
Conformational Restriction of Histamine with a Rigid Bicyclo[3.1.0]hexane Scaffold Provided Selective H3 Receptor Ligands

 ,
, 
Abstract
:
1. Introduction
2. Results
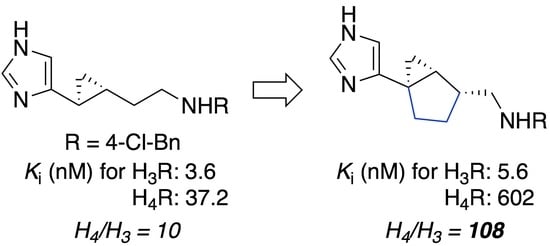
2.1. Design
2.2. Chemistry
2.3. Biological Evaluation
3. Discussion
4. Materials and Methods
4.1. Synthesis
4.2. Biological Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arrang, J.M.; Garbarg, M.; Schwartz, J.C. Auto-inhibition of brain histamine-release mediated by a novel class (H-3) of histamine-receptor. Nature 1983, 302, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Lovenberg, T.W.; Roland, B.L.; Wilson, S.J.; Jiang, X.X.; Pyati, J.; Huvar, A.; Jackson, M.R.; Erlander, M.G. Cloning and functional expression of the human histamine H-3 receptor. Mol. Pharmacol. 1999, 55, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Leurs, R.; Bakker, R.A.; Timmerman, H.; de Esch, I.J.P. The histamine H3 receptor: From gene cloning to H3 receptor drugs. Nat. Rev. Drug Discov. 2005, 4, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Celanire, S.; Wijtmans, M.; Talaga, P.; Leurs, R.; de Esch, I.J.P. Histamine H-3 receptor antagonists reach out for the clinic. Drug Discov. Today 2005, 10, 1613–1627. [Google Scholar] [CrossRef]
- Berlin, M.; Boyce, C.W.; Ruiz Mde, L. Histamine H3 receptor as a drug discovery target. J. Med. Chem. 2011, 54, 26–53. [Google Scholar]
- Romigi, A.; Vitrani, G.; Lo Giudice, T.; Centonze, D.; Franco, V. Profile of pitolisant in the management of narcolepsy: Design, development, and place in therapy. Drug Des. Devel. Ther. 2018, 12, 2665–2675. [Google Scholar] [CrossRef] [Green Version]
- Łażewska, D.; Kieć-Kononowicz, K. Progress in the development of histamine H3 receptor antagonists/inverse agonists: A patent review (2013–2017). Expert Opin. Ther. Pat. 2018, 28, 175–196. [Google Scholar] [CrossRef]
- Liu, C.L.; Ma, X.J.; Jiang, X.X.; Wilson, S.J.; Hofstra, C.L.; Blevitt, J.; Pyati, J.; Li, X.B.; Chai, W.Y.; Carruthers, N.; et al. Cloning and pharmacological characterization of a fourth histamine receptor (H-4) expressed in bone marrow. Mol. Pharmacol. 2001, 59, 420–426. [Google Scholar] [CrossRef]
- Nguyen, T.; Shapiro, D.A.; George, S.R.; Setola, V.; Lee, D.K.; Cheng, R.; Rauser, L.; Lee, S.P.; Lynch, K.R.; Roth, B.L.; et al. Discovery of a novel member of the histamine receptor family. Mol. Pharmacol. 2001, 59, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Mizuno, A.; Matsui, K.; Shuto, S. From peptides to peptidomimetics: A strategy based on the structural features of cyclopropane. Chem. Eur. J. 2017, 23, 14394–14409. [Google Scholar] [CrossRef]
- Kazuta, Y.; Matsuda, A.; Shuto, S. Development of versatile cis- and trans-dicarbon-substituted chiral cyclopropane units: Synthesis of (1S,2R)- and (1R,2R)-2-aminomethyl-1-(1H-imidazol-4-yl)cyclopropanes and their enantiomers as conformationally restricted analogues of histamine. J. Org. Chem. 2002, 67, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Kazuta, Y.; Hirano, K.; Natsume, K.; Yamada, S.; Kimura, R.; Matsumoto, S.-i.; Furuichi, K.; Matsuda, A.; Shuto, S. Cyclopropane-based conformational restriction of histamine. (1S,2S)-2-(2-Aminoethyl)-1-(1H-imidazol-4-yl)cyclopropane, a highly selective agonist for the histamine H3 receptor, having a cis-cyclopropane structure. J. Med. Chem. 2003, 46, 1980–1988. [Google Scholar] [PubMed]
- Watanabe, M.; Yamaguchi, K.; Tang, W.; Yoshida, K.; Silverman, R.B.; Arisawa, M.; Shuto, S. Synthesis of a series of 3,4-methanoarginines as side-chain conformationally restricted analogues of arginine. Bioorg. Med. Chem. 2011, 19, 5984–5988. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Kobayashi, T.; Hirokawa, T.; Yoshida, A.; Ito, Y.; Yamada, S.; Orimoto, N.; Yamasaki, Y.; Arisawa, M.; Shuto, S. Cyclopropane-based stereochemical diversity-oriented conformational restriction strategy: Histamine H3 and/or H4 receptor ligands with the 2,3-methanobutane backbone. Org. Biomol. Chem. 2012, 10, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Nakada, K.; Yoshikawa, M.; Ide, S.; Suemasa, A.; Kawamura, S.; Kobayashi, T.; Masuda, E.; Ito, Y.; Hayakawa, W.; Katayama, T.; et al. Cyclopropane-based conformational restriction of GABA by a stereochemical diversity-oriented strategy: Identification of an efficient lead for potent inhibitors of GABA transports. Bioorg. Med. Chem. 2013, 21, 4938–4950. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, M.; Kazuta, Y.; Hayashi, H.; Yamada, S.; Matsuda, A.; Shuto, S. Stereochemical diversity-oriented conformational restriction strategy. Development of potent histamine H-3 and/or H-4 receptor antagonists with an imidazolylcyclopropane structure. J. Med. Chem. 2006, 49, 5587–5596. [Google Scholar]
