
Adhesion and Stability of Nanocellulose Coatings on Flat Polymer Films and Textiles

 , ,
, ,
Abstract
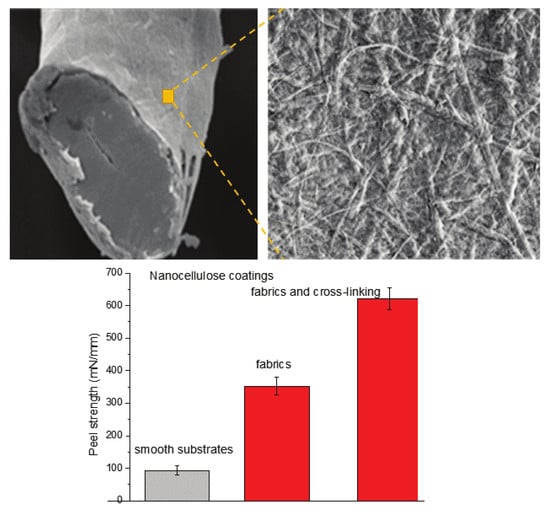
:
1. Introduction
2. Results and Discussion
2.1. Fabrication of Uniform Nanocellulose Coatings on the Surface of Polymeric Materials
2.2. Mechanisms of the Nanocellulose Coating Degradation in a Wet State
2.3. Adhesive Behavior of Nanocellulose Coatings
2.4. Covalent Cross-Linking of Nanocellulose Coatings
3. Materials and Methods
3.1. Materials
3.2. Synthesis of P(GMA-OEGMA) Copolymer
3.3. Preparation of Polymer Substrates for Nanocellulose Deposition
3.4. Nanocellulose Coatings on Si-Wafers and Polymer-Coated Si-Wafers
3.5. Characterization of Coatings
3.6. Structural Characterization
3.7. Cross-Linking of Nanocellulose Coatings
3.8. T-Peel Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Henriksson, M.; Berglund, L.A. Structure and properties of cellulose nanocomposite films containing melamine formaldehyde. J. Appl. Polym. Sci. 2007, 106, 2817–2824. [Google Scholar] [CrossRef]
- Iwamoto, S.; Nakagaito, A.; Yano, H. Nano-fibrillation of pulp fibers for the processing of transparent nanocomposites. Appl. Phys. A Mater. Sci. Process. 2007, 89, 461–466. [Google Scholar] [CrossRef]
- Azzam, F.; Moreau, C.L.; Cousin, F.; Menelle, A.; Bizot, H.; Cathala, B. Cellulose nanofibril-based multilayered thin films: Effect of ionic strength on porosity, swelling, and optical properties. Langmuir 2014, 30, 8091–8100. [Google Scholar] [CrossRef] [PubMed]
- Kalia, S.; Dufresne, A.; Cherian, B.M.; Kaith, B.; Avérous, L.; Njuguna, J.; Nassiopoulos, E. Cellulose-based bio-and nanocomposites: A review. Int. J. Polym. Sci. 2011, 2011, 1–35. [Google Scholar] [CrossRef]
- Daicho, K.; Saito, T.; Fujisawa, S.; Isogai, A. The Crystallinity of Nanocellulose: Dispersion-Induced Disordering of the Grain Boundary in Biologically Structured Cellulose. ACS Appl. Nano Mater. 2018, 1, 5774–5785. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.; Sain, M.; Kortschot, M. Cellulose microfibrils: A novel method of preparation using high shear refining and cryocrushing. Holzforschung 2005, 59, 102–107. [Google Scholar] [CrossRef]
- Johnson, R.K.; Zink-Sharp, A.; Renneckar, S.H.; Glasser, W.G. A new bio-based nanocomposite: Fibrillated TEMPO-oxidized celluloses in hydroxypropylcellulose matrix. Cellulose 2009, 16, 227–238. [Google Scholar] [CrossRef]
- Lavoine, N.; Desloges, I.; Dufresne, A.; Bras, J. Microfibrillated cellulose—Its barrier properties and applications in cellulosic materials: A review. Carbohydr. Polym. 2012, 90, 735–764. [Google Scholar] [CrossRef]
- Nishiyama, Y. Structure and properties of the cellulose microfibril. J. Wood Sci. 2009, 55, 241–249. [Google Scholar] [CrossRef]
- Wang, S.; Cheng, Q. A novel process to isolate fibrils from cellulose fibers by high-intensity ultrasonication, Part 1: Process optimization. J. Appl. Polym. Sci. 2009, 113, 1270–1275. [Google Scholar] [CrossRef]
- Missoum, K.; Belgacem, M.N.; Bras, J. Nanofibrillated cellulose surface modification: A review. Materials 2013, 6, 1745–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Kimura, S.; Nishiyama, Y.; Isogai, A. Cellulose nanofibers prepared by TEMPO-mediated oxidation of native cellulose. Biomacromolecules 2007, 8, 2485–2491. [Google Scholar] [CrossRef]
