
The Reactivity and Stability of Polyoxometalate Water Oxidation Electrocatalysts


Abstract
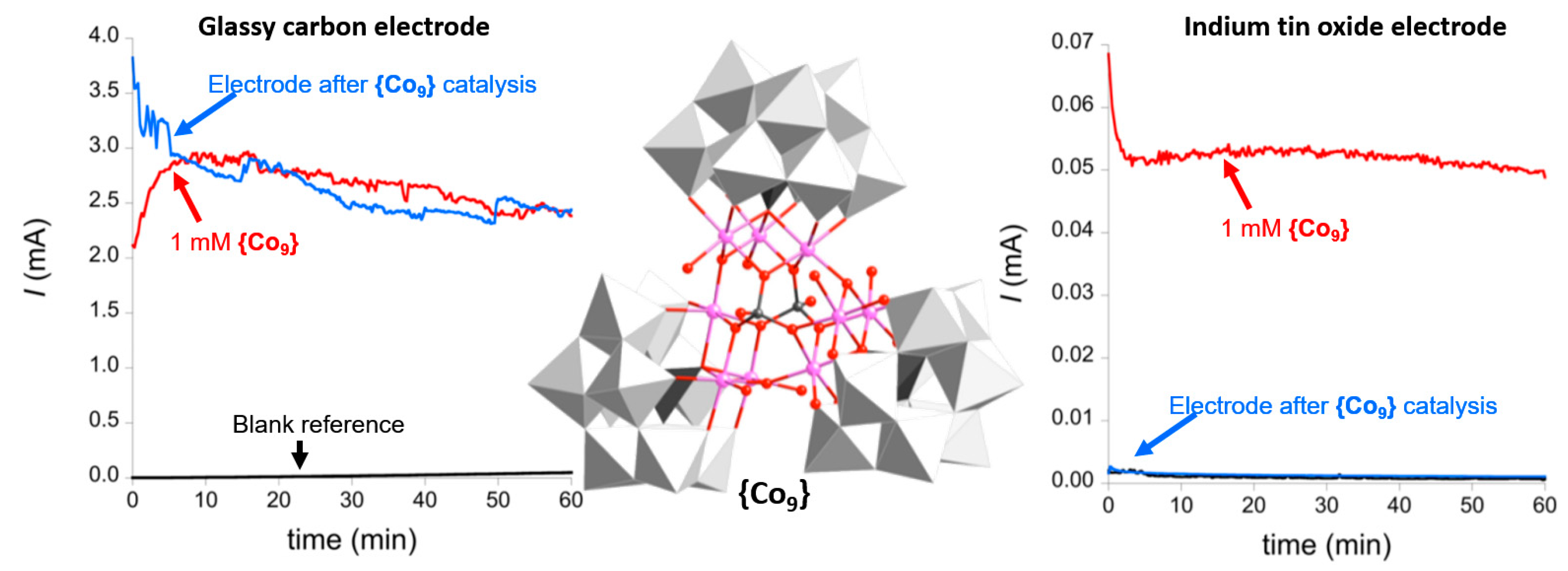
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
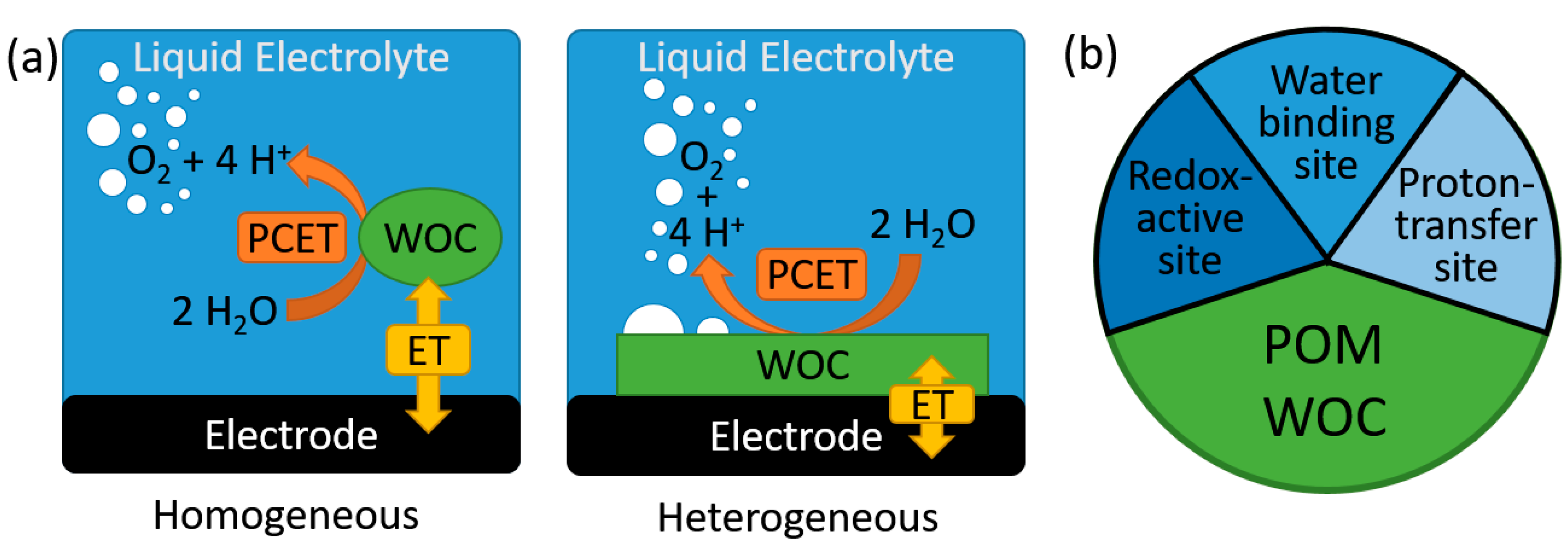
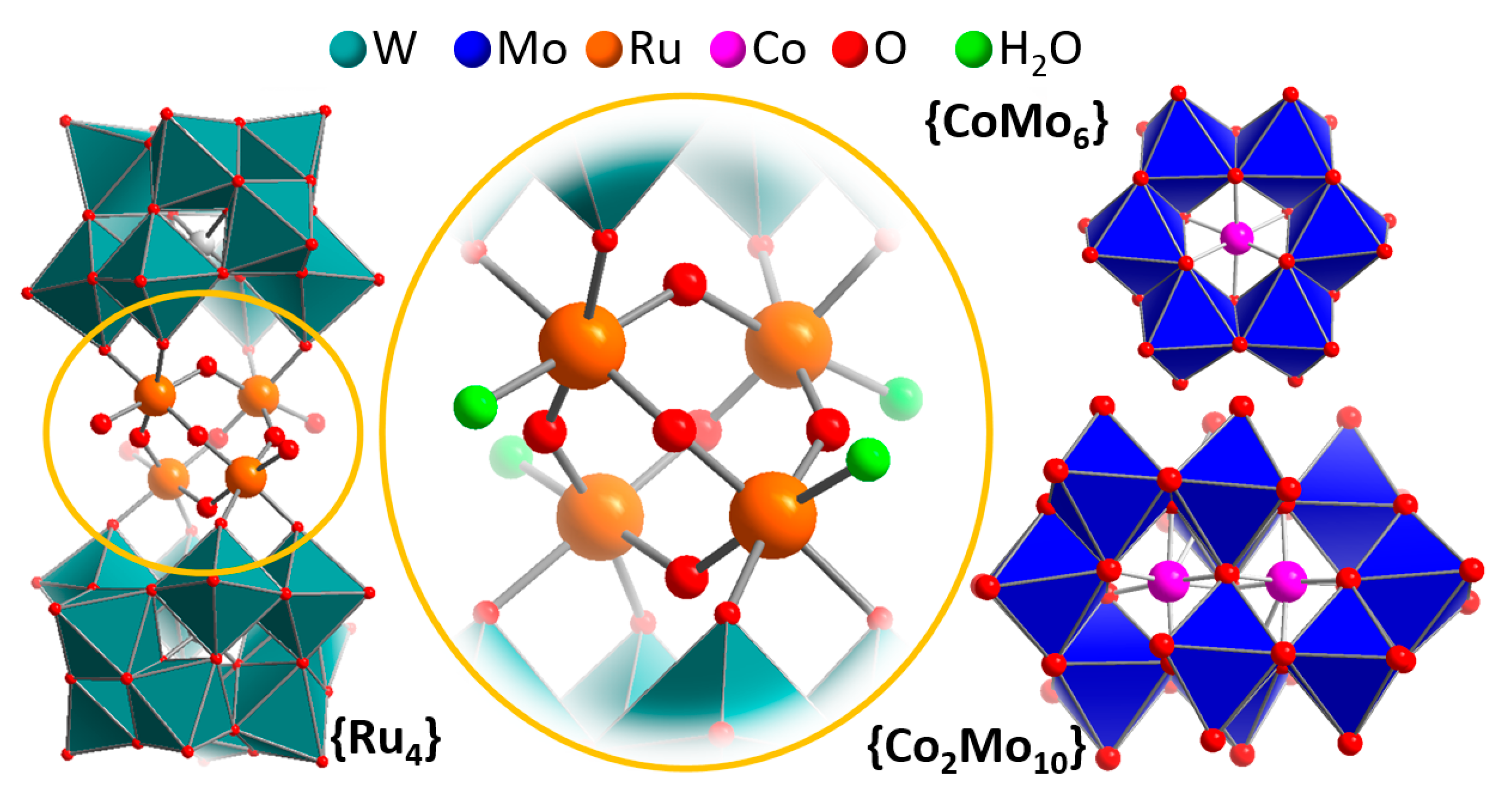
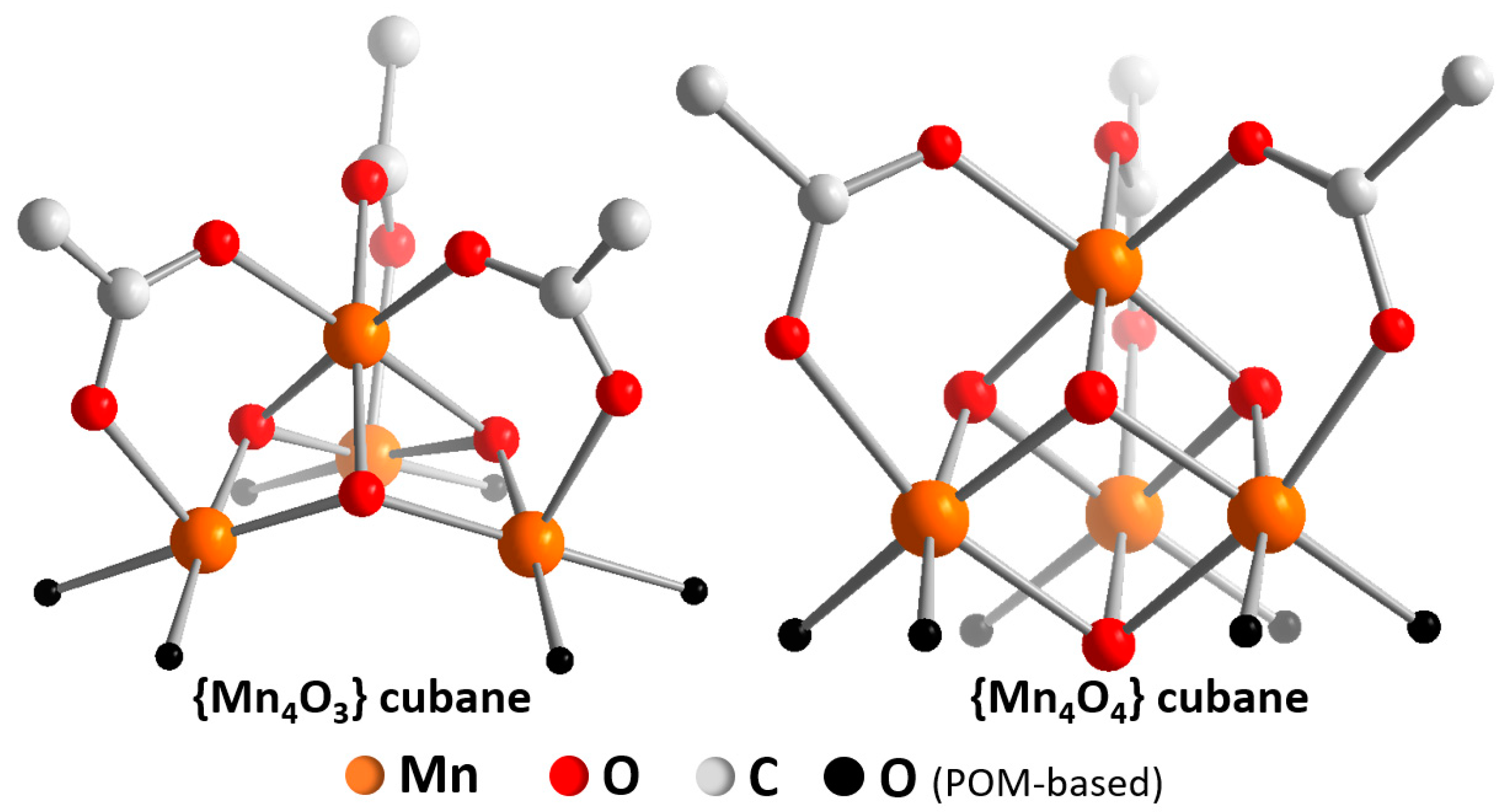
2. Homogeneous Electrocatalytic Water Oxidation by POMs
3. Theoretical Simulations for Mechanistic Insights into POM-WOCs
4. Heterogeneous Electrocatalytic Water Oxidation by POMs
5. Stability of POM-WOCs under Catalytic Operation
6. Outlook and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Du, P.; Eisenberg, R. Catalysts made of earth-abundant elements (Co, Ni, Fe) for water splitting: Recent progress and future challenges. Energy Environ. Sci. 2012, 5, 6012. [Google Scholar] [CrossRef]
- Nocera, D.G. The artificial leaf. Acc. Chem. Res. 2012, 45, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Roger, I.; Shipman, M.A.; Symes, M.D. Earth-abundant catalysts for electrochemical and photoelectrochemical water splitting. Nat. Rev. Chem. 2017, 1, 0003. [Google Scholar] [CrossRef]
- Rausch, B.; Symes, M.D.; Chisholm, G.; Cronin, L. Decoupled catalytic hydrogen evolution from a molecular metal oxide redox mediator in water splitting. Science 2014, 345, 1326–1330. [Google Scholar] [CrossRef] [PubMed]
- Special issue: Electrocatalysis for the generation and consumption of fuels. Nat. Rev. Chem. 2018, 2, 125. [CrossRef] [Green Version]
- Stamenkovic, V.R.; Strmcnik, D.; Lopes, P.P.; Markovic, N.M. Energy and fuels from electrochemical interfaces. Nat. Mater. 2016, 16, 57–69. [Google Scholar] [CrossRef]
- Lee, K.J.; Elgrishi, N.; Kandemir, B.; Dempsey, J.L. Electrochemical and spectroscopic methods for evaluating molecular electrocatalysts. Nat. Rev. Chem. 2017, 1, 39. [Google Scholar] [CrossRef]
- Deng, X.; Petric, A. Geometrical modeling of the triple-phase-boundary in solid oxide fuel cells. J. Power Sources 2005, 140, 297–303. [Google Scholar] [CrossRef]
- Bard, A.J. Inner-Sphere Heterogeneous Electrode Reactions. Electrocatalysis and Photocatalysis: The Challenge. J. Am. Chem. Soc. 2010, 132, 7559–7567. [Google Scholar] [CrossRef]
- Cai, Z.; Bu, X.; Wang, P.; Ho, J.C.; Yang, J.; Wang, X. Recent advances in layered double hydroxide electrocatalysts for the oxygen evolution reaction. J. Mater. Chem. A 2019, 7, 5069–5089. [Google Scholar] [CrossRef]
- Luo, Y.; Zhou, H.; Sun, J.; Qin, F.; Yu, F.; Bao, J.; Yu, Y.; Chen, S.; Ren, Z. Cu Nanowires Shelled with NiFe Layered Double Hydroxide Nanosheets as Bifunctional Electrocatalysts for Overall Water Splitting. Energy Environ. Sci. 2017, 10, 1820–1827. [Google Scholar]
- Subbaraman, R.; Tripkovic, D.; Chang, K.C.; Strmcnik, D.; Paulikas, A.P.; Hirunsit, P.; Chan, M.; Greeley, J.; Stamenkovic, V.; Markovic, N.M. Trends in activity for the water electrolyser reactions on 3d M(Ni,Co,Fe,Mn) hydr(oxy)oxide catalysts. Nat. Mater. 2012, 11, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Carmo, M.; Fritz, D.L.; Mergel, J.; Stolten, D. A comprehensive review on PEM water electrolysis. Int. J. Hydrogen Energy 2013, 38, 4901–4934. [Google Scholar] [CrossRef]
- Marshall, A.; Børresen, B.; Hagen, G.; Tsypkin, M.; Tunold, R. Hydrogen production by advanced proton exchange membrane (PEM) water electrolysers-Reduced energy consumption by improved electrocatalysis. Energy 2007, 32, 431–436. [Google Scholar] [CrossRef]
