
Two Beta-Phosphorylamide Compounds as Ligands for Sm3+, Eu3+, and Tb3+: X-ray Crystallography and Luminescence Properties

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis of the bis-CMPO Ligands 1 and 2, and the 1:1 Ln Complexes
2.2. Characterization of the Ligands and Complexes in the Solid State—IR Spectra and Single Crystal X-Ray Diffraction Studies
2.3. Characterization of the Ln-Ligand Complexes in Solution Using 1H-NMR
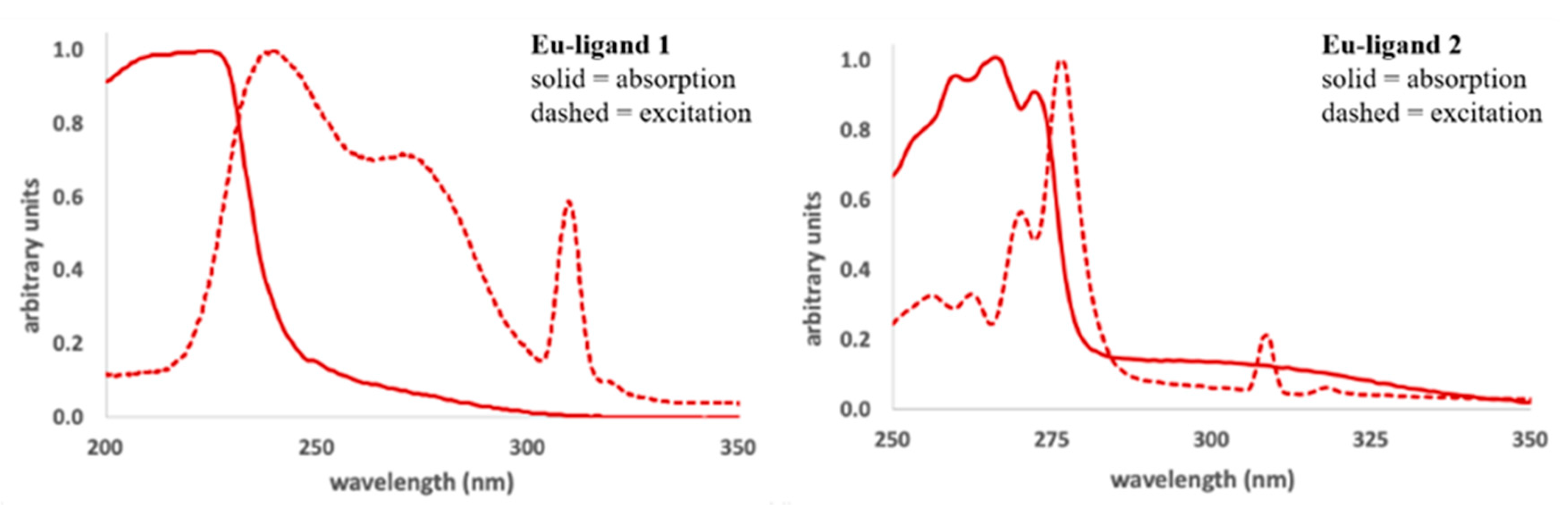
2.4. Characterization of the Ln-Ligand Complexes in Solution Using Luminescence
3. Scope and Outlook
4. Materials and Methods
4.1. General Considerations
4.2. Single Crystal X-Ray Crystallography
4.3. Photophysical Studies
4.4. Synthesis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cotton, S. Lanthanide and Actinide Chemistry; Wiley: Southern Gate, UK, 2006. [Google Scholar]
- Bünzli, J.-C.G.; Piguet, C. Taking Advantage of Luminescent Lanthanide Ions. Chem. Soc. Rev. 2005, 34, 1048–1077. [Google Scholar] [CrossRef] [PubMed]
- Bünzli, J.-C.G. Benefiting from the Unique Properties of Lanthanide Ions. Acc. Chem. Res. 2006, 39, 53–61. [Google Scholar] [CrossRef] [PubMed]
- De Bettencourt-Dias, A. Small Molecule Luminescent Lanthanide Ion Complexes-Photophysical Characterization and Recent Developments. Curr. Org. Chem. 2007, 11, 1460–1480. [Google Scholar] [CrossRef]
- De Bettencourt-Dias, A. Luminescence of Lanthanide Ions in Coordination Compounds and Nanomaterials, 1st ed.; Wiley: Chichester, UK, 2014; p. 384. [Google Scholar]
- Bünzli, J.-C.G. On the design of highly luminescent lanthanide complexes. Coord. Chem. Rev. 2015, 293–294, 19–47. [Google Scholar]
- Hemmilä, I.; Laitala, V. Progress in lanthanides as luminescent probes. J. Fluoresc. 2005, 15, 529–542. [Google Scholar] [CrossRef]
- Bünzli, J.-C.G. Lanthanide Luminescent Bioprobes (LLBs). Chem. Lett. 2009, 38, 104–109. [Google Scholar] [CrossRef]
- Thibon, A.; Pierre, V.C. Principles of Responsive Lanthanide-Based Luminescent Probes. Anal. Bioanal. Chem. 2009, 394, 107–120. [Google Scholar] [CrossRef]
- Parker, D. Luminescent Lanthanide Sensors for pH, pO2 and Selected Anions. Coord. Chem. Rev. 2000, 205, 109–130. [Google Scholar] [CrossRef] [Green Version]
- Spangler, C.; Schäferling, M. Luminescent Chemical and Physical Sensors Based on Lanthanide Complexes. In Lanthanide Luminescence: Photophysical, Analytical and Biological Aspects; Hänninen, P., Härmä, H., Eds.; Springer: Heidelberg, Germany; Dordrecht, The Netherlands; London, UK; New York, NY, USA, 2011; p. 400. [Google Scholar]
- Faulkner, S.; Pope, S.J.A.; Burton-Pye, B.P. Lanthanide complexes for luminescence imaging applications. Appl. Spectrosc. Rev. 2005, 40, 1–31. [Google Scholar] [CrossRef]
