
High Molecular Weight Hyaluronan Suppresses Macrophage M1 Polarization and Enhances IL-10 Production in PM2.5-Induced Lung Inflammation


Abstract
:1. Introduction
2. Results
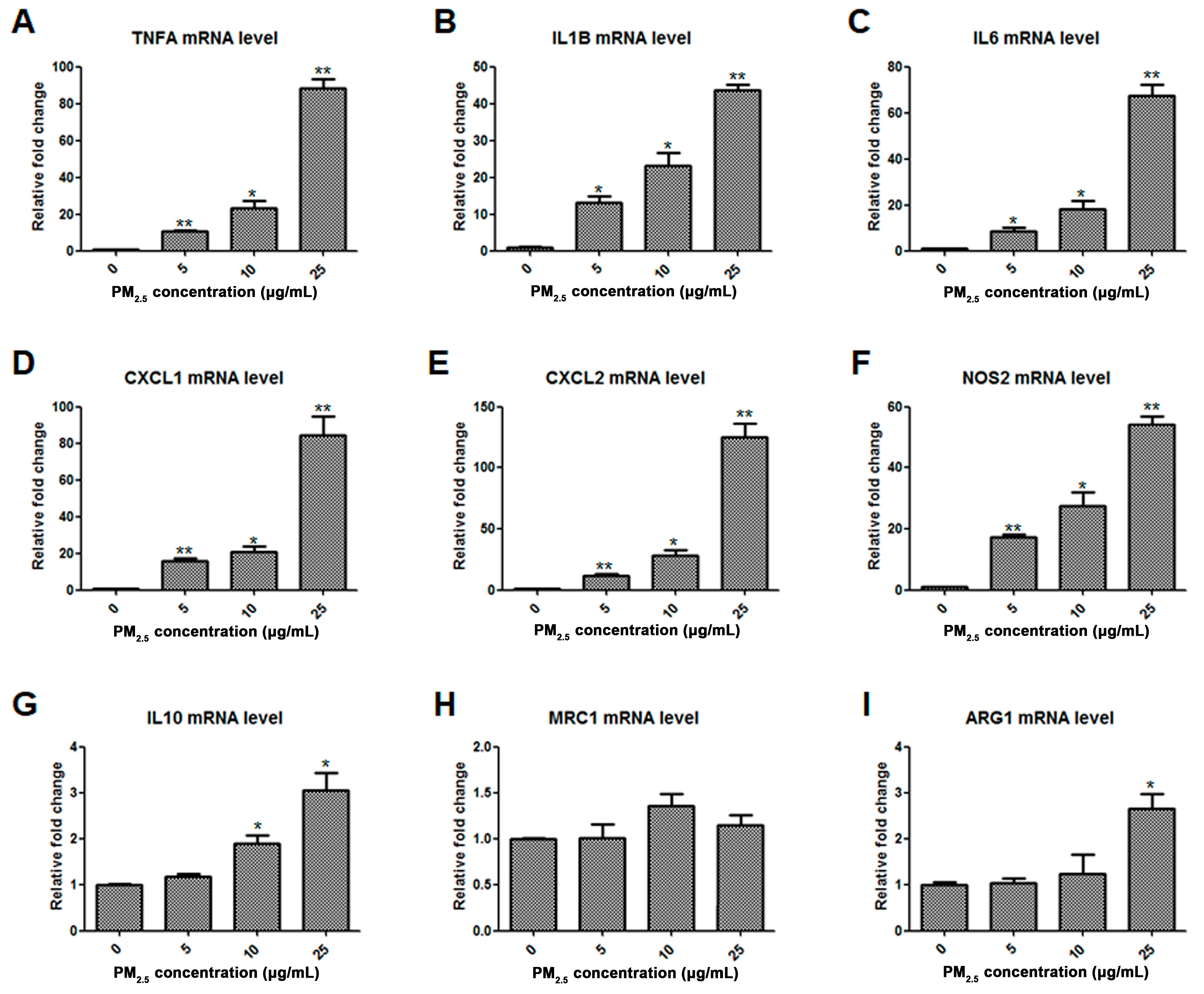
2.1. Assessment of Macrophage Phenotype in Response to PM2.5
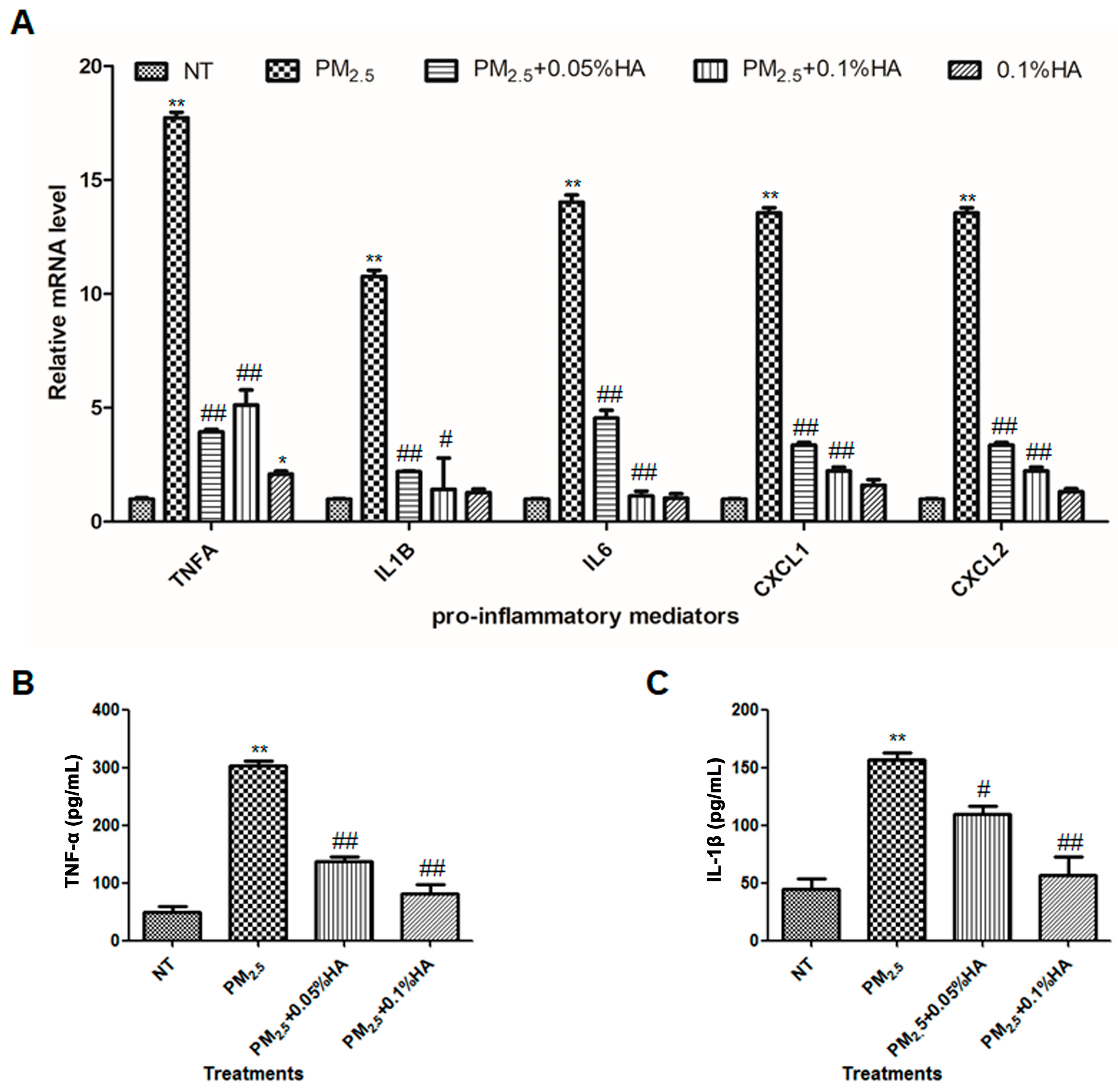
2.2. HMW-HA Attenuated PM2.5-Induced Production of Pro-Inflammatory Mediators
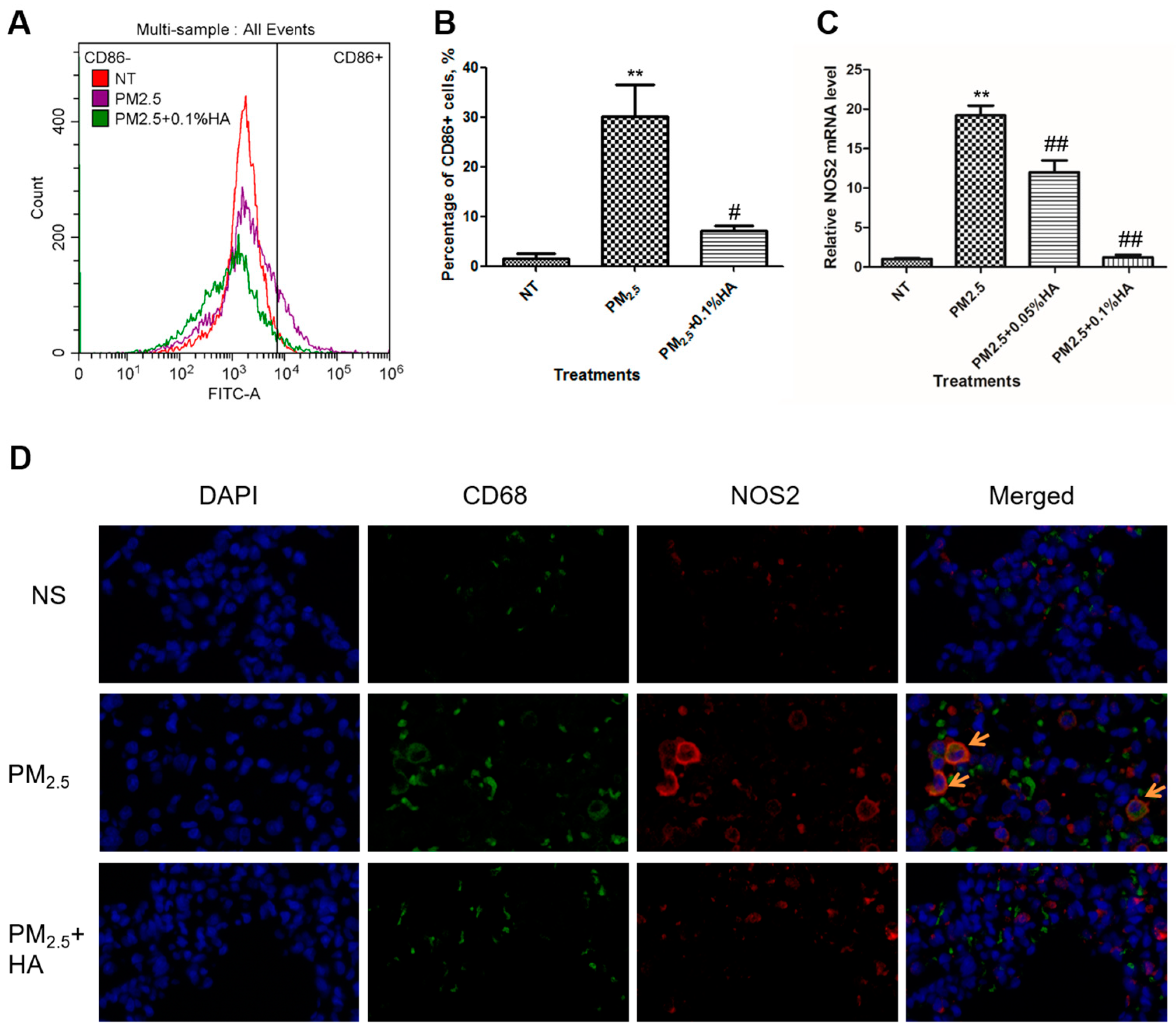
2.3. HMW-HA Negatively Regulated PM2.5-Induced M1 Polarization
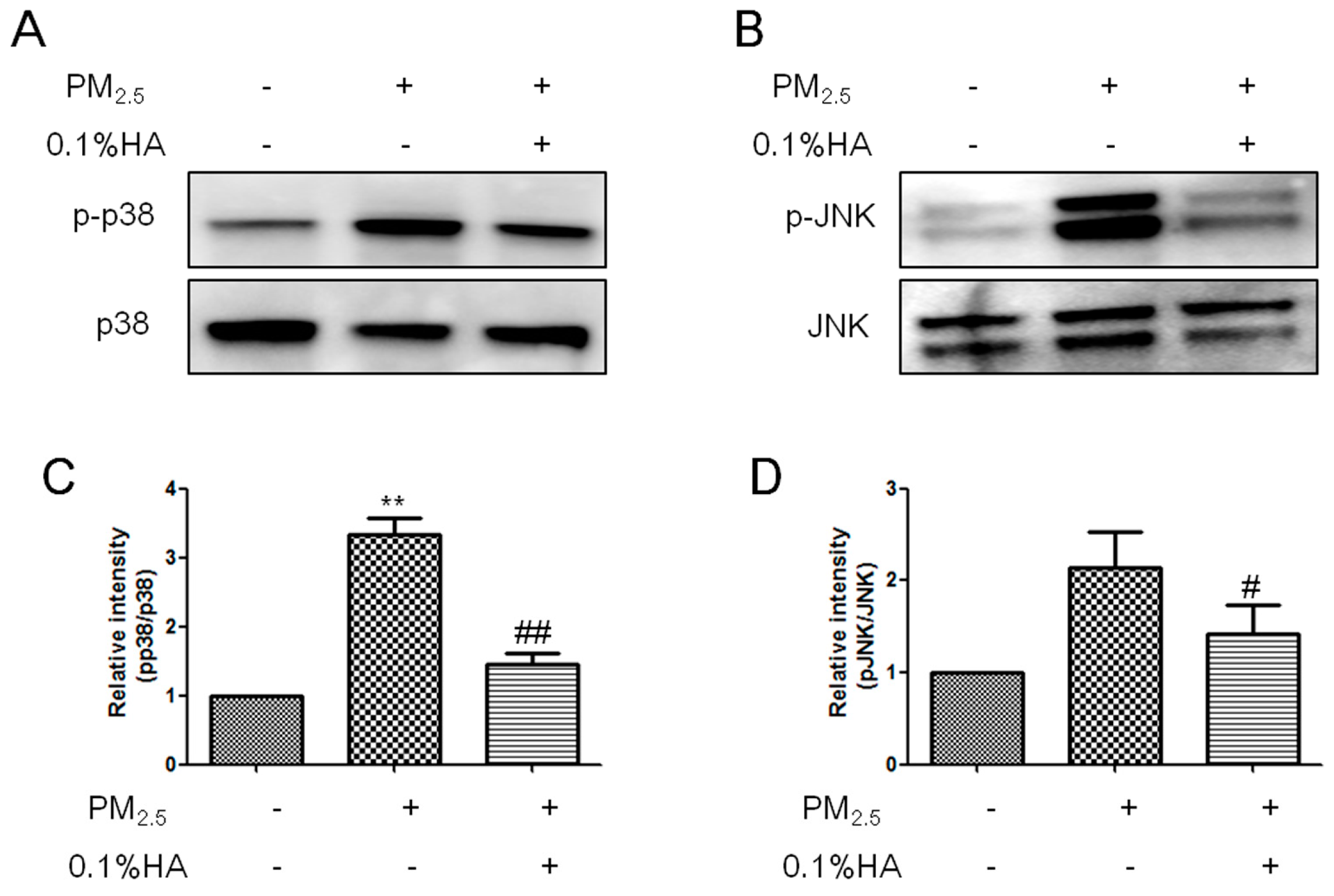
2.4. HMW-HA Inhibited PM2.5-Induced Phosphorylation of JNK and p38
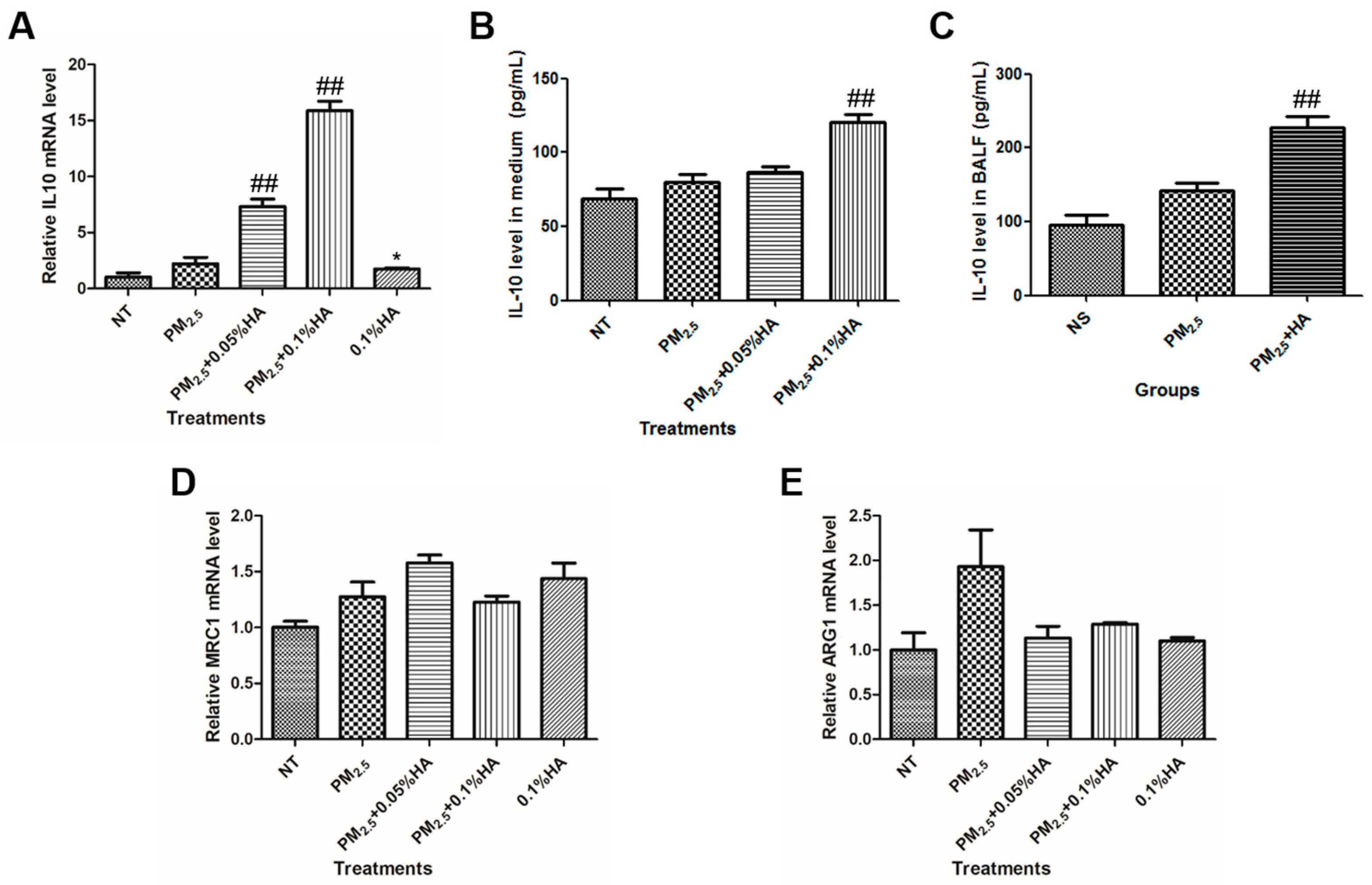
2.5. HMW-HA Stimulated IL-10 Production in the Presence of PM2.5
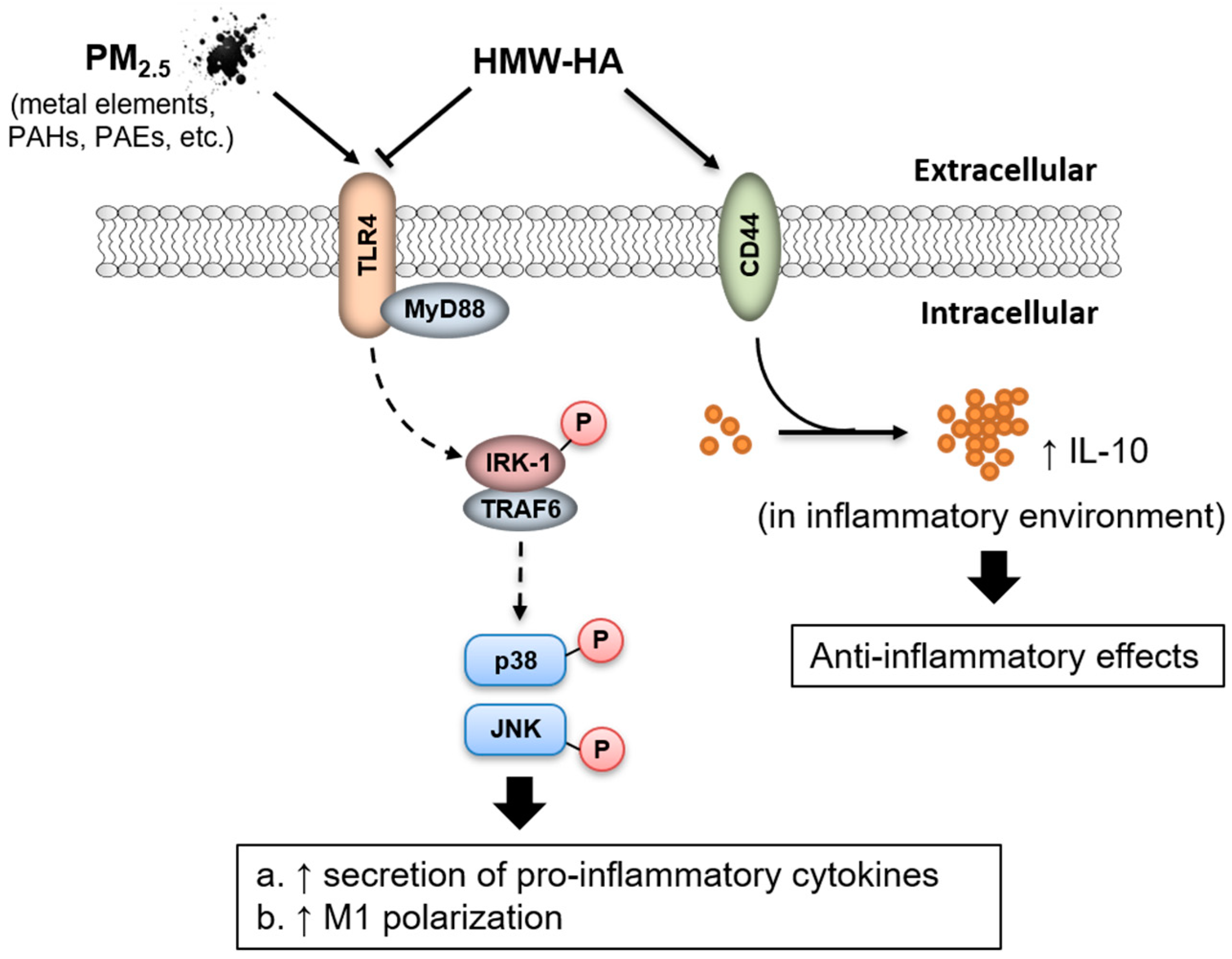
3. Discussion
4. Materials and Methods
4.1. PM2.5 Collection and Preparation
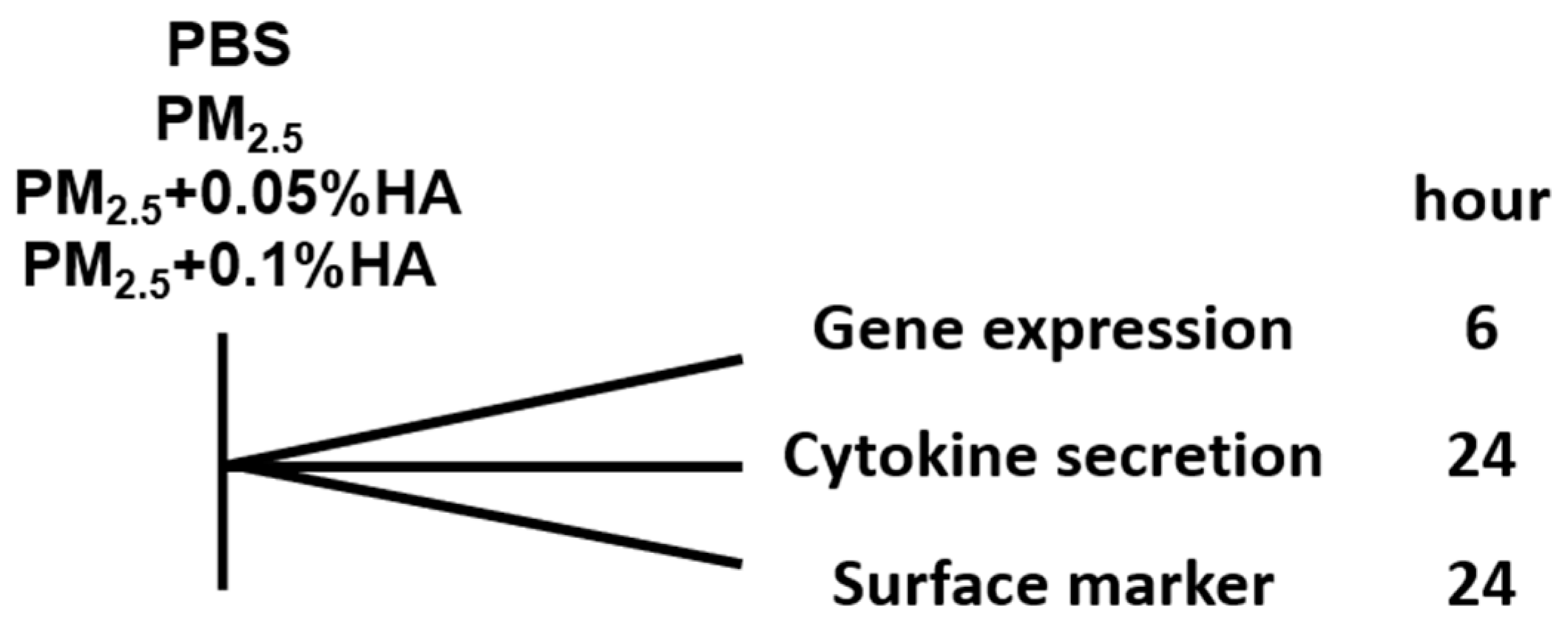
4.2. Cell Culture
4.3. RNA Isolation and Quantitative Real-time RT-PCR
4.4. Western Blot Analysis
4.5. Flow Cytometry
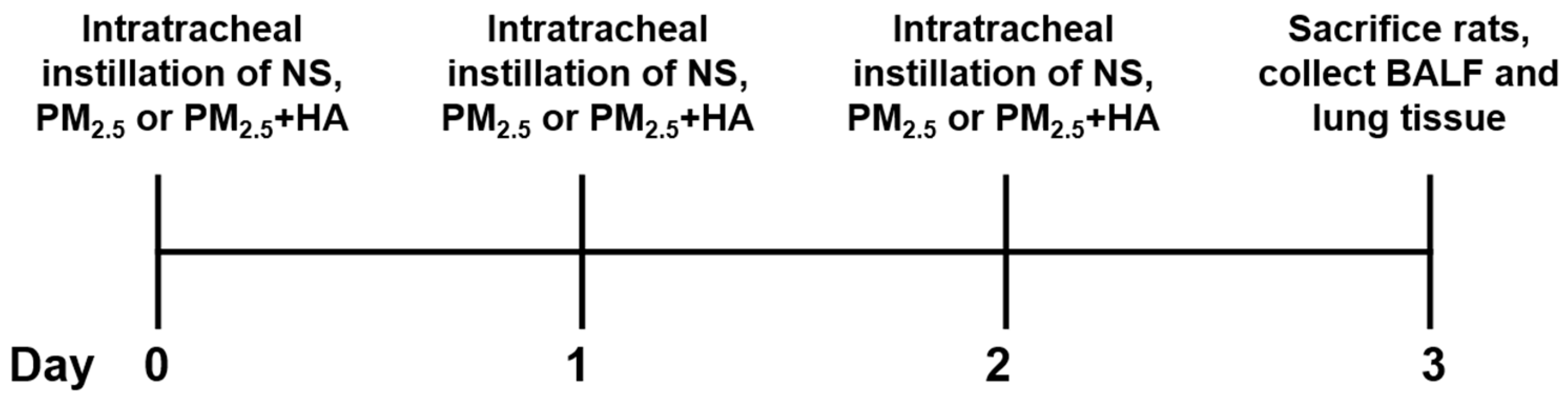
4.6. Collection of BALF and Lung Tissues from Animal Model
4.7. Immunofluoresecence Staining
4.8. ELISA Assay
4.9. Statistical Method
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Feng, S.; Gao, D.; Liao, F.; Zhou, F.; Wang, X. The health effects of ambient PM2.5 and potential mechanisms. Ecotoxicol. Environ. Saf. 2016, 128, 67–74. [Google Scholar] [CrossRef]
- Falcon-Rodriguez, C.I.; Osornio-Vargas, A.R.; Sada-Ovalle, I.; Segura-Medina, P. Aeroparticles, Composition, and Lung Diseases. Front. Immunol. 2016, 7, 3. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.Y.; Huang, D.Y.; Zhang, H.J.; Wang, S.; Chen, X.F. Exposure to particulate matter 2.5 (PM2.5) induced macrophage-dependent inflammation, characterized by increased Th1/Th17 cytokine secretion and cytotoxicity. Int. Immunopharmacol. 2017, 50, 139–145. [Google Scholar] [CrossRef]
