13C CPMAS NMR as a Tool for Full Structural Description of 2-Phenyl Substituted Imidazoles That Overcomes the Effects of Fast Tautomerization



Abstract
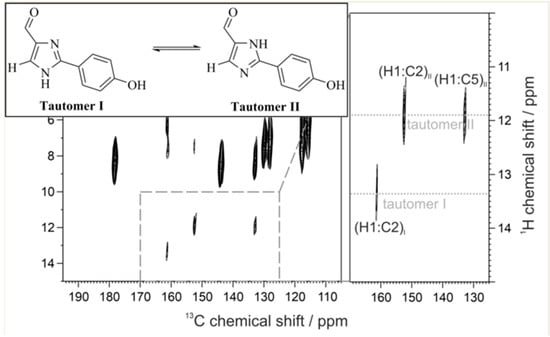
:
1. Introduction
2. Results
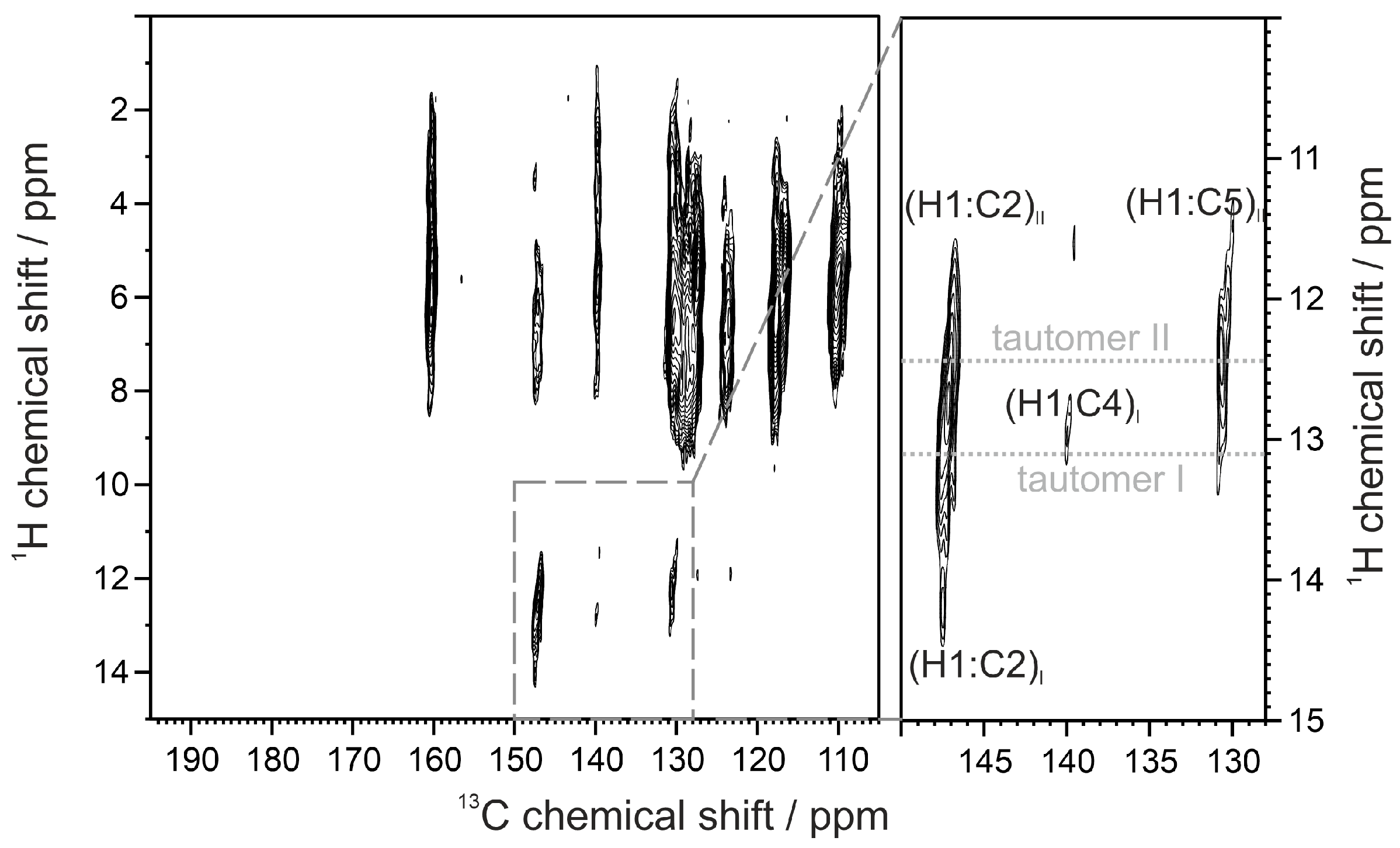
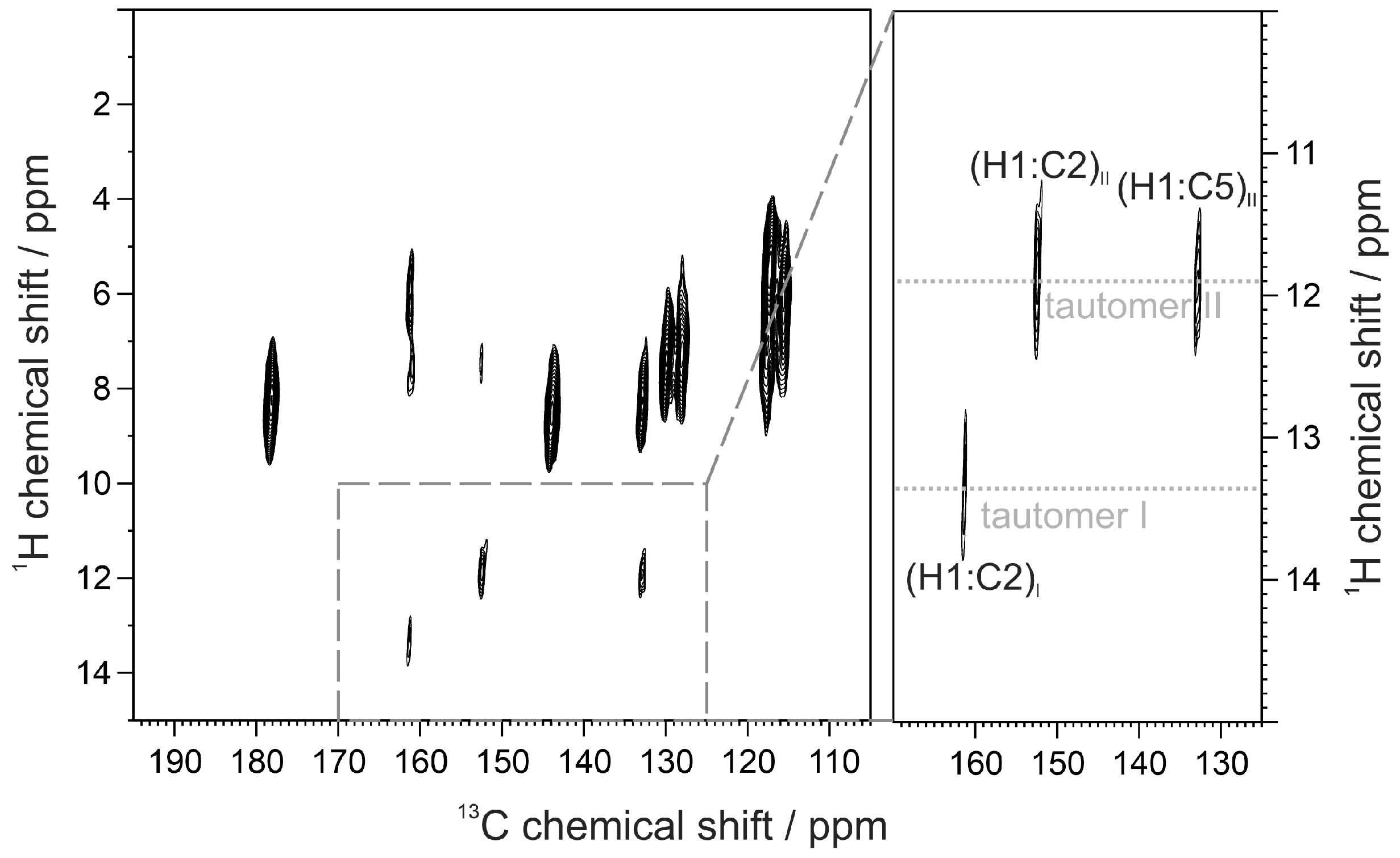
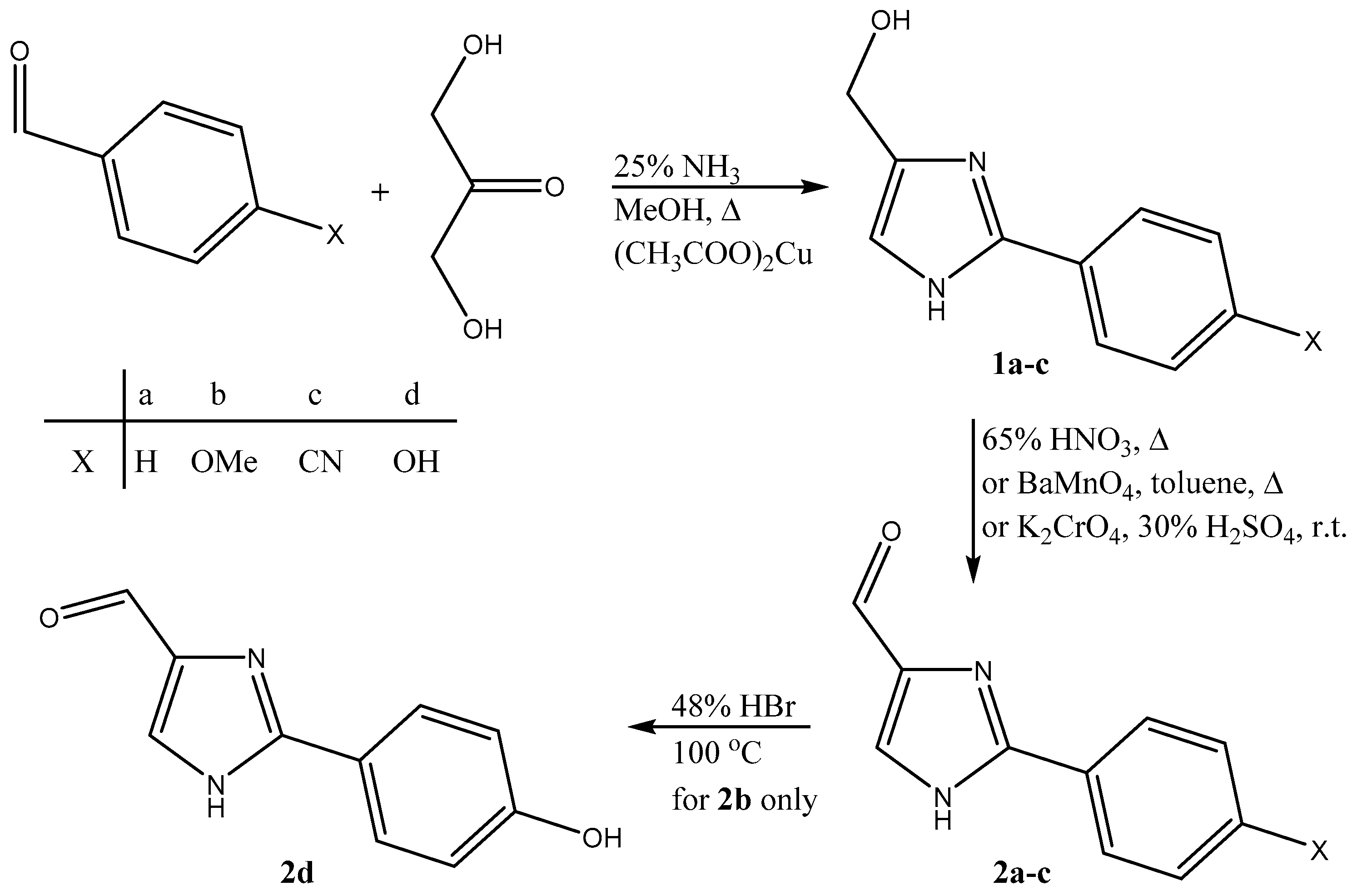
2.1. Synthesis and NMR Measurements in Solution
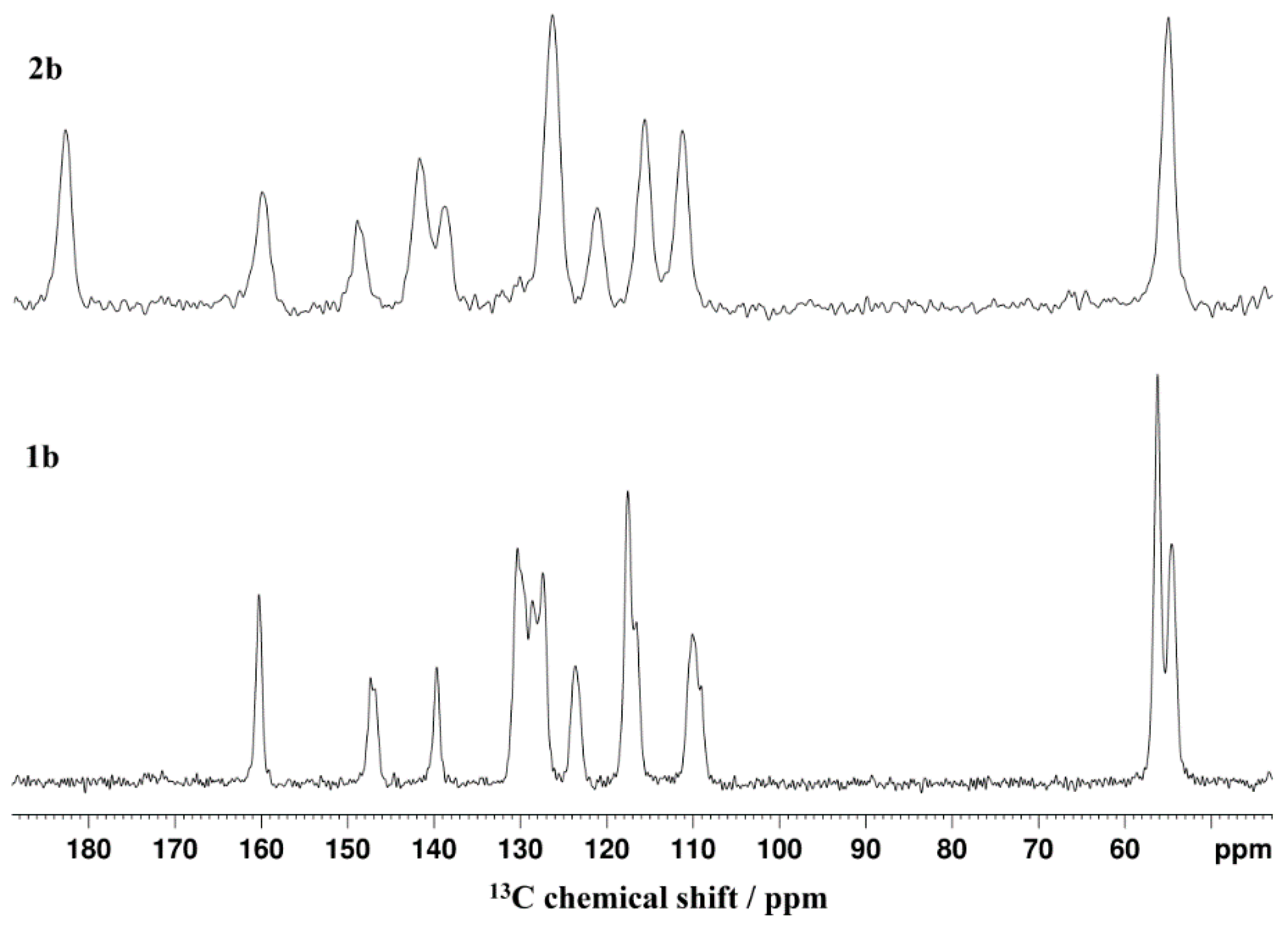
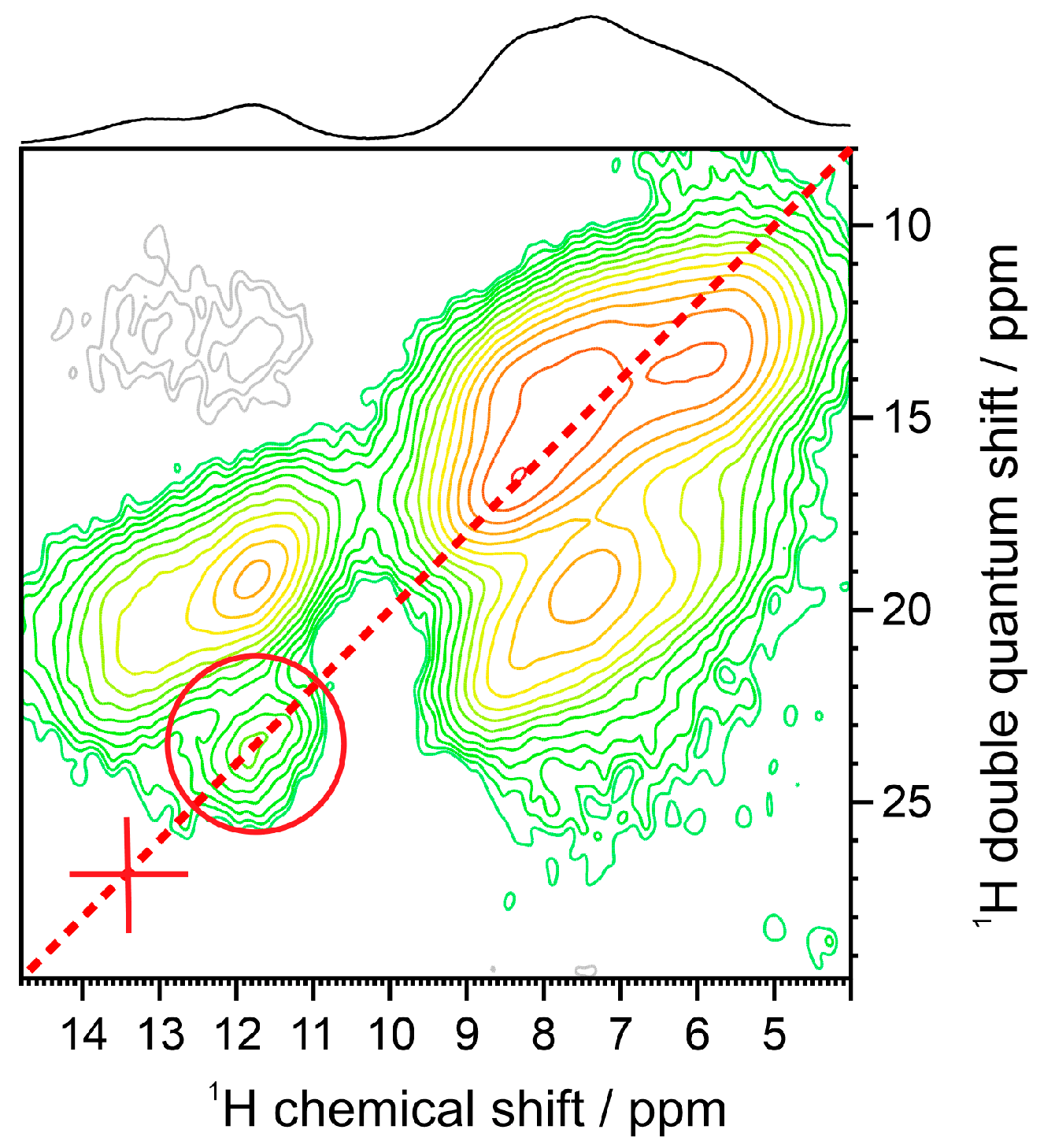
2.2. Solid-State NMR Measurements
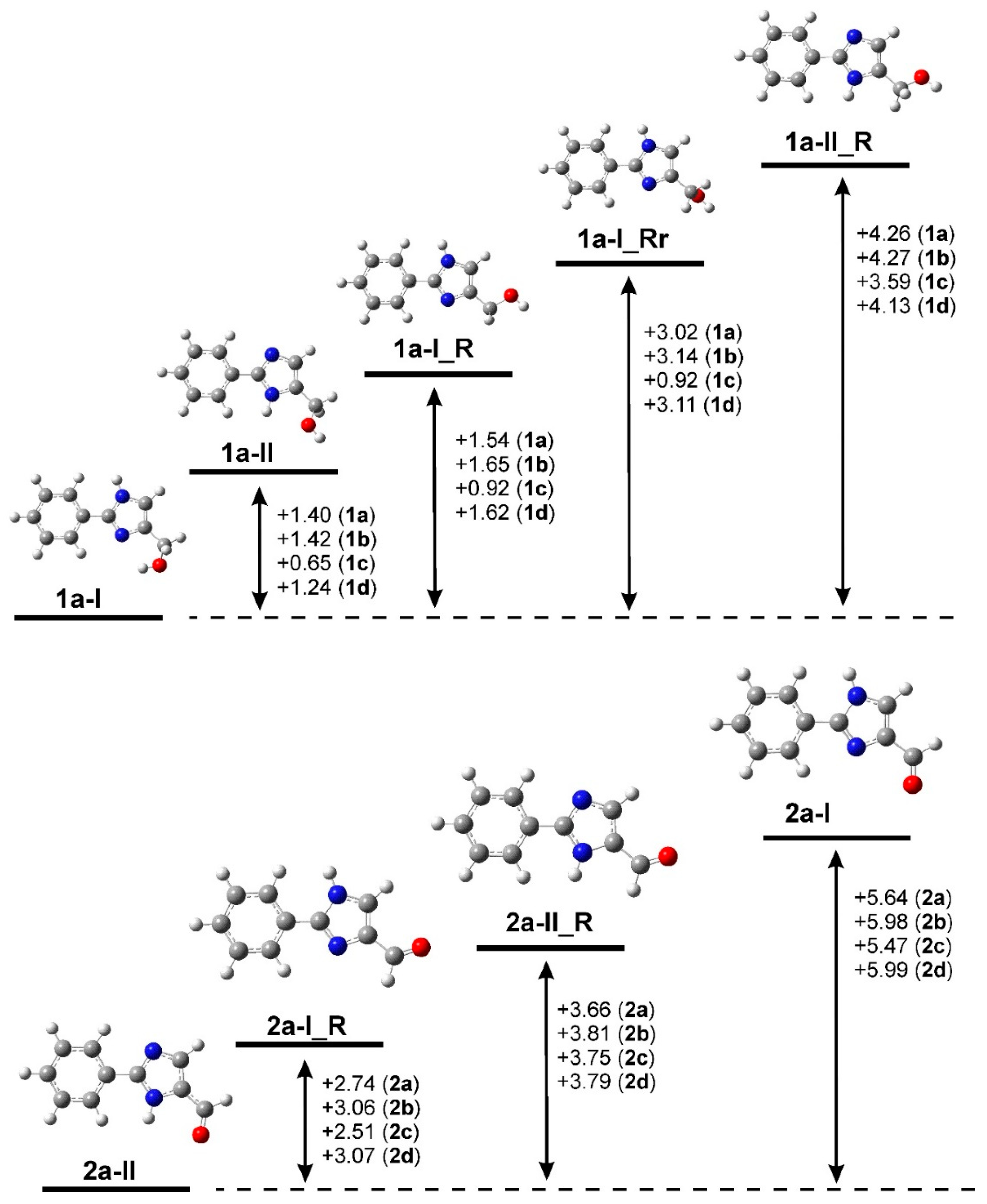
2.3. Quantum Chemical Calculations
3. Discussion
4. Materials and Methods
4.1. Apparatus
4.2. Synthesis
4.3. Computational Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Joule, J.A.; Mills, K. Heterocyclic Chemistry, 5th ed.; John Wiley & Sons Ltd.: Chichester, UK, 2010; p. 462. [Google Scholar]
- Minkin, V.I.; Garnovskii, A.D.; Elguero, J.; Katritzky, A.R.; Denisko, O.V. The tautomerism of heterocycles: Five-membered rings with two or more heteroatoms. In Advances in Heterocyclic Chemistry; Academic Press: San Diego, CA, USA, 2000; Volume 76, pp. 157–323. [Google Scholar]
- Papadopoulos, E.P.; Hollstein, U. Carbon–13 NMR studies of tautomerism in some 2-substituted imidazoles and benzimidazoles. Org. Magn. Resonance 1982, 19, 188–191. [Google Scholar] [CrossRef]
- Bryant, R.G. The NMR time scale. J. Chem. Educ. 1983, 60, 933–935. [Google Scholar] [CrossRef]
- Pinto, D.C.G.A.; Santos, C.M.M.; Silva, A.M.S. Advanced NMR techniques for structure characterization of heterocyclic compounds. In Recent Research Developments in Heterocyclic Chemistry; Research Signpost: Kerala, India, 2007; pp. 397–475. [Google Scholar]
- Benhabbour, S.R.; Chapman, R.P.; Scharfenberger, G.; Meyer, W.H.; Goward, G.R. Study of Imidazole-Based Proton-Conducting Composite Materials Using Solid-State NMR. Chem. Mater. 2005, 17, 1605–1612. [Google Scholar] [CrossRef]