- Patel, P.P.; Datta, M.K.; Velikokhatnyi, O.I.; Kuruba, R.; Damodaran, K.; Jampani, P.; Gattu, B.; Shanthi, P.M.; Damle, S.S.; Kumta, P.N. Noble metal-free bifunctional oxygen evolution and oxygen reduction acidic media electro-catalysts. Sci. Rep. 2016, 6, 28367. [Google Scholar] [CrossRef] [Green Version]
- Vincent, I.; Bessarabov, D. Low cost hydrogen production by anion exchange membrane electrolysis: A review. Renew. Sustain. Energy Rev. 2018, 81, 1690–1704. [Google Scholar] [CrossRef]
- Recatalá, D.; Llusar, R.; Gushchin, A.L.; Kozlova, E.A.; Laricheva, Y.A.; Abramov, P.A.; Sokolov, M.N.; Gómez, R.; Lana-Villarreal, T. Photogeneration of Hydrogen from Water by Hybrid Molybdenum Sulfide Clusters Immobilized on Titania. ChemSusChem 2015, 8, 148–157. [Google Scholar] [CrossRef]
- Cronin, L.; Müller, A. (Eds.) POM-themed issue. Chem. Soc. Rev. 2012, 41, 7325–7648. [Google Scholar]
- Pope, M.T. Heteropoly and Isopoly Oxometalates; Springer: Berlin, Germany, 1983; ISBN 978-3-662-12006-4. [Google Scholar]
- Hill, C.L. POM-themed issue. Chem. Rev. 1998, 98, 1–302. [Google Scholar] [CrossRef] [Green Version]
- Kortz, U. Polyoxometalates. Eur. J. Inorg. Chem. 2009, 2009, 5055–5276. [Google Scholar] [CrossRef]
- Van Eldik, R.; Cronin, L. (Eds.) Polyoxometalate Chemistry; Academic Press: Cambridge, MA, USA, 2017; Volume 69. [Google Scholar]
- Sartorel, A.; Carraro, M.; Toma, F.M.; Prato, M.; Bonchio, M. Shaping the beating heart of artificial photosynthesis: Oxygenic metal oxide nano-clusters. Energy Environ. Sci. 2012, 5, 5592. [Google Scholar] [CrossRef]
- Lv, H.; Geletii, Y.V.; Zhao, C.; Vickers, J.W.; Zhu, G.; Luo, Z.; Song, J.; Lian, T.; Musaev, D.G.; Hill, C.L. Polyoxometalate water oxidation catalysts and the production of green fuel. Chem. Soc. Rev. 2012, 41, 7572–7589. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Güttinger, R.; Moré, R.; Song, F.; Wan, W.; Patzke, G.R. Frontiers of water oxidation: The quest for true catalysts. Chem. Soc. Rev. 2017, 46, 6124–6147. [Google Scholar] [CrossRef] [PubMed]
- Dau, H.; Limberg, C.; Reier, T.; Risch, M.; Roggan, S.; Strasser, P. The Mechanism of Water Oxidation: From Electrolysis via Homogeneous to Biological Catalysis. ChemCatChem 2010, 2, 724–761. [Google Scholar] [CrossRef]
- Blasco-Ahicart, M.; Soriano-Lopez, J.; Carbo, J.J.; Poblet, J.M.; Galan-Mascaros, J.R. Polyoxometalate electrocatalysts based on earthabundant metals for efficient water oxidation in acidic media. Nat. Chem. 2018, 10, 24–30. [Google Scholar] [CrossRef]
- Goberna-Ferrón, S.; Vigara, L.; Soriano-López, J.; Galán-Mascarós, J.R. Identification of a nonanuclear {CoII9} polyoxometalate cluster as a homogeneous catalyst for water oxidation. Inorg. Chem. 2012, 51, 11707–11715. [Google Scholar] [CrossRef]
- Gao, J.; Cao, S.; Tay, Q.; Liu, Y.; Yu, L.; Ye, K.; Mun, P.C.S.; Li, Y.; Rakesh, G.; Loo, S.C.J.; et al. Molecule-based water-oxidation catalysts (WOCs): Cluster-size-dependent dye-sensitized polyoxometalates for visible-light-driven O2 evolution. Sci. Rep. 2013, 3, 1853. [Google Scholar] [CrossRef]
- Basu, O.; Mukhopadhyay, S.; Das, S.K. Cobalt based functional inorganic materials: Electrocatalytic water oxidation. J. Chem. Sci. 2018, 130, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, S.; Debgupta, J.; Singh, C.; Kar, A.; Das, S.K. A Keggin Polyoxometalate Shows Water Oxidation Activity at Neutral pH: POM@ZIF-8, an Efficient and Robust Electrocatalyst. Angew. Chem. Int. Ed. 2018, 57, 1918–1923. [Google Scholar] [CrossRef]