- Ni, L.; Chuang, C.C.; Zuo, L. Fine particulate matter in acute exacerbation of COPD. Front. Physiol. 2015, 6, 294. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Chen, H.; Yang, T.; Rui, W.; Liu, F.; Zhang, F.; Zhao, Y.; Ding, W. Direct effects of airborne PM2.5 exposure on macrophage polarizations. Biochim. Biophys. Acta 2016, 1860, 2835–2843. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Liang, H.; Zen, K. Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 2014, 5, 614. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Myasoedova, V.A.; Revin, V.V.; Orekhov, A.N.; Bobryshev, Y.V. The impact of interferon-regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology 2018, 223, 101–111. [Google Scholar] [CrossRef]
- Sica, A.; Erreni, M.; Allavena, P.; Porta, C. Macrophage polarization in pathology. Cell. Mol. Life Sci. 2015, 72, 4111–4126. [Google Scholar] [CrossRef]
- Saradna, A.; Do, D.C.; Kumar, S.; Fu, Q.L.; Gao, P. Macrophage polarization and allergic asthma. Transl. Res. 2018, 191, 1–14. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Ichinose, T.; Yoshida, Y.; Arashidani, K.; Yoshida, S.; Takano, H.; Sun, G.; Shibamoto, T. Urban PM2.5 exacerbates allergic inflammation in the murine lung via a TLR2/TLR4/MyD88-signaling pathway. Sci. Rep. 2017, 7, 11027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoenfelt, J.; Mitkus, R.J.; Zeisler, R.; Spatz, R.O.; Powell, J.; Fenton, M.J.; Squibb, K.A.; Medvedev, A.E. Involvement of TLR2 and TLR4 in inflammatory immune responses induced by fine and coarse ambient air particulate matter. J. Leukoc. Biol. 2009, 86, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxid. Med. Cell Longev. 2016, 2016, 2795090. [Google Scholar] [CrossRef]