- Saito, T.; Nishiyama, Y.; Putaux, J.-L.; Vignon, M.; Isogai, A. Homogeneous suspensions of individualized microfibrils from TEMPO-catalyzed oxidation of native cellulose. Biomacromolecules 2006, 7, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- Moon, R.J.; Martini, A.; Nairn, J.; Simonsen, J.; Youngblood, J. Cellulose nanomaterials review: Structure, properties and nanocomposites. Chem. Soc. Rev. 2011, 40, 3941–3994. [Google Scholar] [CrossRef] [PubMed]
- Klemm, D.; Kramer, F.; Moritz, S.; Lindström, T.; Ankerfors, M.; Gray, D.; Dorris, A. Nanocelluloses: A new family of nature-based materials. Angew. Chem. Int. Ed. 2011, 50, 5438–5466. [Google Scholar] [CrossRef] [PubMed]
- Atalla, R.H.; Brady, J.W.; Matthews, J.F.; Ding, S.Y.; Himmel, M.E. Structures of plant cell wall celluloses. In Biomass Recalcitrance: Deconstructing the Plant Cell Wall for Bioenergy; Wiley-Blackwell: Chichester, UK, 2008; pp. 188–212. [Google Scholar]
- Salas, C.; Nypelö, T.; Rodriguez-Abreu, C.; Carrillo, C.; Rojas, O.J. Nanocellulose properties and applications in colloids and interfaces. Curr. Opin. Colloid Interface Sci. 2014, 19, 383–396. [Google Scholar] [CrossRef]
- Habibi, Y.; Lucia, L.A.; Rojas, O.J. Cellulose nanocrystals: Chemistry, self-assembly, and applications. Chem. Rev. 2010, 110, 3479–3500. [Google Scholar] [CrossRef]
- Johar, N.; Ahmad, I.; Dufresne, A. Extraction, preparation and characterization of cellulose fibres and nanocrystals from rice husk. Ind. Crops Prod. 2012, 37, 93–99. [Google Scholar] [CrossRef]
- Usov, I.; Nyström, G.; Adamcik, J.; Handschin, S.; Schütz, C.; Fall, A.; Bergström, L.; Mezzenga, R. Understanding nanocellulose chirality and structure-properties relationship at the single fibril level. Nat. Commun. 2015, 6, 7564. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Hsieh, Y.L. Chemically and mechanically isolated nanocellulose and their self-assembled structures. Carbohydr. Polym. 2013, 95, 32–40. [Google Scholar] [CrossRef]
- Zhao, Y.; Simonsen, J.; Cavender, G.; Jung, J.; Fuchigami, L.H. Nano-Cellulose Coatings to Prevent Damage in Foodstuffs. U.S. Patent 10,334,863, 2 July 2019. [Google Scholar]
- Vilarinho, F.; Sanches-Silva, A.; Vaz, M.F.; Farinha, J.P. Nanocellulose: A benefit for green food packaging. Crit. Rev. Food Sci. Nutr. 2016, 58, 1526–1537. [Google Scholar] [CrossRef] [PubMed]
- Rampazzo, R.; Mascheroni, E.; Fasano, F.; Mari, M.; Piergiovanni, L. Strategies for implementing nano-cellulose coatings in flexible packaging. Ital. J. Food Sci. 2015, 13–17. [Google Scholar]
- Li, F.; Biagioni, P.; Bollani, M.; Maccagnan, A.; Piergiovanni, L. Multi-functional coating of cellulose nanocrystals for flexible packaging applications. Cellulose 2013, 20, 2491–2504. [Google Scholar] [CrossRef] [Green Version]
- Aulin, C.; Gallstedt, M.; Lindstrom, T. Oxygen and oil barrier properties of microfibrillated cellulose films and coatings. Cellulose 2010, 17, 559–574. [Google Scholar] [CrossRef]
- Jabbar, A.; Militký, J.; Wiener, J.; Kale, B.M.; Ali, U.; Rwawiire, S. Nanocellulose coated woven jute/green epoxy composites: Characterization of mechanical and dynamic mechanical behavior. Compos. Struct. 2017, 161, 340–349. [Google Scholar] [CrossRef]
- Jiang, F.; Hsieh, Y.L. Amphiphilic superabsorbent cellulose nanofibril aerogels. J. Mater. Chem. A 2014, 2, 6337–6342. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Li, Y.L.; Hong, F.; Tang, S.J.; Wang, Y.Y. Nano-cellulose coating small-caliber artificial blood vessel. Adv. Mater. Res. 2011, 332, 1794–1798. [Google Scholar] [CrossRef]