- Soriano-López, J.; Musaev, D.G.; Hill, C.L.; Galán-Mascarós, J.R.; Carbó, J.J.; Poblet, J.M. Tetracobalt-polyoxometalate catalysts for water oxidation: Key mechanistic details. J. Catal. 2017, 350, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Helm, L.; Merbach, A.E. Water exchange on metal ions: Experiments and simulations. Coord. Chem. Rev. 1999, 187, 151–181. [Google Scholar] [CrossRef]
- Stracke, J.J.; Finke, R.G. Distinguishing homogeneous from heterogeneous water oxidation catalysis when beginning with polyoxometalates. ACS Catal. 2014, 4, 909–933. [Google Scholar] [CrossRef]
- Gersten, S.W.; Samuels, G.J.; Meyer, T.J. Catalytic oxidation of water by an oxo-bridged ruthenium dimer. J. Am. Chem. Soc. 1982, 104, 4029–4030. [Google Scholar] [CrossRef]
- Howells, A.R.; Sankarraj, A.; Shannon, C. A Diruthenium-Substituted Polyoxometalate as an Electrocatalyst for Oxygen Generation. J. Am. Chem. Soc. 2004, 126, 12258–12259. [Google Scholar] [CrossRef]
- Sartorel, A.; Carraro, M.; Scorrano, G.; De Zorzi, R.; Geremia, S.; McDaniel, N.D.; Bernhard, S.; Bonchio, M. Polyoxometalate embedding of a tetraruthenium(IV)-oxo-core by template-directed metalation of [γ-SiW10O36]8−: A totally inorganic oxygen-evolving catalyst. J. Am. Chem. Soc. 2008, 130, 5006–5007. [Google Scholar] [CrossRef] [Green Version]
- Geletii, Y.V.; Botar, B.; Kögerler, P.; Hillesheim, D.A.; Musaev, D.G.; Hill, C.L. An All-Inorganic, Stable, and Highly Active Tetraruthenium Homogeneous Catalyst for Water Oxidation. Angew. Chem. 2008, 120, 3960–3963. [Google Scholar] [CrossRef]
- Galan-Mascarós, J.R. Water Oxidation at Electrodes Modified with Earth-Abundant Transition-Metal Catalysts. ChemElectroChem 2015, 2, 37–50. [Google Scholar] [CrossRef] [Green Version]
- Yin, Q.; Tan, J.M.; Besson, C.; Geletii, Y.V.; Musaev, D.G.; Kuznetsov, A.E.; Luo, Z.; Hardcastle, K.I.; Hill, C.L. A fast soluble carbon-free molecular water oxidation catalyst based on abundant metals. Science 2010, 328, 342–345. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Ding, Y.; Zheng, M.; Chen, H.; Zhao, J. [{β-SiNi2W10O36(OH)2(H2O)}4]24−: A new robust visible light-driven water oxidation catalyst based on nickel-containing polyoxometalate. Chem. Commun. 2016, 52, 14494–14497. [Google Scholar] [CrossRef]
- Tanaka, S.; Annaka, M.; Sakai, K. Visible light-induced water oxidation catalyzed by molybdenum-based polyoxometalates with mono- and dicobalt(III) cores as oxygen-evolving centers. Chem. Commun. 2012, 48, 1653. [Google Scholar] [CrossRef]
- Hetterscheid, D.G.H.; Reek, J.N.H. Mononuclear water oxidation catalysts. Angew. Chem. Int. Ed. 2012, 51, 9740–9747. [Google Scholar] [CrossRef]
- Hetterscheid, D.G.H.; Van Der Vlugt, J.I.; De Bruin, B.; Reek, J.N.H. Water splitting by cooperative catalysis. Angew. Chem. Int. Ed. 2009, 48, 8178–8181. [Google Scholar] [CrossRef]
- Kern, J.; Chatterjee, R.; Young, I.D.; Fuller, F.D.; Lassalle, L.; Ibrahim, M.; Gul, S.; Fransson, T.; Brewster, A.S.; Alonso-Mori, R.; et al. Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 2018, 563, 421–425. [Google Scholar] [CrossRef]
- Schwarz, B.; Forster, J.; Goetz, M.K.; Yücel, D.; Berger, C.; Jacob, T.; Streb, C. Visible-Light-Driven Water Oxidation by a Molecular Manganese Vanadium Oxide Cluster. Angew. Chem. Int. Ed. 2016, 55, 6329–6333. [Google Scholar] [CrossRef] [PubMed]
