
Is It Conjugated or Not? The Theoretical and Experimental Electron Density Map of Bonding in p-CH3CH2COC6H4-C≡C-C≡C-p-C6H4COCH3CH2

 , ,
, ,
Abstract
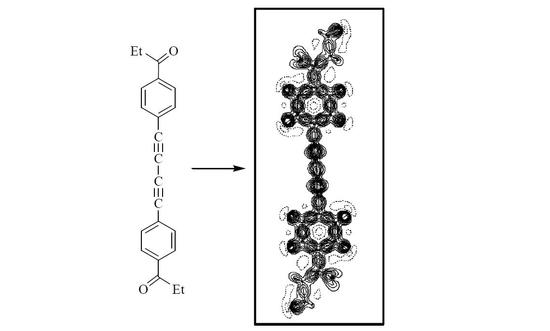
:
1. Introduction
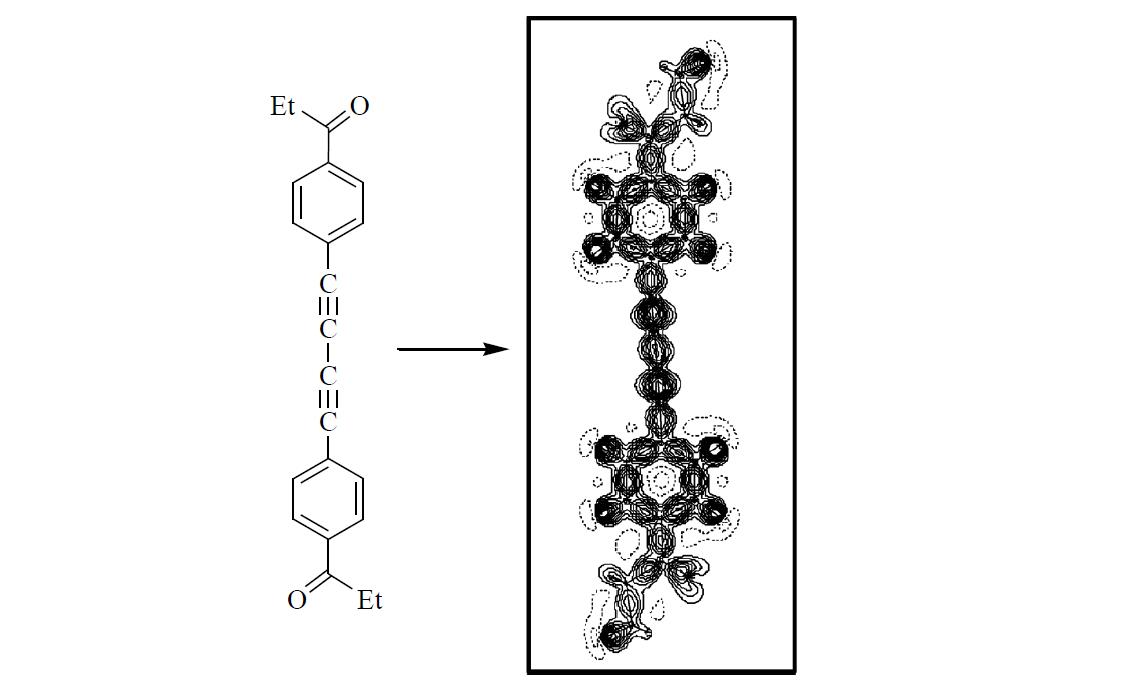
