 2019, 24, 1377. https://doi.org/10.3390/molecules24071377
Armagan G, Sevgili E, Gürkan FT, Köse FA, Bilgiç T, Dagcı T, Saso L. Regulation of the Nrf2 Pathway by Glycogen Synthase Kinase-3β in MPP+-Induced Cell Damage. Molecules. 2019; 24(7):1377. https://doi.org/10.3390/molecules24071377
Chicago/Turabian StyleArmagan, Güliz, Elvin Sevgili, Fulya Tuzcu Gürkan, Fadime Aydın Köse, Tuğçe Bilgiç, Taner Dagcı, and Luciano Saso. 2019. "Regulation of the Nrf2 Pathway by Glycogen Synthase Kinase-3β in MPP+-Induced Cell Damage" Molecules 24, no. 7: 1377. https://doi.org/10.3390/molecules24071377
APA StyleArmagan, G., Sevgili, E., Gürkan, F. T., Köse, F. A., Bilgiç, T., Dagcı, T., & Saso, L. (2019). Regulation of the Nrf2 Pathway by Glycogen Synthase Kinase-3β in MPP+-Induced Cell Damage. Molecules, 24(7), 1377. https://doi.org/10.3390/molecules24071377