- Al-Oweini, R.; Sartorel, A.; Bassil, B.S.; Natali, M.; Berardi, S.; Scandola, F.; Kortz, U.; Bonchio, M. Photocatalytic water oxidation by a mixed-valent MnIII3MnIVO3 manganese oxo core that mimics the natural oxygen-evolving center. Angew. Chem. Int. Ed. 2014, 11182–11185. [Google Scholar] [CrossRef] [PubMed]
- Dismukes, G.C.; Brimblecombe, R.; Felton, G.A.N.; Pryadun, R.S.; Sheats, J.E.; Spiccia, L.; Swiegers, G.F. Development of Bioinspired Mn4O4−Cubane Water Oxidation Catalysts: Lessons from Photosynthesis. Acc. Chem. Res. 2009, 42, 1935–1943. [Google Scholar] [CrossRef]
- Car, P.-E.; Guttentag, M.; Baldridge, K.K.; Alberto, R.; Patzke, G.R. Synthesis and characterization of open and sandwich-type polyoxometalates reveals visible-light-driven water oxidation via POM-photosensitizer complexes. Green Chem. 2012, 14, 1680–1688. [Google Scholar] [CrossRef]
- Sartorel, A.; Miró, P.; Salvadori, E.; Romain, S.; Carraro, M.; Scorrano, G.; Di Valentin, M.; Llobet, A.; Bo, C.; Bonchio, M. Water oxidation at a tetraruthenate core stabilized by polyoxometalate ligands: Experimental and computational evidence to trace the competent intermediates. J. Am. Chem. Soc. 2009, 131, 16051–16053. [Google Scholar] [CrossRef]
- Nishiki, K.; Umehara, N.; Kadota, Y.; López, X.; Poblet, J.M.; Mezui, C.A.; Teillout, A.L.; Mbomekalle, I.M.; De Oliveira, P.; Miyamoto, M.; et al. Preparation of α1- And α2-isomers of mono-Ru-substituted Dawson-type phosphotungstates with an aqua ligand and comparison of their redox potentials, catalytic activities, and thermal stabilities with Keggin-type derivatives. Dalton Trans. 2016, 45, 3715–3726. [Google Scholar] [CrossRef]
- Lang, Z.L.; Yang, G.C.; Ma, N.N.; Wen, S.Z.; Yan, L.K.; Guan, W.; Su, Z.M. DFT characterization on the mechanism of water splitting catalyzed by single-Ru-substituted polyoxometalates. Dalton Trans. 2013, 42, 10617–10625. [Google Scholar] [CrossRef]
- Kuznetsov, A.E.; Geletii, Y.V.; Hill, C.L.; Morokuma, K.; Musaev, D.G. Dioxygen and Water Activation Processes on Multi-Ru-Substituted Polyoxometalates: Comparison with the “Blue-Dimer” Water Oxidation Catalyst. J. Am. Chem. Soc. 2009, 131, 6844–6854. [Google Scholar] [CrossRef] [PubMed]
- Quiñonero, D.; Kaledin, A.L.; Kuznetsov, A.E.; Geletii, Y.V.; Besson, C.; Hill, C.L.; Musaev, D.G. Computational studies of the Geometry and electronic structure of an all-inorganic and homogeneous tetra-Ru-Polyoxotungstate catalyst for water oxidation and its four subsequent one-electron oxidized forms. J. Phys. Chem. A 2010, 114, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Piccinin, S.; Fabris, S. A first principles study of water oxidation catalyzed by a tetraruthenium-oxo core embedded in polyoxometalate ligands. Phys. Chem. Chem. Phys. 2011, 13, 7666–7674. [Google Scholar] [CrossRef] [PubMed]
- Piccinin, S.; Sartorel, A.; Aquilanti, G.; Goldoni, A.; Bonchio, M.; Fabris, S. Water oxidation surface mechanisms replicated by a totally inorganic tetraruthenium–oxo molecular complex. Proc. Natl. Acad. Sci. USA 2013, 110, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccinin, S.; Fabris, S. Water oxidation by Ru-Polyoxometalate Catalysts: Overpotential dependency on the number and charge of the metal centers. Inorganics 2015, 3, 374–387. [Google Scholar] [CrossRef]