- Bünzli, J.-C.G. Lanthanide Luminescence for Biomedical Analyses and Imaging. Chem. Rev. 2010, 110, 2729–2755. [Google Scholar] [CrossRef]
- Sprecher, B.; Xiao, Y.; Walton, A.; Speight, J.; Harris, R.; Kleijn, R.; Visser, G.; Kramer, G.J. Life cycle inventory of the production of rare earths and the subsequent production of NdFeB rare earth permanent magnets. Environ. Sci. Technol. 2014, 48, 3951–3958. [Google Scholar] [CrossRef] [PubMed]
- Binnemans, K. Lanthanide-Based Luminescent Hybrid Materials. Chem. Rev. 2009, 109, 4283–4374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliseeva, S.V.; Bünzli, J.-C.G. Lanthanide Luminescence for Functional Materials and Bio-sciences. Chem. Soc. Rev. 2010, 39, 189–227. [Google Scholar] [CrossRef] [PubMed]
- Jowitt, S.M.; Werner, T.T.; Weng, Z.; Mudd, G.M. Recycling of the rare earth elements. Curr. Opin. Green Sustain. Chem. 2018, 13, 1–7. [Google Scholar] [CrossRef]
- Binnemans, K.; Jones, P.T.; Blanpain, B.; Van Gerven, T.; Yang, Y.; Walton, A.; Buchert, M. Recycling of rare earths: A critical review. J. Clean. Prod. 2013, 51, 1–22. [Google Scholar] [CrossRef]
- Haque, N.; Hughes, A.; Lim, S.; Vernon, C. Rare Earth Elements: Overview of Mining, Mineralogy, Uses, Sustainability and Environmental Impact. Resources 2014, 3, 614–635. [Google Scholar] [CrossRef] [Green Version]
- Kumari, A.; Panda, R.; Jha, M.K.; Kumar, J.R.; Lee, J.Y. Process development to recover rare earth metals from monazite mineral: A review. Miner. Eng. 2015, 79, 102–115. [Google Scholar] [CrossRef]
- Ansari, S.A.; Mohapatra, P.K. A review on solid phase extraction of actinides and lanthanides with amide based extractants. J. Chrom. A 2017, 1499, 1–20. [Google Scholar] [CrossRef]
- Horwitz, E.P.; Kalina, D.C.; Diamond, H.; Vandegrift, G.F.; Schulz, W.W. The TRUEX Process—A Process for the Extraction of the Transuranic Elements from Nitric Acid Wastes Utilizing Modified PUREX Solvent. Solvent Extr. Ion. Exch. 1985, 3, 75–109. [Google Scholar] [CrossRef]
- Hamdouchi, C.; de Blas, J.; del Prado, M.; Gruber, J.; Heinz, B.A.; Vance, L. 2-Amino-3-substituted-6-[(E)-1-phenyl-2-(N-methylcarbamoyl)vinyl]imidazo-[1,2-a]pyridines as a Novel Class of Inhibitors of Human Rhinovirus: Stereospecific Synthesis and Antiviral Activity. J. Med. Chem. 1999, 42, 50–59. [Google Scholar] [CrossRef]
- Arnaud-Neu, F.; Böhmer, V.; Dozol, J.-F.; Grüttner, C.; Jakobi, R.A.; Kraft, D.; Mauprivez, O.; Rouquette, H.; Schwing-Weill, M.-J.; Simon, N.; et al. Calixarenes with diphenylphosphoryl acetamide functions at the upper rim. A new class of highly efficient extractants for lanthanides and actinides. J. Chem. Soc. Perkin Trans. 1996, 2, 1175–1182. [Google Scholar] [CrossRef]