- Wu, X.; Gao, H.; Hou, Y.; Yu, J.; Sun, W.; Wang, Y.; Chen, X.; Feng, Y.; Xu, Q.M. Dihydronortanshinone, a natural product, alleviates LPS-induced inflammatory response through NF-kappaB, mitochondrial ROS, and MAPK pathways. Toxicol. Appl. Pharmacol. 2018, 355, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cowman, M.K. Hyaluronan and Hyaluronan Fragments. Adv. Carbohydr. Chem. Biochem. 2017, 74, 1–59. [Google Scholar] [CrossRef] [PubMed]
- Tavianatou, A.G.; Caon, I.; Franchi, M.; Piperigkou, Z.; Galesso, D.; Karamanos, N.K. Hyaluronan: molecular size-dependent signaling and biological functions in inflammation and cancer. FEBS J. 2019. [Google Scholar] [CrossRef]
- Litwiniuk, M.; Krejner, A.; Speyrer, M.S.; Gauto, A.R.; Grzela, T. Hyaluronic Acid in Inflammation and Tissue Regeneration. Wounds 2016, 28, 78–88. [Google Scholar]
- Hussain, S.; Ji, Z.; Taylor, A.J.; DeGraff, L.M.; George, M.; Tucker, C.J.; Chang, C.H.; Li, R.; Bonner, J.C.; Garantziotis, S. Multiwalled Carbon Nanotube Functionalization with High Molecular Weight Hyaluronan Significantly Reduces Pulmonary Injury. ACS Nano 2016, 10, 7675–7688. [Google Scholar] [CrossRef]
- Lu, K.W.; Goerke, J.; Clements, J.A.; Taeusch, H.W. Hyaluronan reduces surfactant inhibition and improves rat lung function after meconium injury. Pediatr. Res. 2005, 58, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Singleton, P.A.; Mirzapoiazova, T.; Guo, Y.; Sammani, S.; Mambetsariev, N.; Lennon, F.E.; Moreno-Vinasco, L.; Garcia, J.G. High-molecular-weight hyaluronan is a novel inhibitor of pulmonary vascular leakiness. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L639–L651. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Ichinose, T.; Kobayashi, M.; Arashidani, K.; Yoshida, S.; Nishikawa, M.; Takano, H.; Sun, G.; Shibamoto, T. Differences in allergic inflammatory responses between urban PM2.5 and fine particle derived from desert-dust in murine lungs. Toxicol. Appl. Pharmacol. 2016, 297, 41–55. [Google Scholar] [CrossRef]