- Zhao, L.; Smolarkiewicz, I.; Limbach, H.H.; Breitzke, H.; Pogorzelec-Glaser, K.; Pankiewicz, R.; Tritt-Goc, J.; Gutmann, T.; Buntkowsky, G. Imidazole-Doped Cellulose as Membrane for Fuel Cells: Structural and Dynamic Insights from Solid-State NMR. J. Phys. Chem. C 2016, 120, 19574–19585. [Google Scholar] [CrossRef]
- Pardeshi, S.D.; Sathe, P.A.; Vadagaonkar, K.S.; Chaskar, A.C. One-Pot Protocol for the Synthesis of Imidazoles and Quinoxalines using NBS. Adv. Synth. Catal. 2017, 359, 4217–4226. [Google Scholar] [CrossRef]
- Al-Wabli, R.I.; Al-Ghamdi, A.R.; Ghabbour, H.A.; Al-Agamy, M.H.; Attia, M.I. Synthesis and Spectroscopic Identification of Certain Imidazole-Semicarbazone Conjugates Bearing Benzodioxole Moieties: New Antifungal Agents. Molecules 2019, 24, 200. [Google Scholar] [CrossRef] [Green Version]
- Kalaria, P.N.; Karad, S.C.; Raval, D.K. A review on diverse heterocyclic compounds as the privileged scaffolds in antimalarial drug discovery. Eur. J. Med. Chem. 2018, 158, 917–936. [Google Scholar] [CrossRef]
- Tripathi, A.C.; Upadhyay, S.; Paliwal, S.; Saraf, S.K. Privileged scaffolds as MAO inhibitors: Retrospect and prospects. Eur. J. Med. Chem. 2018, 145, 445–497. [Google Scholar] [CrossRef]
- Shimahara, H.; Yoshida, T.; Shibata, Y.; Shimizu, M.; Kyogoku, Y.; Sakiyama, F.; Nakazawa, T.; Tate, S.I.; Ohki, S.Y.; Kato, T.; et al. Tautomerism of histidine 64 associated with proton transfer in catalysis of carbonic anhydrase. J. Biol. Chem. 2007, 282, 9646–9656. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Chen, S.; Li, R.; Cui, W.; Jiang, H.; Ling, Y.; Yang, Z.; Hu, W. Imidazole-based pinanamine derivatives: Discovery of dual inhibitors of the wild-type and drug-resistant mutant of the influenza A virus. Eur. J. Med. Chem. 2016, 108, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Mihorianu, M.; Franz, M.H.; Jones, P.G.; Freytag, M.; Kelter, G.; Fiebig, H.-H.; Tamm, M.; Neda, I. N-Heterocyclic carbenes derived from imidazo-[1,5-a]pyridines related to natural products: Synthesis, structure and potential biological activity of some corresponding gold(I) and silver(I) complexes. Appl. Organomet. Chem. 2016, 30, 581–589. [Google Scholar] [CrossRef]