- VanderWeide, A.I.; Staples, R.J.; Biros, S.M. Crystal structures of two bis-carbamoylmethylphosphine oxide (CMPO) compounds. Acta Cryst. 2019, E75, 991–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoscup, J.A.; Staples, R.J.; Biros, S.M. Crystal structure of a samarium(III) nitrate chain cross-linked by a bis-carbamoylmethylphosphine oxide ligand. Acta Cryst. 2014, E70, 188–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spek, A.L. PLATON. Utrecht University, The Netherlands. 1980. Available online: https://www.platonsoft.nl/platon/ (accessed on 26 June 2020).
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Cryst. 2002, 36, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. 2009, D65, 148–155. [Google Scholar] [CrossRef]
- Spek, A.L. What makes a crystal structure report valid? Inorg. Chim. Acta 2018, 470, 232–237. [Google Scholar] [CrossRef]
- Spek, A.L. checkCIF validation ALERTS: What they mean and how to respond. Acta Cryst. 2020, E76, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Sartain, H.T.; McGraw, S.N.; Lawrence, C.T.; Werner, E.J.; Biros, S.M. A Novel Tripodal CMPO Ligand: Affinity for f-Elements, Computational Investigations and Luminescence Properties. Inorg. Chim. Acta 2015, 426, 126–135. [Google Scholar] [CrossRef]
- Coburn, K.M.; Hardy, D.A.; Patterson, M.G.; McGraw, S.N.; Peruzzi, M.T.; Boucher, F.; Beelen, B.; Sartain, H.T.; Neils, T.; Lawrence, C.L.; et al. f-Element Coordination and Extraction Selectivity of a Carbamoylmethylphosphine Oxide Ligand Based on a Tripodal Phosphine Oxide Scaffold. Inorg. Chim. Acta 2016, 449, 96–106. [Google Scholar] [CrossRef]
- Patterson, M.G.; Mulville, A.K.; Connor, E.K.; Henry, A.T.; Hudson, M.J.; Tissue, K.; Biros, S.M.; Werner, E.J. Lanthanide extraction selectivity of a tripodal carbamoylmethylphosphine oxide ligand system. Dalton Trans. 2018, 47, 14318–14326. [Google Scholar] [CrossRef]
- Cable, M.L.; Levine, D.J.; Kirby, J.P.; Gray, H.B.; Ponce, A. Luminescent Lanthanide Sensors. Adv. Inorg. Chem. 2011, 63, 1–45. [Google Scholar]
- De Bettencourt-Dias, A.; Rossini, J.S.K. Ligand design for luminescent lanthanide-containing metallopolymers. Inorg. Chem. 2016, 55, 9954–9963. [Google Scholar] [CrossRef] [PubMed]
- Weissman, S.I. Intramolecular Energy Transfer: The Fluorescence of Complexes of Europium. J. Chem. Phys. 1942, 10, 214–217. [Google Scholar] [CrossRef]
- 1.61, C. v. Software for the CCD Detector Systems for Determining Data Collection Parameters; Bruker Analytical X-ray Systems: Madison, WI, USA, 2009.