- Johnson, P.; Arif, A.A.; Lee-Sayer, S.S.M.; Dong, Y. Hyaluronan and Its Interactions With Immune Cells in the Healthy and Inflamed Lung. Front. Immunol. 2018, 9, 2787. [Google Scholar] [CrossRef]
- Ruppert, S.M.; Hawn, T.R.; Arrigoni, A.; Wight, T.N.; Bollyky, P.L. Tissue integrity signals communicated by high-molecular weight hyaluronan and the resolution of inflammation. Immunol. Res. 2014, 58, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Shi, Q.; Zhang, L.; Zhao, H. High molecular weight hyaluronan attenuates fine particulate matter-induced acute lung injury through inhibition of ROS-ASK1-p38/JNK-mediated epithelial apoptosis. Environ. Toxicol. Pharmacol. 2018, 59, 190–198. [Google Scholar] [CrossRef] [PubMed]
- Rayahin, J.E.; Buhrman, J.S.; Zhang, Y.; Koh, T.J.; Gemeinhart, R.A. High and low molecular weight hyaluronic acid differentially influence macrophage activation. ACS Biomater. Sci. Eng. 2015, 1, 481–493. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.P.; Zhang, X.; Tan, Q.L.; Xu, W.X.; Zhou, C.Y.; Luo, M.; Li, X.; Huang, R.Y.; Zeng, X. NF-kappaB pathways are involved in M1 polarization of RAW 264.7 macrophage by polyporus polysaccharide in the tumor microenvironment. PLoS ONE 2017, 12, e0188317. [Google Scholar] [CrossRef]
- Tian, L.; Li, W.; Yang, L.; Chang, N.; Fan, X.; Ji, X.; Xie, J.; Li, L. Cannabinoid Receptor 1 Participates in Liver Inflammation by Promoting M1 Macrophage Polarization via RhoA/NF-kappaB p65 and ERK1/2 Pathways, Respectively, in Mouse Liver Fibrogenesis. Front. Immunol. 2017, 8, 1214. [Google Scholar] [CrossRef] [PubMed]