- Su, X.F.; Guan, W.; Yan, L.K.; Lang, Z.L.; Su, Z.M. Evidence of two-state reactivity in water oxidation catalyzed by polyoxometalate-based complex [Mn3(H2O)3(SbW9O33)2]12−. J. Catal. 2019, 376, 146–149. [Google Scholar] [CrossRef]
- Su, X.-F.; Yan, L.-K.; Su, Z.-M. Theoretical Insight into the Performance of MnII/III-Monosubstituted Heteropolytungstates as Water Oxidation Catalysts. Inorg. Chem. 2019, 4. [Google Scholar] [CrossRef]
- Ma, C.; Piccinin, S.; Fabris, S. Interface structure and reactivity of water-oxidation Ru-polyoxometalate catalysts on functionalized graphene electrodes. Phys. Chem. Chem. Phys. 2014, 16, 5333–5341. [Google Scholar] [CrossRef]
- Lee, G.Y.; Kim, I.; Lim, J.; Yang, M.Y.; Choi, D.S.; Gu, Y.; Oh, Y.; Kang, S.H.; Nam, Y.S.; Kim, S.O. Spontaneous linker-free binding of polyoxometalates on nitrogen-doped carbon nanotubes for efficient water oxidation. J. Mater. Chem. A 2017, 5, 1941–1947. [Google Scholar] [CrossRef]
- Paille, G.; Gomez-Mingot, M.; Roch-Marchal, C.; Lassalle-Kaiser, B.; Mialane, P.; Fontecave, M.; Mellot-Draznieks, C.; Dolbecq, A. A Fully Noble Metal-Free Photosystem Based on Cobalt-Polyoxometalates Immobilized in a Porphyrinic Metal-Organic Framework for Water Oxidation. J. Am. Chem. Soc. 2018, 140, 3613–3618. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Choi, K.; Oh, H.; Kim, C.; Jeon, D.; Lee, C.; Lee, J.H.; Ryu, J. Cobalt polyoxometalate-derived CoWO4 oxygen-evolving catalysts for efficient electrochemical and photoelectrochemical water oxidation. J. Catal. 2018, 367, 212–220. [Google Scholar] [CrossRef]
- López, X.; Carbó, J.J.; Bo, C.; Poblet, J.M. Structure, properties and reactivity of polyoxometalates: A theoretical perspective. Chem. Soc. Rev. 2012, 41, 7537–7571. [Google Scholar] [CrossRef] [PubMed]
- Barnett, S.M.; Goldberg, K.I.; Mayer, J.M. A soluble copper-bipyridine water-oxidation electrocatalyst. Nat. Chem. 2012, 4, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Duan, L.; Bozoglian, F.; Mandal, S.; Stewart, B.; Privalov, T.; Llobet, A.; Sun, L. A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat. Chem. 2012, 4, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Geletii, Y.V.; Yin, Q.; Hou, Y.; Huang, Z.; Ma, H.; Song, J.; Besson, C.; Luo, Z.; Cao, R.; O’Halloran, K.P.; et al. Polyoxometalates in the design of effective and tunable water oxidation catalysts. Isr. J. Chem. 2011, 51, 238–246. [Google Scholar] [CrossRef]
- Yi, Y.; Weinberg, G.; Prenzel, M.; Greiner, M.; Heumann, S.; Becker, S.; Schlögl, R. Electrochemical corrosion of a glassy carbon electrode. Catal. Today 2017, 295, 32–40. [Google Scholar] [CrossRef]
- Yi, Y.; Tornow, J.; Willinger, E.; Willinger, M.G.; Ranjan, C.; Schlögl, R. Electrochemical Degradation of Multiwall Carbon Nanotubes at High Anodic Potential for Oxygen Evolution in Acidic Media. ChemElectroChem 2015, 2, 1929–1937. [Google Scholar] [CrossRef]
- Gao, G.; Vecitis, C.D. Electrocatalysis aqueous phenol with carbon nanotubes networks as anodes: Electrodes passivation and regeneration and prevention. Electrochim. Acta 2013, 98, 131–138. [Google Scholar] [CrossRef]
- Evolution, E.O.; Wang, H.; You, W.; Meng, B. A New 2D Layered CoII Complex Based on Monosubstituted Keggin Anions and its. J. Clust. Sci. 2010, 21, 857–865. [Google Scholar]
- Soriano-López, J.; Goberna-Ferrón, S.; Vigara, L.; Carbó, J.J.; Poblet, J.M.; Galán-Mascarós, J.R. Cobalt polyoxometalates as heterogeneous water oxidation catalysts. Inorg. Chem. 2013, 52, 4753–4755. [Google Scholar] [CrossRef]