- Maftei, E.; Maftei, C.V.; Jones, P.G.; Freytag, M.; Franz, M.H.; Kelter, G.; Fiebig, H.-H.; Tamm, M.; Neda, I. Trifluoromethylpyridine-Substituted N-Heterocyclic Carbenes Related to Natural Products: Synthesis, Structure, and Potential Antitumor Activity of some Corresponding Gold(I), Rhodium(I), and Iridium(I) Complexes. Helv. Chimica Acta 2016, 99, 469–481. [Google Scholar] [CrossRef]
- Parík, P.; Chlupatý, T. New Chiral and Achiral Imines and Bisimines Derived from 2-Phenyl-1Himidazole-4-carbaldehyde. Synthesis, Structural Studies and Complexation Stability Constants. J. Heterocycl. Chem. 2014, 51, 695–701. [Google Scholar] [CrossRef]
- Procopiou, P.A.; Anderson, N.A.; Barrett, J.; Barrett, T.N.; Crawford, M.H.J.; Fallon, B.J.; Hancock, A.P.; Le, J.; Lemma, S.; Marshall, R.P.; et al. Discovery of (S)-3-(3-(3,5-Dimethyl-1H-pyrazol-1-yl)phenyl)-4-((R)-3-(2-(5,6,7,8-tetrahydro-1,8-naphthyridin-2-yl)ethyl)pyrrolidin-1-yl)butanoic Acid, a Nonpeptidic αvβ6 Integrin Inhibitor for the Inhaled Treatment of Idiopathic Pulmonary Fibrosis. J. Med. Chem. 2018, 61, 8417–8443. [Google Scholar]
- Shen, H.; Xie, Z. Atom-Economical Synthesis of N-Heterocycles via Cascade Inter-/Intramolecular C-N Bond-Forming Reactions Catalyzed by Ti Amides. J. Am. Chem. Soc. 2010, 132, 11473–11480. [Google Scholar] [CrossRef]
- Prakash, P.; Aparna, P.S.; Jijy, E.; Santhini, P.V.; Varughese, S.; Radhakrishnan, K.V. Rodium(III)-Catalyzed C–H Activation of Phenylazoles toward C–N Bond Cleavage of Diazabicyclic Olefins: A Facile Access to Mono- and Biscyclopentenyl-Functionalized Aza-Heteroaromatics. SYNLETT 2014, 25, 0275–0279. [Google Scholar]
- Nieto, C.I.; Cabildo, P.; Garcia, M.A.; Claramunt, R.M.; Alkorta, I.; Elguero, J. An experimental and theoretical NMR study of NH-benzimidazoles in solution and in the solid state: Proton transfer and tautomerism. Beilstein J. Org. Chem. 2014, 10, 1620–1629. [Google Scholar] [CrossRef]