- Kou, X.X.; Li, C.S.; He, D.Q.; Wang, X.D.; Hao, T.; Meng, Z.; Zhou, Y.H.; Gan, Y.H. Estradiol promotes M1-like macrophage activation through cadherin-11 to aggravate temporomandibular joint inflammation in rats. J. Immunol. 2015, 194, 2810–2818. [Google Scholar] [CrossRef]
- Zhong, J.; Wang, H.; Chen, W.; Sun, Z.; Chen, J.; Xu, Y.; Weng, M.; Shi, Q.; Ma, D.; Miao, C. Ubiquitylation of MFHAS1 by the ubiquitin ligase praja2 promotes M1 macrophage polarization by activating JNK and p38 pathways. Cell Death Dis. 2017, 8, e2763. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Ichinose, T.; Yoshida, S.; Ito, T.; He, C.; Yoshida, Y.; Arashidani, K.; Takano, H.; Sun, G.; Shibamoto, T. PM2.5-induced lung inflammation in mice: Differences of inflammatory response in macrophages and type II alveolar cells. J. Appl. Toxicol. 2017, 37, 1203–1218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zeng, X.; Li, Y.; Zhao, W.; Chen, Z.; Du, Q.; Zhou, F.; Ji, N.; Huang, M. Exposure to Ambient Particles Alters the Evolution of Macrophage Phenotype and Amplifies the Inducible Release of Eotaxin-1 in Allergen-Sensitized Mice. J. Biomed. Nanotechnol. 2019, 15, 382–395. [Google Scholar] [CrossRef]
- Zhu, X.; Zhao, P.; Lu, Y.; Huo, L.; Bai, M.; Yu, F.; Tie, Y. Potential injurious effects of the fine particulate PM2.5 on the progression of atherosclerosis in apoE-deficient mice by activating platelets and leukocytes. Arch. Med. Sci. 2019, 15, 250–261. [Google Scholar] [CrossRef]
- Krocova, Z.; Macela, A.; Kroca, M.; Hernychova, L. The immunomodulatory effect(s) of lead and cadmium on the cells of immune system in vitro. Toxicol. In Vitro 2000, 14, 33–40. [Google Scholar] [CrossRef]