- Ullah, H.; Santos, H.A.; Khan, T. Applications of bacterial cellulose in food, cosmetics and drug delivery. Cellulose 2016, 23, 2291–2314. [Google Scholar] [CrossRef]
- Zhou, J.; Hsieh, Y.L. Nanocellulose aerogel-based porous coaxial fibers for thermal insulation. Nano Energy 2020, 68, 9. [Google Scholar] [CrossRef] [Green Version]
- Osong, S.H.; Norgren, S.; Engstrand, P. Processing of wood-based microfibrillated cellulose and nanofibrillated cellulose, and applications relating to papermaking: A review. Cellulose 2016, 23, 93–123. [Google Scholar] [CrossRef]
- Carpenter, A.W.; de Lannoy, C.F.; Wiesner, M.R. Cellulose nanomaterials in water treatment technologies. Environ. Sci. Technol. 2015, 49, 5277–5287. [Google Scholar] [CrossRef]
- Nemoto, J.; Saito, T.; Isogai, A. Simple freeze-drying procedure for producing nanocellulose aerogel-containing, high-performance air filters. ACS Appl. Mater. Interfaces 2015, 7, 19809–19815. [Google Scholar] [CrossRef] [PubMed]
- Tao, X. Handbook of Smart Textiles; Springer: Singapore, 2015. [Google Scholar]
- Song, J.H.; Murphy, R.J.; Narayan, R.; Davies, G.B.H. Biodegradable and Compostable Alternatives to Conventional Plastics. Philos. Trans. R. Soc. B-Biol. Sci. 2009, 364, 2127–2139. [Google Scholar] [CrossRef] [PubMed]
- Minko, S.; Sharma, S.; Hardin, I.; Luzinov, I.; Daubenmire, S.W.; Zakharchenko, A.; Saremi, R.; Kim, Y.S. Textile Dyeing Using Nanocellulosic Fibers. U.S. Patent US20160010275A1, 29 November 2016. [Google Scholar]
- Kim, Y.; McCoy, L.T.; Lee, E.; Lee, H.; Saremi, R.; Feit, C.; Hardin, I.R.; Sharma, S.; Mani, S.; Minko, S. Environmentally sound textile dyeing technology with nanofibrillated cellulose. Green Chem. 2017, 19, 4031–4035. [Google Scholar] [CrossRef]
- Liyanapathiranage, A.; Peña, M.J.; Sharma, S.; Minko, S. Nanocellulose-Based Sustainable Dyeing of Cotton Textiles with Minimized Water Pollution. ACS Omega 2020, 5, 9196–9203. [Google Scholar] [CrossRef] [PubMed]
- Gardner, D.J.; Oporto, G.S.; Mills, R.; Samir, M.A.S.A. Adhesion and surface issues in cellulose and nanocellulose. J. Adhes. Sci. Technol. 2008, 22, 545–567. [Google Scholar] [CrossRef] [Green Version]
- Sinko, R.; Qin, X.; Keten, S. Interfacial mechanics of cellulose nanocrystals. MRS Bull. 2015, 40, 340–348. [Google Scholar] [CrossRef]
- d’Eon, J.; Zhang, W.; Chen, L.; Berry, R.M.; Zhao, B.X. Coating cellulose nanocrystals on polypropylene and its film adhesion and mechanical properties. Cellulose 2017, 24, 1877–1888. [Google Scholar] [CrossRef]
- Hossain, L.; Raghuwanshi, V.S.; Tanner, J.; Wu, C.M.; Kleinerman, O.; Cohen, Y.; Garnier, G. Structure and swelling of cross-linked nanocellulose foams. J. Colloid Interface Sci. 2020, 568, 234–244. [Google Scholar] [CrossRef]
- Aulin, C.; Ahola, S.; Josefsson, P.; Nishino, T.; Hirose, Y.; Osterberg, M.; Wagberg, L. Nanoscale Cellulose Films with Different Crystallinities and Mesostructures-Their Surface Properties and Interaction with Water. Langmuir 2009, 25, 7675–7685. [Google Scholar] [CrossRef]
- Ahola, S.; Salmi, J.; Johansson, L.-S.; Laine, J.; Österberg, M. Model films from native cellulose nanofibrils. Preparation, swelling, and surface interactions. Biomacromolecules 2008, 9, 1273–1282. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Notley, S.M.; Wågberg, L. Cellulose thin films: Degree of cellulose ordering and its influence on adhesion. Biomacromolecules 2007, 8, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Fält, S.; Wågberg, L.; Vesterlind, E.-L.; Larsson, P.T. Model films of cellulose ID–improved preparation method and characterization of the cellulose film. Cellulose 2004, 11, 151–162. [Google Scholar] [CrossRef]