- Enchev, V.; Markova, N.; Marinov, M.; Stoyanov, N.; Rogojerov, M.; Ugrinov, A.; Wawer, I.; Pisklak, D.M. 2-Methylthio-imidazolins: A rare case of different tautomeric forms in solid state and in solution. Struct. Chem. 2016, 28, 757–772. [Google Scholar] [CrossRef]
- Pinto, J.; Silva, V.L.M.; Silva, A.M.S.; Claramunt, R.M.; Sanz, D.; Torralba, M.C.; Torres, M.R.; Reviriego, F.; Alkorta, I.; Elguero, J. The structure of azines derived from C-formyl-1H-imidazoles in solution and in the solid state: Tautomerism, configurational and conformational studies. Magn. Reson. Chem. 2013, 51, 203–221. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Hong, M. Protonation, tautomerization, and rotameric structure of histidine: A comprehensive study by magic-angle-spinning solid-state NMR. J. Am. Chem. Soc. 2011, 133, 1534–1544. [Google Scholar] [CrossRef] [Green Version]
- Ahmedova, A.; Paradowska, K.; Wawer, I. 1H, 13C MAS NMR and DFT GIAO study of quercetin and its complex with Al(III) in solid state. J. Inorg. Biochem. 2012, 110, 27–35. [Google Scholar] [CrossRef]
- Ahmedova, A.; Marinova, P.; Paradowska, K.; Marinov, M.; Wawer, I.; Mitewa, M. Structure of 2,4-dithiohydantoin complexes with copper and nickel: Solid-state NMR as verification method. Polyhedron 2010, 29, 1639–1645. [Google Scholar] [CrossRef]
- Ahmedova, A.; Marinova, P.; Paradowska, K.; Stoyanov, N.; Wawer, I.; Mitewa, M. Spectroscopic aspects of the coordination modes of 2,4-dithiohydantoins: Experimental and theoretical study on copper and nickel complexes of cyclohexanespiro-5-(2,4-dithiohydantoin). Inorg. Chim. Acta 2010, 363, 3919–3925. [Google Scholar] [CrossRef]
- Ahmedova, A.; Marinova, P.; Ciattini, S.; Stoyanov, N.; Springborg, M.; Mitewa, M. A combined experimental and theoretical approach for structural study on a new cinnamoyl derivative of 2-acetyl-1,3-indandione and its metal(II) complexes. Struct. Chem. 2009, 20, 101–111. [Google Scholar] [CrossRef]
- Siskos, M.G.; Choudhary, M.I.; Gerothanassis, I.P. Hydrogen Atomic Positions of O-H...O Hydrogen Bonds in Solution and in the Solid State: The Synergy of Quantum Chemical Calculations with (1)H-NMR Chemical Shifts and X-ray Diffraction Methods. Molecules 2017, 22, 415. [Google Scholar] [CrossRef]