- Misra, A.; Kozma, K.; Streb, C.; Nyman, M. Beyond Charge Balance: Counter-Cations in Polyoxometalate Chemistry. Angew. Chem. Int. Ed. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCrory, C.C.L.; Jung, S.; Ferrer, I.M.; Chatman, S.M.; Peters, J.C.; Jaramillo, T.F. Benchmarking Hydrogen Evolving Reaction and Oxygen Evolving Reaction Electrocatalysts for Solar Water Splitting Devices. J. Am. Chem. Soc. 2015, 137, 4347–4357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
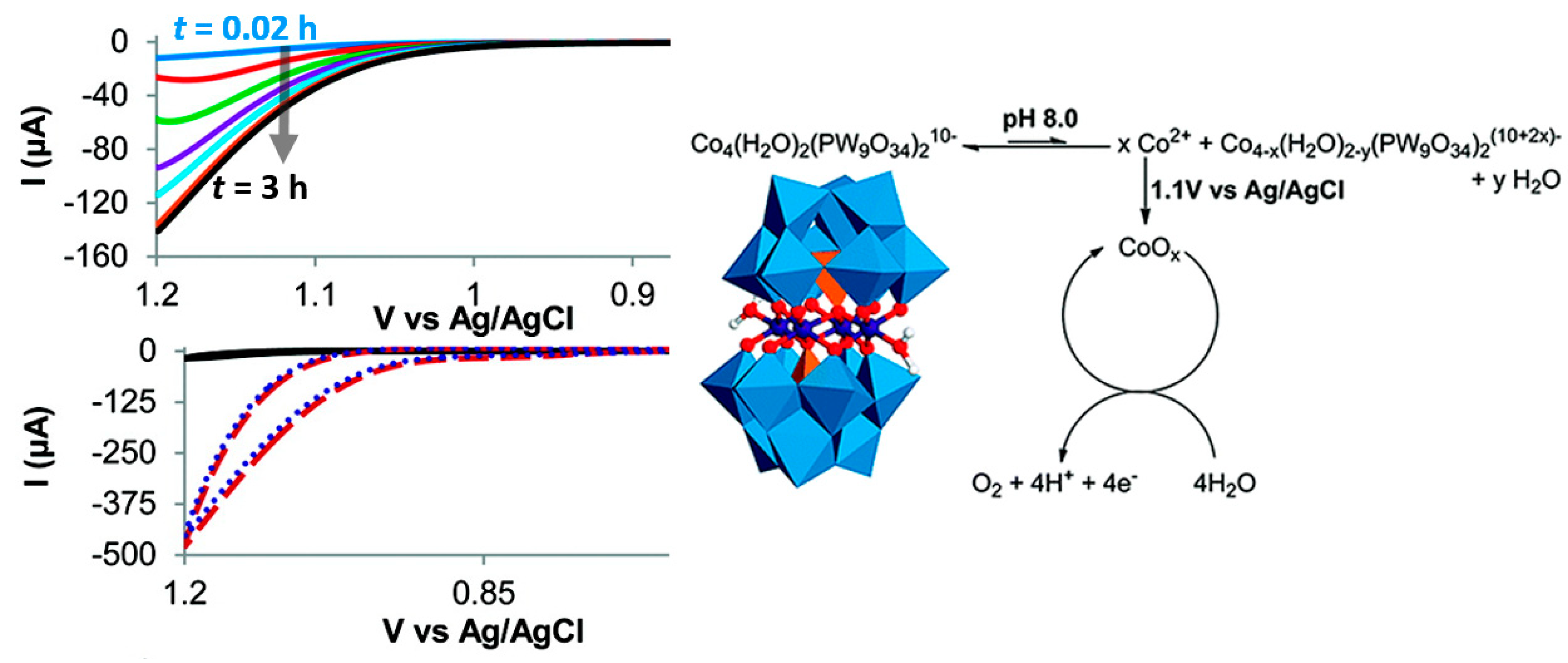
- Berardi, S.; Sartorel, A.; Bonchio, M. Is [Co4(H2O)2(α-PW9O34)2]10− a genuine molecular catalyst in Photochemical water oxidation? Answer from time-resolved hole-scavenging experiment. Chem. Commun. 2012, 48, 8808–8810. [Google Scholar]
- Stracke, J.J.; Finke, R.G. Electrocatalytic Water Oxidation Beginning with the Cobalt Polyoxometalate [Co4(H2O)2(PW9O34)2]10–: Identification of Heterogeneous CoOx as the Dominant Catalyst. J. Am. Chem. Soc. 2011, 133, 10–13. [Google Scholar] [CrossRef]
- Toma, F.M.; Sartorel, A.; Iurlo, M.; Carraro, M.; Parisse, P.; MacCato, C.; Rapino, S.; Gonzalez, B.R.; Amenitsch, H.; Da Ros, T.; et al. Efficient water oxidation at carbon nanotube-polyoxometalate electrocatalytic interfaces. Nat. Chem. 2010, 2, 826–831. [Google Scholar] [CrossRef]
- Toma, F.M.; Sartorel, A.; Carraro, M.; Bonchio, M.; Prato, M. Dendron-functionalized multiwalled carbon nanotubes incorporating polyoxometalates for water-splitting catalysis. Pure Appl. Chem. 2011, 83, 1529–1542. [Google Scholar] [CrossRef]
- Anwar, N.; Sartorel, A.; Yaqub, M.; Wearen, K.; Laffir, F.; Armstrong, G.; Dickinson, C.; Bonchio, M.; McCormac, T. Surface immobilization of a tetra-ruthenium substituted polyoxometalate water oxidation catalyst through the employment of conducting polypyrrole and the layer-by-layer (LBL) technique. ACS Appl. Mater. Interfaces 2014, 6, 8022–8031. [Google Scholar] [CrossRef]
- Brownson, D.A.C.; Kampouris, D.K.; Banks, C.E. An overview of graphene in energy production and storage applications. J. Power Sources 2011, 196, 4873–4885. [Google Scholar] [CrossRef]
- Feng, J.; Liang, Y.; Wang, H.; Li, Y.; Zhang, B.; Zhou, J.; Wang, J.; Regier, T. Engineering Manganese Oxide/Nanocarbon Hybrid Materials for Oxygen Reduction Electrocatalysis. Nano Res. 2012, 5, 718–725. [Google Scholar] [CrossRef]
- Yoo, E.; Okata, T.; Akita, T.; Kohyama, M. Enhanced Electrocatalytic Activity of Pt Subnanoclusters on Graphene. Nano Lett. 2009, 2, 4–8. [Google Scholar]
- Kou, R.; Shao, Y.; Wang, D.; Engelhard, M.H.; Hun, J.; Wang, J.; Viswanathan, V.V.; Wang, C.; Lin, Y.; Wang, Y.; et al. Electrochemistry Communications Enhanced activity and stability of Pt catalysts on functionalized graphene sheets for electrocatalytic oxygen reduction. Electrochem. Commun. 2009, 11, 954–957. [Google Scholar] [CrossRef]
- Guo, S.X.; Liu, Y.; Lee, C.Y.; Bond, A.M.; Zhang, J.; Geletii, Y.V.; Hill, C.L. Graphene-supported [{Ru4O4(OH)2(H2O)4}(γ-SiW10O36)2]10− for highly efficient electrocatalytic water oxidation. Energy Environ. Sci. 2013, 6, 2654–2663. [Google Scholar] [CrossRef]