- Gunnars, S.; Wågberg, L.; Stuart, M.C. Model films of cellulose: I. Method development and initial results. Cellulose 2002, 9, 239–249. [Google Scholar] [CrossRef]
- Hoeger, I.; Rojas, O.J.; Efimenko, K.; Velev, O.D.; Kelley, S.S. Ultrathin film coatings of aligned cellulose nanocrystals from a convective-shear assembly system and their surface mechanical properties. Soft Matter 2011, 7, 1957–1967. [Google Scholar] [CrossRef]
- Notley, S.M.; Eriksson, M.; Wågberg, L.; Beck, S.; Gray, D.G. Surface forces measurements of spin-coated cellulose thin films with different crystallinity. Langmuir 2006, 22, 3154–3160. [Google Scholar] [CrossRef] [PubMed]
- Sczech, R.; Riegler, H. Molecularly smooth cellulose surfaces for adhesion studies. J. Colloid Interface Sci. 2006, 301, 376–385. [Google Scholar] [CrossRef]
- Wågberg, L.; Decher, G.; Norgren, M.; Lindström, T.; Ankerfors, M.; Axnäs, K. The build-up of polyelectrolyte multilayers of microfibrillated cellulose and cationic polyelectrolytes. Langmuir 2008, 24, 784–795. [Google Scholar] [CrossRef]
- Yokota, S.; Kitaoka, T.; Wariishi, H. Surface morphology of cellulose films prepared by spin coating on silicon oxide substrates pretreated with cationic polyelectrolyte. Appl. Surf. Sci. 2007, 253, 4208–4214. [Google Scholar] [CrossRef]
- Savchak, M.; Borodinov, N.; Burtovyy, R.; Anayee, M.; Hu, K.; Ma, R.; Grant, A.; Li, H.; Cutshall, D.B.; Wen, Y.; et al. Highly conductive and transparent reduced graphene oxide nanoscale films via thermal conversion of polymer-encapsulated graphene oxide sheets. ACS Appl. Mater. Interfaces 2018, 10, 3975–3985. [Google Scholar] [CrossRef]
- Doszlop, S.; Vargha, V.; Horkay, F. Reactions of epoxy with other functional groups and the arising sec-hydroxyl groups. Periodica Polytechn. Chem. Eng. 1978, 22, 253–275. [Google Scholar]
- Cay, A.; Atrav, R.; Duran, K. Effects of warp-weft density variation and fabric porosity of the cotton fabrics on their colour in reactive dyeing. Fibres Text. East. Eur. 2007, 60, 91–94. [Google Scholar]
- Allen, T. Non-aqueous ester cross-linking of cotton cellulose. Text. Res. J. 1964, 34, 331–336. [Google Scholar] [CrossRef]
- Chen, D.; Yang, C.Q.; Qiu, X. Aqueous polymerization of maleic acid and cross-linking of cotton cellulose by poly (maleic acid). Ind. Eng. Chem. Res. 2005, 44, 7921–7927. [Google Scholar] [CrossRef]
- Gagliardi, D.; Shippee, F. Crosslinking of cellulose with polycarboxylic acids. Am. Dyest. Rep. 1963, 52, 74–77. [Google Scholar]
- Rowland, S.P.; Welch, C.M.; Brannan, M.A.F.; Gallagher, D.M. Introduction of ester cross links into cotton cellulose by a rapid curing process. Text. Res. J. 1967, 37, 933–941. [Google Scholar] [CrossRef]
- Welch, C. Formaldehyde Free Durable Press Finishing in Surface Characterization of Fibres and Textiles; Marcel Dekker: New York, NY, USA, 2000. [Google Scholar]
- Welch, C.M. Tetracarboxylic acids as formaldehyde-free durable press finishing agents: Part i: Catalyst, additive, and durability studies. Text. Res. J. 1988, 58, 480–486. [Google Scholar] [CrossRef]
- Yang, C.Q.; Chen, D.; Guan, J.; He, Q. Cross-linking cotton cellulose by the combination of maleic acid and sodium hypophosphite. 1. Fabric wrinkle resistance. Ind. Eng. Chem. Res. 2010, 49, 8325–8332. [Google Scholar] [CrossRef]