- Matsumoto, N.; Mizuguchi, Y.; Mago, G.; Eguchi, S.; Miyasaka, H.; Nakashima, T.; Tuchagues, J.-P. Proton-Dependent Monomer-Oligomer Interconversion of Metal Complexes. Angew. Chem. Int. Ed. Engl. 1997, 36, 1860–1862. [Google Scholar] [CrossRef]
- Matsumoto, N.; Motoda, Y.; Matsuo, T.; Nakashima, T.; Re, N.; Dahan, F.; Tuchagues, J.-P. pH-Dependent Monomer T Oligomer Interconversion of Copper(II) Complexes with N-(2-R-imidazol-4-ylmethylidene)-2-aminoethylpyridine (R) Methyl, Phenyl). Inorg. Chem. 1999, 38, 1165–1173. [Google Scholar] [CrossRef]
- Wang, Z. Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 2951–2955. [Google Scholar]
- Baldwin, J.; Christy, M.; Denny, G.; Habecker, C.; Freedman, M.; Lyle, P.; Ponticello, G.; Varga, S.; Gross, D.; Sweet, C. Selective Adrenoceptor Antagonists: Examples of the 2-[4-[3-(Substitutedamino)-2-hydroxypropoxy]phenyl]imidazole Class. 2. J. Med. Chem. 1986, 29, 1065–1080. [Google Scholar] [CrossRef]
- Kótai, L.; Sajó, I.; Gács, I.; Sharmaa, P.; Banerji, K. Convenient Routes for the Preparation of Barium Permanganate and other Permanganate Salts. Z. Anorg. Allg. Chem. 2007, 633, 1257–1260. [Google Scholar] [CrossRef]
- Antonov, V.; Nedyalkova, M.; Tzvetkova, P.; Ahmedova, A. Solid State Structure Prediction Through DFT Calculations and 13C NMR Measurements: Case Study of Spiro-2, 4-dithiohydantoins. Z. für. Phys. Chem. 2016, 230, 909–930. [Google Scholar] [CrossRef]
- Lawson, A. The Reaction of Imidates with α- Amino-acetals and α-Amino-aldehydes. J. Chem. Soc. 1957, 4225–4228. [Google Scholar] [CrossRef]
- Dziuron, P.; Schunack, W. Eine neue Synthese von Imidazolderivaten 1. Mitt.: Zur Darstellung Zsubstituierter Imidazol-4alkohole. Arch. Pharmaz. 1973, 306, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Davood, A.; Alipour, E.; Shafiee, A. Efficient Synthesis of Imidazole Derivatives: An Important Synthon for the Preparation of Biologically Active Compounds. Turk. J. Chem. 2008, 32, 389–395. [Google Scholar]