- A. Software for the CCD Detector System, v2010.11-3; Bruker Analytical X-ray Systems: Madison, WI, USA, 2010.
- Software for the Integration of CCD Detector System, 7.68A, S. v; Bruker Analytical X-ray Systems: Madison, WI, USA, 2010.
- Blessing, R.H. SADABS v2.008/2 Program for absorption correction using Bruker-AXS CCD based on the method of Robert Blessing. Acta Cryst. 1995, A51, 33–38. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained, refinement program for the modern computing environment-Olex2 dissected. Acta Cryst. 2015, A71, 59–75. [Google Scholar]
Sample Availability: Samples of the compounds 1 and 2 are available from the authors upon request. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stretch | Ligand 1 | Sm-1 | Eu-1 | Tb-1 | Ligand 2 | Sm-2 | Eu-2 | Tb-2 |
|---|---|---|---|---|---|---|---|---|
| C=O | 1666 | 1626 | 1627 | 1627 | 1661 | 1623 | 1621 | 1621 |
| P=O | 1203 | 1191 | 1191 | 1191 | 1173 | 1157 | 1156 | 1156 |
| P-O | 1016 | 1014 | 1013 | 1014 | ----- | ----- | ----- | ----- |
| N-H | 3268 | 3317 | 3328 | 3321 | 3271 | weak | weak | weak |
| Structure Number | 2 | [Sm(NO3)3(1)]2• (CH3CN) | [Tb(NO3)3(1)]2•(CH3CN) | [Sm(NO3)3(2)]2•H2O | [Tb(NO3)3(2)(H2O)2]2 | [Tb(NO3)3(2)(MeOH)]2 |
|---|---|---|---|---|---|---|
| CCDC number | 2003372 | 2003370 | 2003373 | 2003369 | 2003374 | 2003371 |
| Empirical formula | C30H30N2O4P2 | C32H66N12O34P4Sm2 | C32H66N12O34P4Tb2 | C60H64N10O28P4Sm2 | C60H72N10O32P4Tb2 | C64H76N10O30P4Tb2 |
| Formula weight | 544.50 | 1587.54 | 1604.68 | 1797.79 | 1886.99 | 1907.06 |
| Temperature/K | 173(2) | 173(2) | 173(2) | 173(2) | 173(2) | 173(2) |
| Crystal system | monoclinic | monoclinic | monoclinic | monoclinic | triclinic | tetragonal |
| Space group | P21/c | C2/c | C2/c | P21/n | P-1 | P-421c |
| a/Å | 5.65550(10) | 32.318(11) | 32.3189(5) | 10.5235(15) | 11.438(2) | 18.2898(4) |
| b/Å | 28.8994(5) | 12.538(4) | 12.5255(2) | 18.133(3) | 12.724(2) | 18.2898(4) |
| c/Å | 8.4517(2) | 16.447(6) | 16.4158(3) | 19.488(3) | 14.681(3) | 27.4073(6) |
| α/° | 90 | 90 | 90 | 90 | 91.784(2) | 90 |
| β/° | 109.4290(10) | 116.217(4) | 116.1620(10) | 96.944(12) | 106.593(2) | 90 |
| γ/° | 90 | 90 | 90 | 90 | 113.139(2) | 90 |
| Volume/Å3 | 1302.69(5) | 5979(4) | 5964.48(18) | 3691.6(9) | 1857.8(6) | 9168.2(4) |
| Z | 2 | 4 | 4 | 2 | 1 | 4 |
| ρcalcg/cm3 | 1.388 | 1.764 | 1.787 | 1.617 | 1.687 | 1.382 |