- Yang, C.Q.; Wang, X. Formation of cyclic anhydride intermediates and esterification of cotton cellulose by multifunctional carboxylic acids: An infrared spectroscopy study. Text. Res. J. 1996, 66, 595–603. [Google Scholar] [CrossRef]
- Yadavalli, N.S.; Borodinov, N.; Choudhury, C.K.; Quiñones-Ruiz, T.; Laradji, A.M.; Tu, S.; Lednev, I.K.; Kuksenok, O.; Luzinov, I.; Minko, S. Thermal Stabilization of Enzymes with Molecular Brushes. ACS Catalysis 2017, 7, 8675–8684. [Google Scholar] [CrossRef]
- Borodinov, N.; Gil, D.; Savchak, M.; Gross, C.E.; Yadavalli, N.S.; Ma, R.; Tsukruk, V.V.; Minko, S.; Vertegel, A.; Luzinov, I. En route to practicality of the polymer grafting technology: One-step interfacial modification with amphiphilic molecular brushes. ACS Appl. Mater. Interfaces 2018, 10, 13941–13952. [Google Scholar] [CrossRef] [PubMed]
- McCormick, C.L. Novel Cellulose Solutions. U.S. Patent 4,278,790, 14 July 1981. [Google Scholar]
- McCormick, C.L.; Callais, P.A.; Hutchinson, B.H., Jr. Solution studies of cellulose in lithium chloride and N, N-dimethylacetamide. Macromolecules 1985, 18, 2394–2401. [Google Scholar] [CrossRef]
- Mayer, E. Porometry Characterization of Filtration Media. Filtn News 2002, 20, 1–7. [Google Scholar]
- Kawashita, L.F.; Moore, D.R. ICPeel Digitised Stress-Strain. Available online: http://www.imperial.ac.uk/media/imperial-college/research-centres-and-groups/adhesion-and-adhesives-group/17285696.XLS (accessed on 8 February 2017).
Sample Availability: Samples of the nanocellulose coatings labeled with reactive dyes are available from the authors for a limited period. |













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Film Thickness, H, and A fraction of Washed-Out Coating from Different Substrates, F (10% Error) | |||||
|---|---|---|---|---|---|---|
| CL | PET | PA 6,6 | ||||
| H, nm | F, % | H, nm | F, % | H, nm | F, % | |
| NFC | 50 | 68 | non-uniform | 71 | non-uniform | 40 |
| PEI-NFC | 46 | 22 | 42 | 18 | - | - |
| NFC+PEI | 20 | 70 | 77 | 81 | 53 | 37 |
| NFC+P(GMA-OEGMA) | 33 | 77 | - | - | 401 | 25 |
| NCC | 21 | 5 | non-uniform | 93 | non-uniform | 93 |
| PEI-NCC | 89 | 53 | 113 | 60 | - | - |
| NCC+PEI | 40 | 25 | 77 | 12 | 65 | 77 |
| NCC+P(GMA-OEGMA) | 8 | 11 | 80 | 1 | 133 | 2 |
| Samples | Warp Density, Yarns/cm | Weft Density, Yarns/cm | MFPD, µm |
|---|---|---|---|
| Cotton | 33 | 29 | 67 |
| Polyester | 22 | 17 | 18 |
| Nylon | 20 | 11 | 64 |
| Cotton/Polyester (50%/50%) | 21 | 19 | 40 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saremi, R.; Borodinov, N.; Laradji, A.M.; Sharma, S.; Luzinov, I.; Minko, S. Adhesion and Stability of Nanocellulose Coatings on Flat Polymer Films and Textiles. Molecules 2020, 25, 3238. https://doi.org/10.3390/molecules25143238
Saremi R, Borodinov N, Laradji AM, Sharma S, Luzinov I, Minko S. Adhesion and Stability of Nanocellulose Coatings on Flat Polymer Films and Textiles. Molecules. 2020; 25(14):3238. https://doi.org/10.3390/molecules25143238
Chicago/Turabian StyleSaremi, Raha, Nikolay Borodinov, Amine Mohamed Laradji, Suraj Sharma, Igor Luzinov, and Sergiy Minko. 2020. "Adhesion and Stability of Nanocellulose Coatings on Flat Polymer Films and Textiles" Molecules 25, no. 14: 3238. https://doi.org/10.3390/molecules25143238
APA StyleSaremi, R., Borodinov, N., Laradji, A. M., Sharma, S., Luzinov, I., & Minko, S. (2020). Adhesion and Stability of Nanocellulose Coatings on Flat Polymer Films and Textiles. Molecules, 25(14), 3238. https://doi.org/10.3390/molecules25143238