- Paul, R.; Menschik, J. Imidazo [1,5-d]-AS-triazine-4(3H)-ones and Thiones. U.S. Patent 843,174, 15 August 1978. [Google Scholar]
- Maślewski, P.; Wyrzykowski, D.; Witwicki, M.; Dołęga, A. Histaminol and Its Complexes with Copper(II)-Studies in Solid State and Solution. Eur. J. Inorg. Chem. 2018, 2018, 1399–1408. [Google Scholar] [CrossRef]
- Kubicki, M. Two tautomers in one crystal: 4(5)-nitro-5(4)-methoxyimidazole. Acta Crystallogr. Sect. B Struct. Sci. 2004, 60, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Drew, M.G.B.; Das, D.; De, S.; Naskar, J.P.; Datta, D. Solid State annular tautomerism in a molecule containing two imidazole moieties. J. Chem. Crystallogr. 2008, 38, 507–512. [Google Scholar] [CrossRef]
- Gottlieb, H.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Dziuron, P.; Schunack, W. Imidazol-4-carbinole aus Iminoestern und Dihydroxyaceton 2. Mitt. iiber Imidazolsynthesen mit fliissigem Ammoniak. Arch. Pharm. 1974, 307, 470–473. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1998, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and Lee, Yang and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* basis set for third-row atoms. J. Comp. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
Sample Availability: Samples of all compounds are available from the authors. |









{kind=link}
{kind=link}
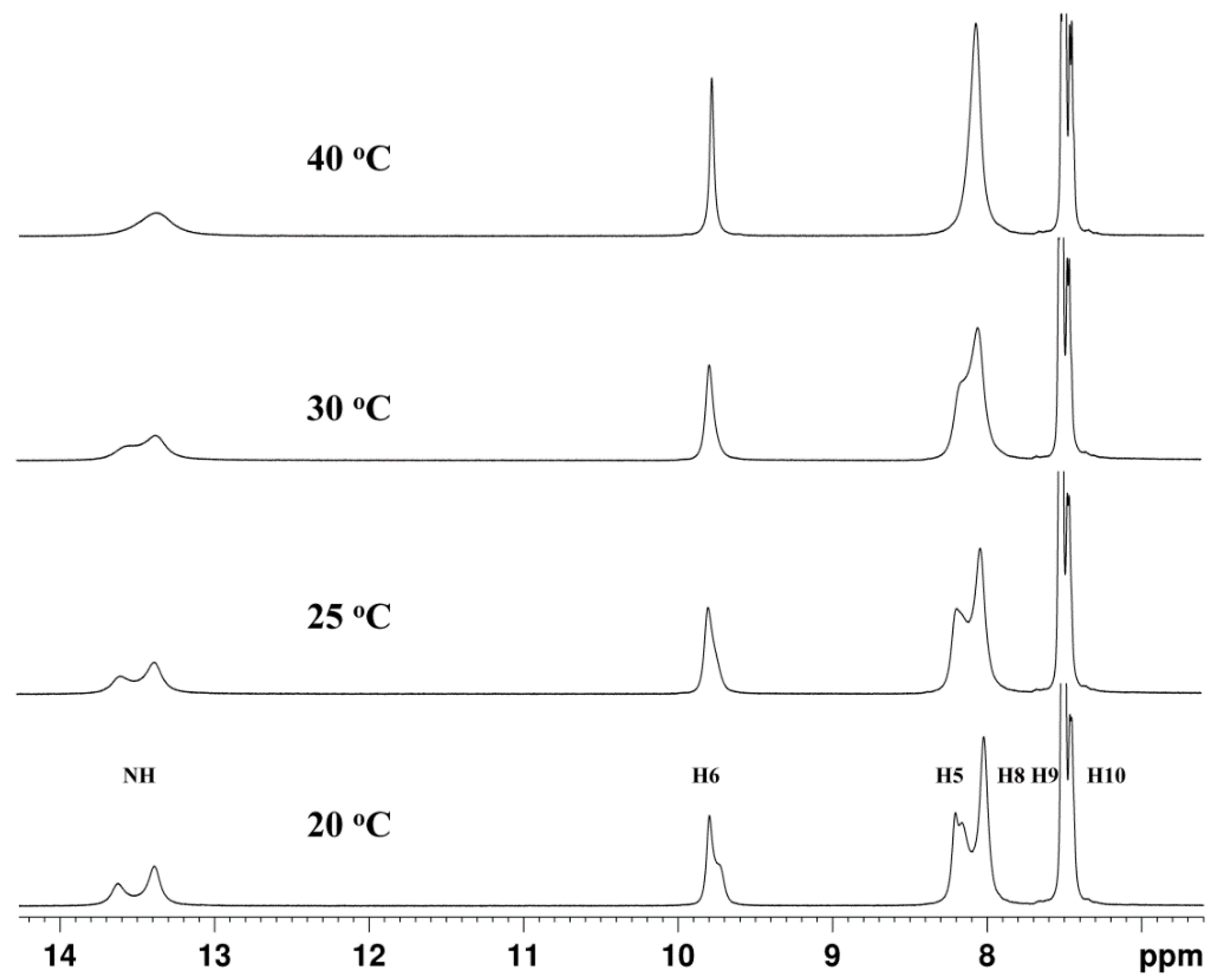{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number H-Atom | H5 | H6 | H8 | H9 | H10 (*-OMe) | OH | NH | |
|---|---|---|---|---|---|---|---|---|
| Compound | ||||||||
| 1a | 6.99 (s) | 4.43 (s) | 7.39–7.44 (m, 2H) | 7.90–7.94 (m, 2H) | 7.31 (dddd) | 4.97 (bs) | 12.38 (bs) | |
| 1b | 6.92 (bs) | 4.41 (s) | 7.85 (d, 2H) | 6.98 (d, 2H) | * 3.78 (s) | 4.93 (bs) | 12.24 (bs) | |
| 1c | 7.19 (bs) 6.97 (bs) | 4.43 (bs) 4.48 (bs) | 8.09 (bs, 4H) | 7.88 (d, 4H) | ─ | 4.94 (bs) 5.20 (bs) | 12.71 (bs) 12.85 (bs) | |
| 2a | 7.90–8.21 (bs) | 9.79 (bs) | 7.90–8.21 (bs) | 7.49–7.53 (m) | 7.41–7.47 (m) | ─ | 13.36 13.58 | |