- Riemschneider, S.; Kohlschmidt, J.; Fueldner, C.; Esser, C.; Hauschildt, S.; Lehmann, J. Aryl hydrocarbon receptor activation by benzo(a)pyrene inhibits proliferation of myeloid precursor cells and alters the differentiation state as well as the functional phenotype of murine bone marrow-derived macrophages. Toxicol. Lett. 2018, 296, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.F.; Nielsen, C.H.; Brorson, M.M.; Frederiksen, H.; Hartoft-Nielsen, M.L.; Rasmussen, A.K.; Bendtzen, K.; Feldt-Rasmussen, U. Influence of phthalates on in vitro innate and adaptive immune responses. PLoS ONE 2015, 10, e0131168. [Google Scholar] [CrossRef]
- Rendra, E.; Riabov, V.; Mossel, D.M.; Sevastyanova, T.; Harmsen, M.C.; Kzhyshkowska, J. Reactive oxygen species (ROS) in macrophage activation and function in diabetes. Immunobiology 2018. [Google Scholar] [CrossRef] [PubMed]
- Quinn, S.R.; O’Neill, L.A. The role of microRNAs in the control and mechanism of action of IL-10. Curr. Top. Microbiol. Immunol. 2014, 380, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Caputi, A. JNKs, insulin resistance and inflammation: A possible link between NAFLD and coronary artery disease. World J. Gastroenterol. 2011, 17, 3785–3794. [Google Scholar] [CrossRef]
- Yang, Y.; Kim, S.C.; Yu, T.; Yi, Y.S.; Rhee, M.H.; Sung, G.H.; Yoo, B.C.; Cho, J.Y. Functional roles of p38 mitogen-activated protein kinase in macrophage-mediated inflammatory responses. Mediat. Inflamm. 2014, 2014, 352371. [Google Scholar] [CrossRef]
- Saraiva, M.; O’Garra, A. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Burmeister, A.R.; Marriott, I. The Interleukin-10 Family of Cytokines and Their Role in the CNS. Front. Cell. Neurosci. 2018, 12, 458. [Google Scholar] [CrossRef]
- Gebe, J.A.; Yadava, K.; Ruppert, S.M.; Marshall, P.; Hill, P.; Falk, B.A.; Sweere, J.M.; Han, H.; Kaber, G.; Harten, I.A.; et al. Modified High-Molecular-Weight Hyaluronan Promotes Allergen-Specific Immune Tolerance. Am. J. Respir. Cell. Mol. Biol. 2017, 56, 109–120. [Google Scholar] [CrossRef] [Green Version]