| μ/mm−1 | 1.848 | 2.154 | 13.430 | 13.393 | 2.068 | 8.794 |
| F(000) | 572.0 | 3192.0 | 3216.0 | 1804.0 | 948.0 | 3840.0 |
| Crystal size/mm3 | 0.169 × 0.093 × 0.035 | 0.279 × 0.218 × 0.082 | 0.264 × 0.173 × 0.103 | 0.262 × 0.126 × 0.063 | 0.187 × 0.11 × 0.038 | 0.284 × 0.2 × 0.183 |
| Radiation | CuKα (λ = 1.54178) | MoKα (λ = 0.71073) | CuKα (λ = 1.54178) | CuKα (λ = 1.54178) | MoKα (λ = 0.71073) | CuKα (λ = 1.54178) |
| 2Θ range for data collection/° | 6.116 to 136.61 | 3.54 to 52.802 | 6.094 to 140.374 | 6.68 to 143.44 | 3.528 to 50.95 | 5.81 to 136.676 |
| Index ranges | −6 ≤ h ≤ 6, −34 ≤ k ≤ 34, −10 ≤ l ≤ 10 | −40 ≤ h ≤ 40, −15 ≤ k ≤ 15, −20 ≤ l ≤ 20 | −39 ≤ h ≤ 39, −15 ≤ k ≤ 15, −17 ≤ l ≤ 19 | −12 ≤ h ≤ 12, −22 ≤ k ≤ 22, −23 ≤ l ≤ 21 | −13 ≤ h ≤ 13, −12 ≤ k ≤ 15, −17 ≤ l ≤ 16 | −22 ≤ h ≤ 22, −21 ≤ k ≤ 22, −33 ≤ l ≤ 33 |
| Reflections collected | 17,978 | 26,038 | 44,653 | 24,709 | 18,678 | 111,590 |
| Independent reflections | 2388 [Rint = 0.0757, Rsigma = 0.0373] | 6120 [Rint = 0.0770, Rsigma = 0.0704] | 5582 [Rint = 0.0588, Rsigma = 0.0348] | 6996 [Rint = 0.1460, Rsigma = 0.1397] | 6825 [Rint = 0.0585, Rsigma = 0.0735] | 8408 [Rint = 0.1335, Rsigma = 0.0634] |
| Data/restraints/parameters | 2388/0/213 | 6120/1/402 | 5582/0/402 | 6996/48/472 | 6825/0/492 | 8408/165/536 |
| Goodness-of-fit on F2 | 1.022 | 1.058 | 1.086 | 1.020 | 1.024 | 1.094 |
| Final R indexes [I> = 2σ (I)] | R1 = 0.0468, wR2 = 0.1144 | R1 = 0.0532, wR2 = 0.1296 | R1 = 0.0287, wR2 = 0.0636 | R1 = 0.0722, wR2 = 0.1524 | R1 = 0.0626, wR2 = 0.1578 | R1 = 0.0638, wR2 = 0.1895 |
| Final R indexes [all data] | R1 = 0.0610, wR2 = 0.1234 | R1 = 0.0776, wR2 = 0.1487 | R1 = 0.0336, wR2 = 0.0653 | R1 = 0.1453, wR2 = 0.1815 | R1 = 0.0791, wR2 = 0.1717 | R1 = 0.0827, wR2 = 0.2052 |
| Largest diff. peak/hole/e Å−3 | 0.67/−0.34 | 3.54/−0.79 | 0.57/−0.75 | 0.91/−0.91 | 3.52/−1.60 | 1.10/−0.77 |
| 0.023(4) |
| Ligand 1 [25] | [Sm(NO3)3(1)]2• (CH3CN) | [Sm(NO3)3(1)]2(polymer) [26] | [Tb(NO3)3(1)]2• (CH3CN) | |
|---|---|---|---|---|
| ligand | ||||
| C=O | 1.226(2) | 1.242(7), 1.242(7) | 1.245(5), 1.249(4) | 1.246(4), 1.245(4) |
| P=O | 1.474(2) | 1.473(4), 1.478(4) | 1.482(3), 1.485(3) | 1.477(2), 1.478(3) |
| P-O | 1.5791(16), 1.5619(15) | 1.551(5), 1.552(5), 1.551(5), 1.563(5) | 1.515(4), 1.537(4), 1.553(3), 1.542(3) | 1.554(2), 1.560(3), 1.564(3), 1.561(3) |