| 2b | 7.95 (bs) 8.10 (bs) | 9.68 (bs) 9.74 (bs) | 7.95 (bs) 8.10 (bs) | 7.06 (d, 4H) | * 3.81 (s) | ─ | 13.19 (bs) 13.39 (bs) | |
| 2c | 8.21 (bs) | 9.81 (s) | 8.21 (bs) | 7.97 (d, 2H) | ─ | ─ | 13.73 (bs) | |
| 2d | 8.01 (bs) | 9.64 (bs) 9.70 (bs) | 7.90 (bs, 4H) | 6.85 (d, 4H) | ─ | 9.92 (bs) | 12.11 (bs) 13.14 (bs) | |
| Number C-Atom | C2 | C4 | C5 | C6 | C7 | C8 | C9 | C10 | CN or Me | |
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | ||||||||||
| 1a–I | 139.20 | 141.11 | 105.47 | 58.72 | 125.38 | 122.19 115.83 | 124.09 127.73 | 122.28 | ─ | |
| 1a–II | 139.61 | 122.08 | 124.99 | 58.31 | 126.35 | 116.07 122.19 | 122.45 123.79 | 121.58 | ─ | |
| 1a CP-MAS | 149.4 | 141.6 | 115.8 | 54.8 | 128.9 | 127.9 | 128.9 | 124.6 | ||
| 1b–I | 139.17 | 140.68 | 104.75 | 58.74 | 118.74 | 124.01 117.22 | 114.08 104.04 | 153.17 | 52.52 | |
| 1b–II | 139.77 | 122.03 | 124.24 | 58.33 | 119.59 | 117.73 123.97 | 112.14 105.56 | 152.65 | 52.43 | |
| 1b CP-MAS | 146.6(I) 146.9(II) | 139.3(I) 109.6(II) | 108.7(I) 130.0(II) | 54.2 | 123.3 | 127.0 128.2 129.4 | 117.2 116.4 130.0 | 159.9 | 55.8 | |
| 1c–I | 137.34 | 141.36 | 109.43 | 62.02 | 128.93 | 122.13 115.35 | 128.80 127.97 | 108.83 | 109.84 | |
| 1c–II | 137.72 | 122.31 | 127.08 | 58.09 | 129.20 | 115.89 122.02 | 127.94 128.75 | 108.83 | 109.92 | |
| 1c CP-MAS | 145.7 | 142.4 | 127.3 | 55.3 | 132.8 | 127.3 | 132.8 | 110.7 | 120.3 | |
| 1d–I | 139.01 | 140.70 | 104.76 | 58.73 | 118.56 | 117.05 124.85 | 107.79 111.62 | 150.24 | ─ | |
| 1d–II | 139.62 | 121.97 | 124.31 | 58.30 | 119.52 | 118.33 123.91 | 109.53 109.44 | 149.75 | ─ | |
| Number C-Atom | C2 | C4 | C5 | C6 | C7 | C8 | C9 | C10 | CN or Me | |
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | ||||||||||
| 2a–I_R | 140.24 | 140.03 | 114.51 | 178.14 | 124.96 | 122.82 116.13 | 124.38 122.78 | 123.18 | ─ | |
| 2a–II | 143.56 | 133.74 | 129.12 | 168.45 | 124.41 | 117.86 124.25 | 123.08 124.18 | 124.29 | ─ | |
| 2a CP-MAS | 150.0 | 129.4 | 139.7 | 183.9 | 129.4 | 126.8 | 129.4 | 129.4 | ─ | |
| 2b–I_R | 140.22 | 139.84 | 114.05 | 178.15 | 118.11 | 124.56 117.57 | 114.43 104.09 | 153.82 | 52.62 | |