- Wu, J.; Liao, L.; Yan, W.; Xue, Y.; Sun, Y.; Yan, X.; Chen, Y.; Xie, Y. Polyoxometalates immobilized in ordered mesoporous carbon nitride as highly efficient water oxidation catalysts. ChemSusChem 2012, 5, 1207–1212. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Liu, R.; Biskupek, J.; Kaiser, U.; Song, Y.-F.; Streb, C. Modular Design of Noble-Metal-Free Mixed Metal Oxide Electrocatalysts for Complete Water Splitting. Angew. Chem. Int. Ed. 2019, 58, 4644–4648. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Hu, J.; Diao, H.; Schwarz, B.; Streb, C.; Song, Y.-F. Robust Polyoxometalate/Nickel Foam Composite Electrodes for Sustained Electrochemical Oxygen Evolution at High pH. Angew. Chem. Int. Ed. 2017, 56, 4941–4944. [Google Scholar] [CrossRef]
- Lv, H.; Song, J.; Geletii, Y.V.; Vickers, J.W.; Sumliner, J.M.; Musaev, D.G.; Kögerler, P.; Zhuk, P.F.; Bacsa, J.; Zhu, G.; et al. An exceptionally fast homogeneous carbon-free cobalt-based water oxidation catalyst. J. Am. Chem. Soc. 2014, 136, 9268–9271. [Google Scholar] [CrossRef]
- Folkman, S.J.; Finke, R.G. Electrochemical Water Oxidation Catalysis Beginning with Co(II) Polyoxometalates: The Case of the Precatalyst Co4V2W18O6810–. ACS Catal. 2017, 7, 7–16. [Google Scholar] [CrossRef]
- Weinstock, I.A.; Barbuzzi, E.M.G.; Wemple, M.W.; Cowan, J.J.; Reiner, R.S.; Sonnen, D.M.; Heintz, R.A.; Bond, J.S.; Hill, C.L. Equilibrating metal-oxide cluster ensembles for oxidation reactions using oxygen in water. Nature 2001, 414, 191–195. [Google Scholar] [CrossRef]
- Kanan, M.W.; Nocera, D.G. In Situ Formation of an Oxygen evolvin Catalyst in Neutral Water Containing Phosphate and Co2+. Science 2008, 321, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Krolicka, A.; Bobrowski, A.; Kalcher, K.; Mocak, J.; Svancara, I.; Vytras, K. Study on catalytic adsorptive stripping voltammetry of trace cobalt at bismuth film electrodes. Electroanalysis 2003, 15, 1859–1863. [Google Scholar] [CrossRef]
- Lutterman, D.A.; Surendranath, Y.; Nocera, D.G. A self-healing oxygen-evolving catalyst. J. Am. Chem. Soc. 2009, 131, 3838–3839. [Google Scholar] [CrossRef] [PubMed]
- Kanan, M.W.; Surendranath, Y.; Nocera, D.G. Cobalt-phosphate oxygen-evolving compound. Chem. Soc. Rev. 2009, 38, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Vickers, J.W.; Lv, H.; Sumliner, J.M.; Zhu, G.; Luo, Z.; Musaev, D.G.; Geletii, Y.V.; Hill, C.L. Differentiating homogeneous and heterogeneous water oxidation catalysis: Confirmation that [Co4(H2O)2(α-PW9O34)2]10− is a molecular water oxidation catalyst. J. Am. Chem. Soc. 2013, 135, 14110–14118. [Google Scholar] [CrossRef] [PubMed]
- Sumliner, J.M.; Lv, H.; Fielden, J.; Geletii, Y.V.; Hill, C.L. Polyoxometalate multi-electron-transfer catalytic systems for water splitting. Eur. J. Inorg. Chem. 2014, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Folkman, S.J.; Soriano-Lopez, J.; Galán-Mascarós, J.R.; Finke, R.G. Electrochemically Driven Water-Oxidation Catalysis Beginning with Six Exemplary Cobalt Polyoxometalates: Is It Molecular, Homogeneous Catalysis or Electrode-Bound, Heterogeneous CoOx Catalysis? J. Am. Chem. Soc. 2018, 140, 12040–12055. [Google Scholar] [CrossRef] [Green Version]
- Folkman, S.J.; Kirner, J.T.; Finke, R.G. Cobalt Polyoxometalate Co4V2W18O6810–: A Critical Investigation of Its Synthesis, Purity, and Observed 51V Quadrupolar NMR. Inorg. Chem. 2016, 55, 5343–5355. [Google Scholar] [CrossRef]
- Ullman, A.M.; Liu, Y.; Huynh, M.; Bediako, D.K.; Wang, H.; Anderson, B.L.; Powers, D.C.; Breen, J.J.; Abruña, H.D.; Nocera, D.G. Water Oxidation Catalysis by Co(II) Impurities in Co(III)4O4 Cubanes. J. Am. Chem. Soc. 2014, 136, 17681–17688. [Google Scholar] [CrossRef] [Green Version]