- Kiss, R.; Keseru, G.M. Novel histamine H4 receptor ligands and their potential therapeutic applications: An update. Expert Opin. Ther. Pat. 2014, 24, 1185–1197. [Google Scholar] [CrossRef]
- Watanabe, M.; Hirokawa, T.; Kobayashi, T.; Yoshida, A.; Ito, Y.; Yamada, S.; Orimoto, N.; Yamasaki, Y.; Arisawa, M.; Shuto, S. Investigation of the bioactive conformation of histamine H3 receptor antagonists by the cyclopropylic strain-based conformational restriction strategy. J. Med. Chem. 2010, 53, 3585–3593. [Google Scholar]
- Kobayashi, T.; Suemasa, A.; Igawa, A.; Ide, S.; Fukuda, H.; Abe, H.; Arisawa, M.; Minami, M.; Shuto, S. Conformationally restricted GABA with bicyclo[3.1.0]hexane backbone as the first highly selective BGT-1 inhibitor. ACS Med. Chem. Lett. 2014, 5, 889–893. [Google Scholar] [CrossRef] [Green Version]
- Horne, D.A.; Yakusijin, K.; Büchi, G. A two-step synthesis of imidazoles from aldehydes via 4-tosyloxazolines. Heterocycles 1994, 39, 139–153. [Google Scholar]
- Shuto, S.; Ono, S.; Imoto, H.; Yoshii, K.; Matsuda, A. Synthesis and biological activity of conformationally restricted analogues of milnacipran: (1S,2R)-1-phenyl-2-[(R)-1-amino-2-propynyl]-N,N-diethylcyclopropanecarboxamide is a novel class of NMDA receptor channel blocker. J. Med. Chem. 1998, 41, 3507–3514. [Google Scholar] [PubMed]
- Kawamura, S.; Unno, Y.; Tanaka, M.; Sasaki, T.; Yamano, A.; Hirokawa, T.; Kameda, T.; Asai, A.; Arisawa, M.; Shuto, S. Investigation of the noncovalent binding mode of covalent proteasome inhibitors around the transition state by combined use of cyclopropylic strain-based conformational restriction and computational modeling. J. Med. Chem. 2013, 56, 5829–5842. [Google Scholar] [PubMed]
- Mizuno, A.; Miura, S.; Watanabe, M.; Ito, Y.; Yamada, S.; Odagami, T.; Kogami, Y.; Arisawa, M.; Shuto, S. Three-dimensional structural diversity-oriented peptidomimetics based on the cyclopropylic strain. Org. Lett. 2013, 15, 1686–1689. [Google Scholar] [CrossRef]
- Matsui, K.; Kido, Y.; Watari, R.; Kashima, Y.; Yoshida, Y.; Shuto, S. Highly conformationally restricted cyclopropane tethers with three-dimensional structural diversity drastically enhance the cell permeability of cyclic peptides. Chem. Eur. J. 2017, 23, 3034–3041. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Form | H3, Ki (nM) | H4, Ki (nM) | Selectivity Ratio (H4/H3) |
|---|---|---|---|---|
| 6 | anti | 31.6 ± 8.2 | 501 ± 201 | 16 |
| 7 | syn | 5.6 ± 1.6 | 602 ± 343 | 108 |
| ent-6 | anti | 21.9 ± 6.0 | 416 ± 155 | 19 |
| ent-7 | syn | 14.8 ± 3.8 | 69.2 ± 21 | 4.7 |
| 3 b | – | 3.6 ± 0.4 | 37.2 ± 2.7 | 10 |
| ent-3 b | – | 8.4 ± 1.5 | 7.6 ± 0.4 | 0.9 |
| 4 | anti | 6.7 ± 0.4 c | 9.0 ± 1.0 | 1.3 |
| 5 | syn | 63.0 ± 6.1 c | 76.5 ± 14 | 1.2 |
| Thioperamide b | – | 51 ± 3.8 | 124 ± 14 | 2.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watanabe, M.; Kobayashi, T.; Ito, Y.; Yamada, S.; Shuto, S. Conformational Restriction of Histamine with a Rigid Bicyclo[3.1.0]hexane Scaffold Provided Selective H3 Receptor Ligands. Molecules 2020, 25, 3562. https://doi.org/10.3390/molecules25163562
Watanabe M, Kobayashi T, Ito Y, Yamada S, Shuto S. Conformational Restriction of Histamine with a Rigid Bicyclo[3.1.0]hexane Scaffold Provided Selective H3 Receptor Ligands. Molecules. 2020; 25(16):3562. https://doi.org/10.3390/molecules25163562
Chicago/Turabian StyleWatanabe, Mizuki, Takaaki Kobayashi, Yoshihiko Ito, Shizuo Yamada, and Satoshi Shuto. 2020. "Conformational Restriction of Histamine with a Rigid Bicyclo[3.1.0]hexane Scaffold Provided Selective H3 Receptor Ligands" Molecules 25, no. 16: 3562. https://doi.org/10.3390/molecules25163562
APA StyleWatanabe, M., Kobayashi, T., Ito, Y., Yamada, S., & Shuto, S. (2020). Conformational Restriction of Histamine with a Rigid Bicyclo[3.1.0]hexane Scaffold Provided Selective H3 Receptor Ligands. Molecules, 25(16), 3562. https://doi.org/10.3390/molecules25163562