| 2b–II | 143.55 | 133.93 | 128.87 | 167.97 | 117.46 | 119.69 125.96 | 104.58 114.15 | 154.71 | 52.53 | |
| 2b CP-MAS | 149.2 | 142.3 | 139.1 | 183.6 | 121.5 | 126.6 | 111.7 116.0 | 160.3 | 55.2 | |
| 2c–I_R | 138.92 | 140.70 | 115.73 | 177.92 | 127.82 | 122.90 116.12 | 129.23 128.15 | 110.78 | 109.38 | |
| 2c–II | 141.86 | 133.61 | 129.79 | 169.24 | 127.39 | 117.84 124.19 | 128.25 129.04 | 111.80 | 109.37 | |
| 2c CP-MAS | 146.7 (I) 147.0 (II) | 131.4 | 141.5 (I) 131.2 (II) | 186.8 | 131.4 | 125.3 127.0 | 131.4 133.8 | 112.3 | 119.9 | |
| 2d–I_R | 140.06 | 139.85 | 114.05 | 178.13 | 117.94 | 125.47 117.39 | 111.98 107.80 | 150.89 | ─ | |
| 2d–II | 143.34 | 133.81 | 128.83 | 168.01 | 117.38 | 119.43 126.90 | 108.29 111.75 | 151.79 | ─ | |
| 2d CP-MAS | 152.4 (II) 161.0 (I) | 144.1 | 132.9 (II) | 178.1 | 117.6 | 128.1 130.0 | 1c15.7 117.6 | 161.2 | ─ | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burdzhiev, N.; Ahmedova, A.; Borrisov, B.; Graf, R. 13C CPMAS NMR as a Tool for Full Structural Description of 2-Phenyl Substituted Imidazoles That Overcomes the Effects of Fast Tautomerization. Molecules 2020, 25, 3770. https://doi.org/10.3390/molecules25173770
Burdzhiev N, Ahmedova A, Borrisov B, Graf R. 13C CPMAS NMR as a Tool for Full Structural Description of 2-Phenyl Substituted Imidazoles That Overcomes the Effects of Fast Tautomerization. Molecules. 2020; 25(17):3770. https://doi.org/10.3390/molecules25173770
Chicago/Turabian StyleBurdzhiev, Nikola, Anife Ahmedova, Boris Borrisov, and Robert Graf. 2020. "13C CPMAS NMR as a Tool for Full Structural Description of 2-Phenyl Substituted Imidazoles That Overcomes the Effects of Fast Tautomerization" Molecules 25, no. 17: 3770. https://doi.org/10.3390/molecules25173770
APA StyleBurdzhiev, N., Ahmedova, A., Borrisov, B., & Graf, R. (2020). 13C CPMAS NMR as a Tool for Full Structural Description of 2-Phenyl Substituted Imidazoles That Overcomes the Effects of Fast Tautomerization. Molecules, 25(17), 3770. https://doi.org/10.3390/molecules25173770