- Bollyky, P.L.; Wu, R.P.; Falk, B.A.; Lord, J.D.; Long, S.A.; Preisinger, A.; Teng, B.; Holt, G.E.; Standifer, N.E.; Braun, K.R.; et al. ECM components guide IL-10 producing regulatory T-cell (TR1) induction from effector memory T-cell precursors. Proc. Natl. Acad. Sci. USA 2011, 108, 7938–7943. [Google Scholar] [CrossRef] [Green Version]
- King, A.; Balaji, S.; Le, L.D.; Marsh, E.; Crombleholme, T.M.; Keswani, S.G. Interleukin-10 regulates fetal extracellular matrix hyaluronan production. J. Pediatr. Surg. 2013, 48, 1211–1217. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, A.; Ito, M.; Chikuma, S.; Akanuma, T.; Nakatsukasa, H. Negative Regulation of Cytokine Signaling in Immunity. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef]
- Dong, Y.; Arif, A.; Olsson, M.; Cali, V.; Hardman, B.; Dosanjh, M.; Lauer, M.; Midura, R.J.; Hascall, V.C.; Brown, K.L.; et al. Endotoxin free hyaluronan and hyaluronan fragments do not stimulate TNF-alpha, interleukin-12 or upregulate co-stimulatory molecules in dendritic cells or macrophages. Sci. Rep. 2016, 6, 36928. [Google Scholar] [CrossRef]
- Qadri, M.; Almadani, S.; Jay, G.D.; Elsaid, K.A. Role of CD44 in Regulating TLR2 Activation of Human Macrophages and Downstream Expression of Proinflammatory Cytokines. J. Immunol. 2018, 200, 758–767. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Primer Sequence (5′→3′) |
|---|---|
| TNFA | Forward primer: CCCAGACCCTCACACTCAGAT Reverse primer: TTGTCCCTTGAAGAGAACCTG |
| IL1B | Forward primer: ATCATCCCACGAGTCACAGAGG Reverse primer: TACCTATGTCTTGCCCGTGGAG |
| IL6 | Forward primer: ACTTCACAGAGGATACCAC Reverse primer: GCATCATCGCTGTTCATAC |