| Ln-O(C) | ----- | 2.387(4), 2.417(4) | 2.387(3), 2.382(3) | 2.378(2), 2.400(2) |
| Ln-O(P) | ----- | 2.400(4), 2.434(4) | 2.344(3), 2.340(3) | 2.390(2), 2.424(2) |
| C(O)-C-P(O) | 110.9(2) | 110.6(4), 113.1(4) | 109.7(3), 110.1(3) | 110.4(2), 111.2(2) |
| O(C)-Ln-O(P) | ----- | 75.24(14), 73.53(15) | 74.17(10), 74.70(9) | 75.44(8), 74.06(8) |
| inner sphere water | ||||
| Ln-O(water) | ----- | ----- | 2.413(3) | ----- |
| Ligand 2 | [Sm(NO3)3(2)]2•H2O | [Tb(NO3)3(2)(MeOH)]2 | [Tb(NO3)3(2)(H2O)2]2 | |
|---|---|---|---|---|
| ligand | ||||
| C=O | 1.224(3) | 1.251(11), 1.253(13) | 1.245(15), 1.255(14) | 1.262(8), 1.266(9) |
| P=O | 1.4912(18) | 1.511(7), 1.508(7) | 1.505(9) | 1.511(5), 1.509(5) |
| Ln-O(C) | ----- | 2.514(7), 2.396(7) | 2.365(8), 2.372(8) | 2.412(5), 2.352(5) |
| Ln-O(P) | ----- | 2.360(7), 2.387(7) | 2.332(8) | 2.277(5), 2.310(5) |
| C(O)-C-P(O) | 111.80(17) | 113.0(7), 115.0(8) | 111.7(9) | 112.8(5), 111.3(5) |
| O(C)-Ln-O(P) | ----- | 76.1(2), 74.5(2) | 75.3(3) | 79.05(17), 77.64(18) |
| inner sphere water | ||||
| Ln-O(water) | ----- | ----- | ----- | 2.356(5), 2.414(6) |
| Ligand 2 Complex | τACN/ms |
|---|---|
| Eu | 1.35 ± 0.08 (at 616 nm) |
| Tb | 1.83 ± 0.05 (at 545 nm) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lear, A.R.; Lenters, J.; Patterson, M.G.; Staples, R.J.; Werner, E.J.; Biros, S.M. Two Beta-Phosphorylamide Compounds as Ligands for Sm3+, Eu3+, and Tb3+: X-ray Crystallography and Luminescence Properties. Molecules 2020, 25, 2971. https://doi.org/10.3390/molecules25132971
Lear AR, Lenters J, Patterson MG, Staples RJ, Werner EJ, Biros SM. Two Beta-Phosphorylamide Compounds as Ligands for Sm3+, Eu3+, and Tb3+: X-ray Crystallography and Luminescence Properties. Molecules. 2020; 25(13):2971. https://doi.org/10.3390/molecules25132971
Chicago/Turabian StyleLear, Alan R., Jonah Lenters, Michael G. Patterson, Richard J. Staples, Eric J. Werner, and Shannon M. Biros. 2020. "Two Beta-Phosphorylamide Compounds as Ligands for Sm3+, Eu3+, and Tb3+: X-ray Crystallography and Luminescence Properties" Molecules 25, no. 13: 2971. https://doi.org/10.3390/molecules25132971
APA StyleLear, A. R., Lenters, J., Patterson, M. G., Staples, R. J., Werner, E. J., & Biros, S. M. (2020). Two Beta-Phosphorylamide Compounds as Ligands for Sm3+, Eu3+, and Tb3+: X-ray Crystallography and Luminescence Properties. Molecules, 25(13), 2971. https://doi.org/10.3390/molecules25132971