- Surendranath, Y.; Lutterman, D.A.; Liu, Y.; Nocera, D.G. Nucleation, Growth, and Repair of a Cobalt-Based Oxygen Evolving Catalyst. J. Am. Chem. Soc. 2012, 134, 6326–6336. [Google Scholar] [CrossRef]
- Schwarz, B.; Forster, J.; Anjass, M.H.; Daboss, S.; Kranz, C.; Streb, C. From molecular to colloidal manganese vanadium oxides for water oxidation catalysis. Chem. Commun. 2017, 53, 11576–11579. [Google Scholar] [CrossRef]
- Wang, Y.; Weinstock, I.A. Polyoxometalate-decorated nanoparticles. Chem. Soc. Rev. 2012, 41, 7479. [Google Scholar] [CrossRef]
- Chakraborty, B.; Gan-Or, G.; Raula, M.; Gadot, E.; Weinstock, I.A. Design of an inherently-stable water oxidation catalyst. Nat. Commun. 2018, 9, 4896. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, B.; Gan-Or, G.; Duan, Y.; Raula, M.; Weinstock, I.A. Visible-Light-Driven Water Oxidation with a Polyoxometalate-Complexed Hematite Core of 275 Iron Atoms. Angew. Chem. Int. Ed. 2019, 58, 6584–6589. [Google Scholar] [CrossRef]
- Tsubaki, S.; Hayakawa, S.; Ueda, T.; Fujii, S.; Suzuki, E.; Zhang, J.; Bond, A.; Wada, Y. Radio frequency alternating electromagnetic field enhanced tetraruthenium polyoxometalate electrocatalytic water oxidation. Chem. Commun. 2019, 55, 1032–1035. [Google Scholar] [CrossRef] [PubMed]
- Garcés-Pineda, F.A.; Blasco-Ahicart, M.; Nieto-Castro, D.; López, N.; Galán-Mascarós, J.R. Direct magnetic enhancement of electrocatalytic water oxidation in alkaline media. Nat. Energy 2019, 4, 519–525. [Google Scholar] [CrossRef]
- Xing, X.; Liu, R.; Cao, K.; Kaiser, U.; Zhang, G.; Streb, C. Manganese Vanadium Oxide–N-Doped Reduced Graphene Oxide Composites as Oxygen Reduction and Oxygen Evolution Electrocatalysts. ACS Appl. Mater. Interfaces 2018, 10, 44511–44517. [Google Scholar] [CrossRef] [PubMed]
- Lauinger, S.M.; Piercy, B.D.; Li, W.; Yin, Q.; Collins-Wildman, D.L.; Glass, E.N.; Losego, M.D.; Wang, D.; Geletii, Y.V.; Hill, C.L. Stabilization of Polyoxometalate Water Oxidation Catalysts on Hematite by Atomic Layer Deposition. ACS Appl. Mater. Interfaces 2017, 9, 35048–35056. [Google Scholar] [CrossRef]
- Solarska, R.; Bienkowski, K.; Zoladek, S.; Majcher, A.; Stefaniuk, T.; Kulesza, P.J.; Augustynski, J. Enhanced Water Splitting at Thin Film Tungsten Trioxide Photoanodes Bearing Plasmonic Gold-Polyoxometalate Particles. Angew. Chem. Int. Ed. 2014, 53, 14196–14200. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, D.; Trentin, I.; Schwiedrzik, L.; González, L.; Streb, C. The Reactivity and Stability of Polyoxometalate Water Oxidation Electrocatalysts. Molecules 2020, 25, 157. https://doi.org/10.3390/molecules25010157
Gao D, Trentin I, Schwiedrzik L, González L, Streb C. The Reactivity and Stability of Polyoxometalate Water Oxidation Electrocatalysts. Molecules. 2020; 25(1):157. https://doi.org/10.3390/molecules25010157
Chicago/Turabian StyleGao, Dandan, Ivan Trentin, Ludwig Schwiedrzik, Leticia González, and Carsten Streb. 2020. "The Reactivity and Stability of Polyoxometalate Water Oxidation Electrocatalysts" Molecules 25, no. 1: 157. https://doi.org/10.3390/molecules25010157
APA StyleGao, D., Trentin, I., Schwiedrzik, L., González, L., & Streb, C. (2020). The Reactivity and Stability of Polyoxometalate Water Oxidation Electrocatalysts. Molecules, 25(1), 157. https://doi.org/10.3390/molecules25010157