| CXCL1 | Forward primer: GCAGACAGTGGCAGGGATT Reverse primer: GGACACCCTTTAGCATCTTTT |
| CXCL2 | Forward primer: TGCTCAAGACTCCAACCACTC Reverse primer: ACAACAACCCCTGTACCCTG |
| NOS2 | Forward primer: CTATTCCCAGCCCAACAACA Reverse primer: CTGGAACATTCTGTGCAGTC |
| IL10 | Forward primer: GAAAAATTGAACCACCCGGCA Reverse primer: TTCCAAGGAGTTGCTCCCGT |
| MRC1 | Forward primer: CAAGGAAGGTTGGCATTTGT Reverse primer: GGAACGTGTGCTCTGAGTT |
| ARG1 | Forward primer: ACAAGACAGGGCTACTTTCAGG Reverse primer: ACAAGACAAGGTCAACGCCA |
| SOCS1 | Forward primer: CCTTCGACTGCCTCTTCGAG Reverse primer: AGTCACGGAGTACCGGGTTA |
| Endogenous control (ACTB) | Forward primer: AGCCATGTACGTAGCCATCC Reverse primer: ACCCTCATAGATGGGCACAG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, Q.; Zhao, L.; Xu, C.; Zhang, L.; Zhao, H. High Molecular Weight Hyaluronan Suppresses Macrophage M1 Polarization and Enhances IL-10 Production in PM2.5-Induced Lung Inflammation. Molecules 2019, 24, 1766. https://doi.org/10.3390/molecules24091766
Shi Q, Zhao L, Xu C, Zhang L, Zhao H. High Molecular Weight Hyaluronan Suppresses Macrophage M1 Polarization and Enhances IL-10 Production in PM2.5-Induced Lung Inflammation. Molecules. 2019; 24(9):1766. https://doi.org/10.3390/molecules24091766
Chicago/Turabian StyleShi, Qiwen, Lan Zhao, Chenming Xu, Leifang Zhang, and Hang Zhao. 2019. "High Molecular Weight Hyaluronan Suppresses Macrophage M1 Polarization and Enhances IL-10 Production in PM2.5-Induced Lung Inflammation" Molecules 24, no. 9: 1766. https://doi.org/10.3390/molecules24091766
APA StyleShi, Q., Zhao, L., Xu, C., Zhang, L., & Zhao, H. (2019). High Molecular Weight Hyaluronan Suppresses Macrophage M1 Polarization and Enhances IL-10 Production in PM2.5-Induced Lung Inflammation. Molecules, 24(9), 1766. https://doi.org/10.3390/molecules24091766