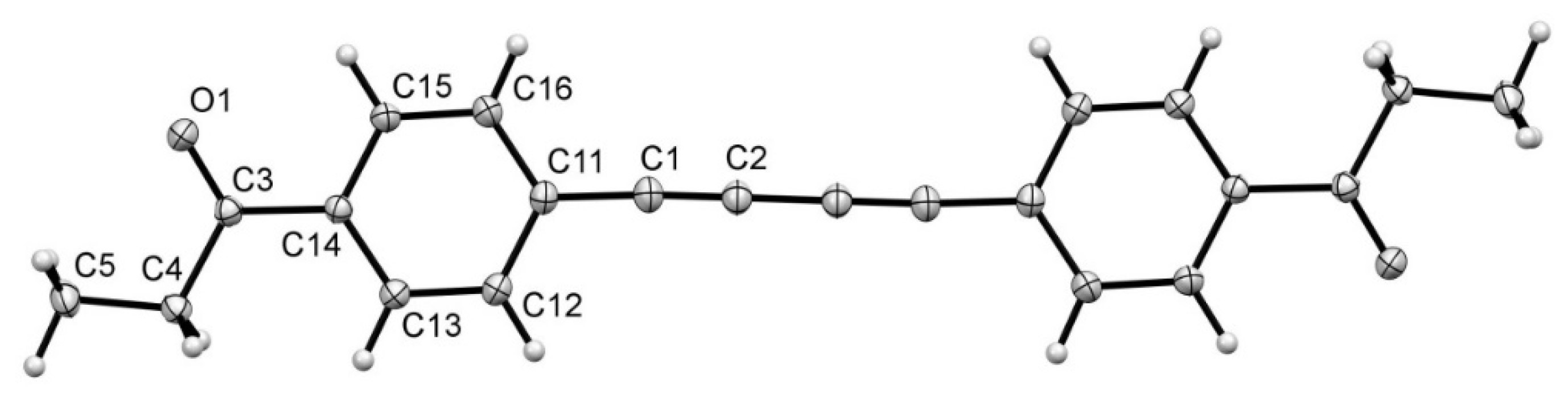
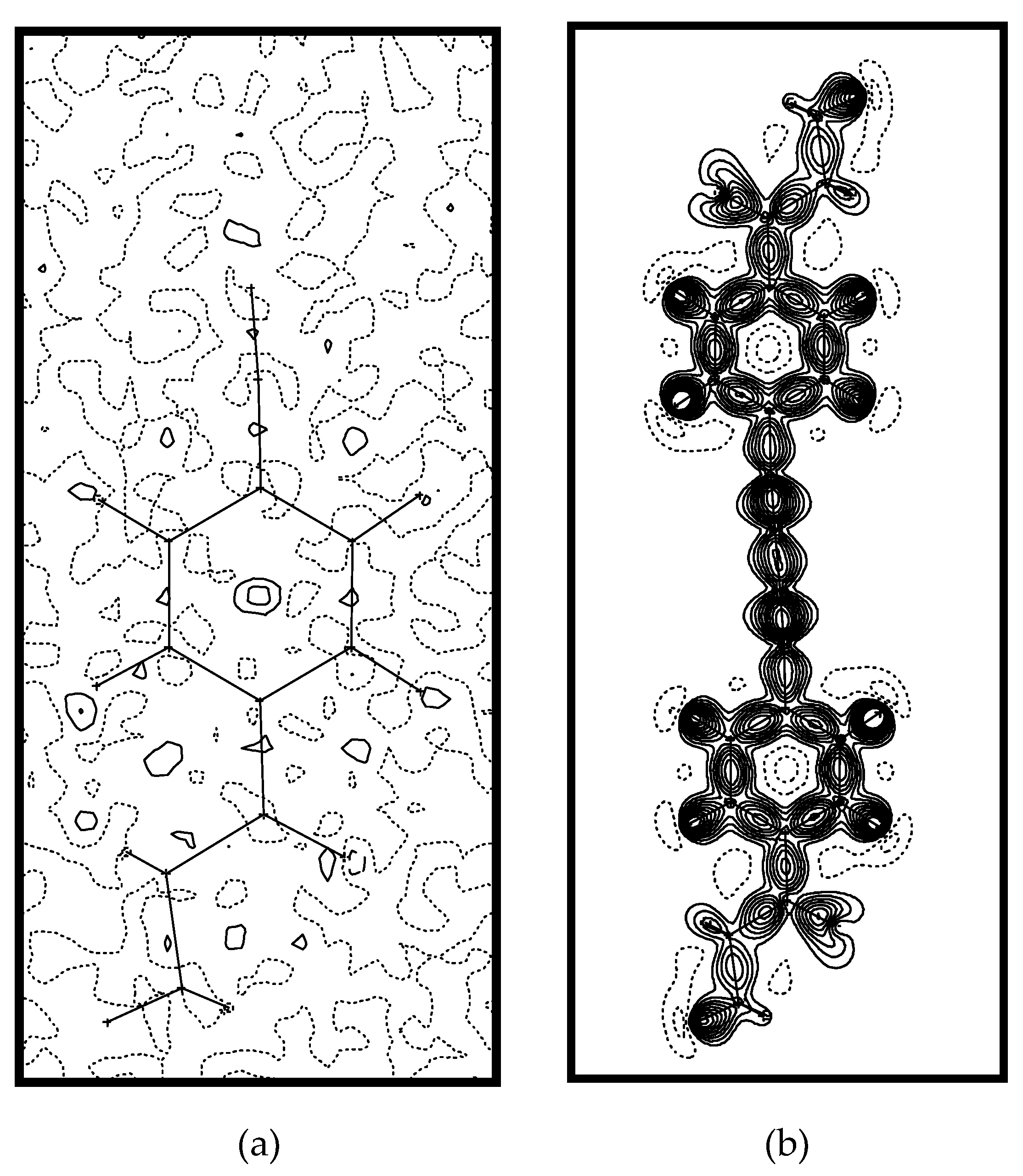
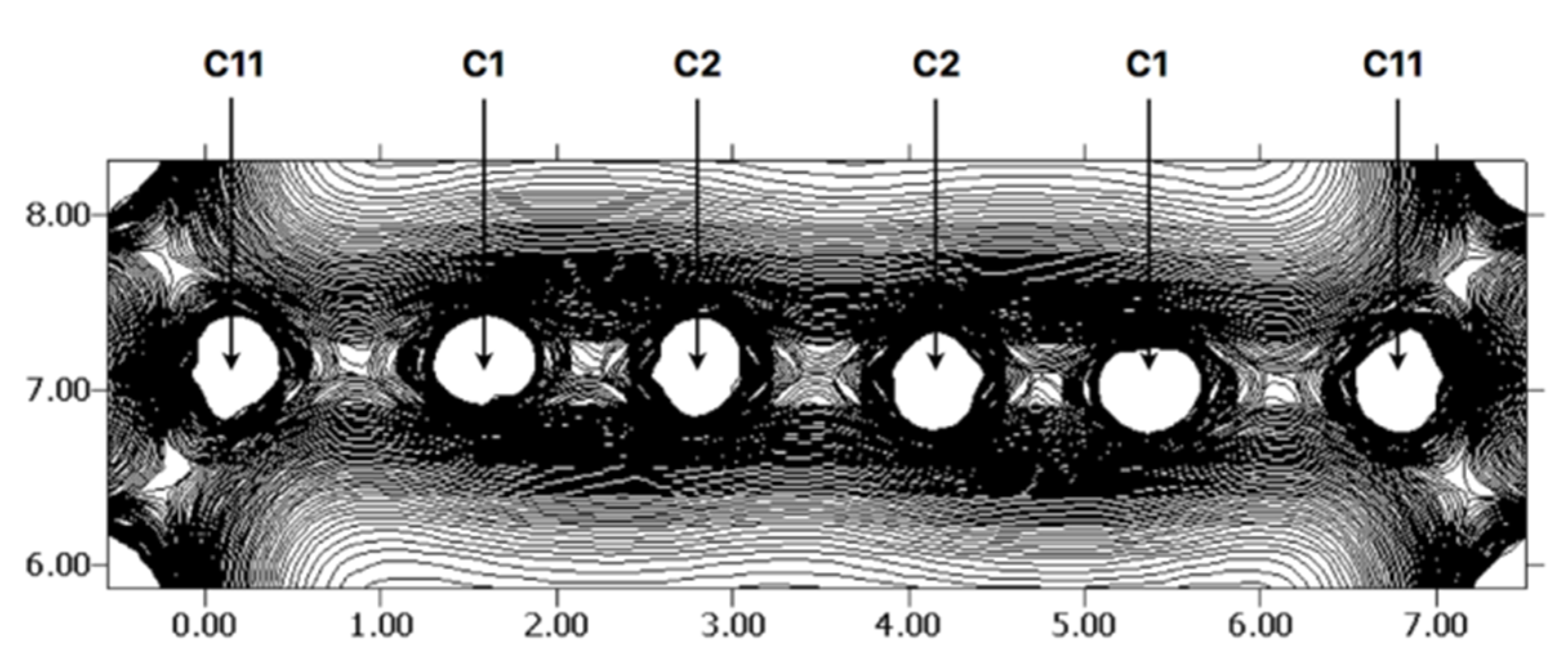
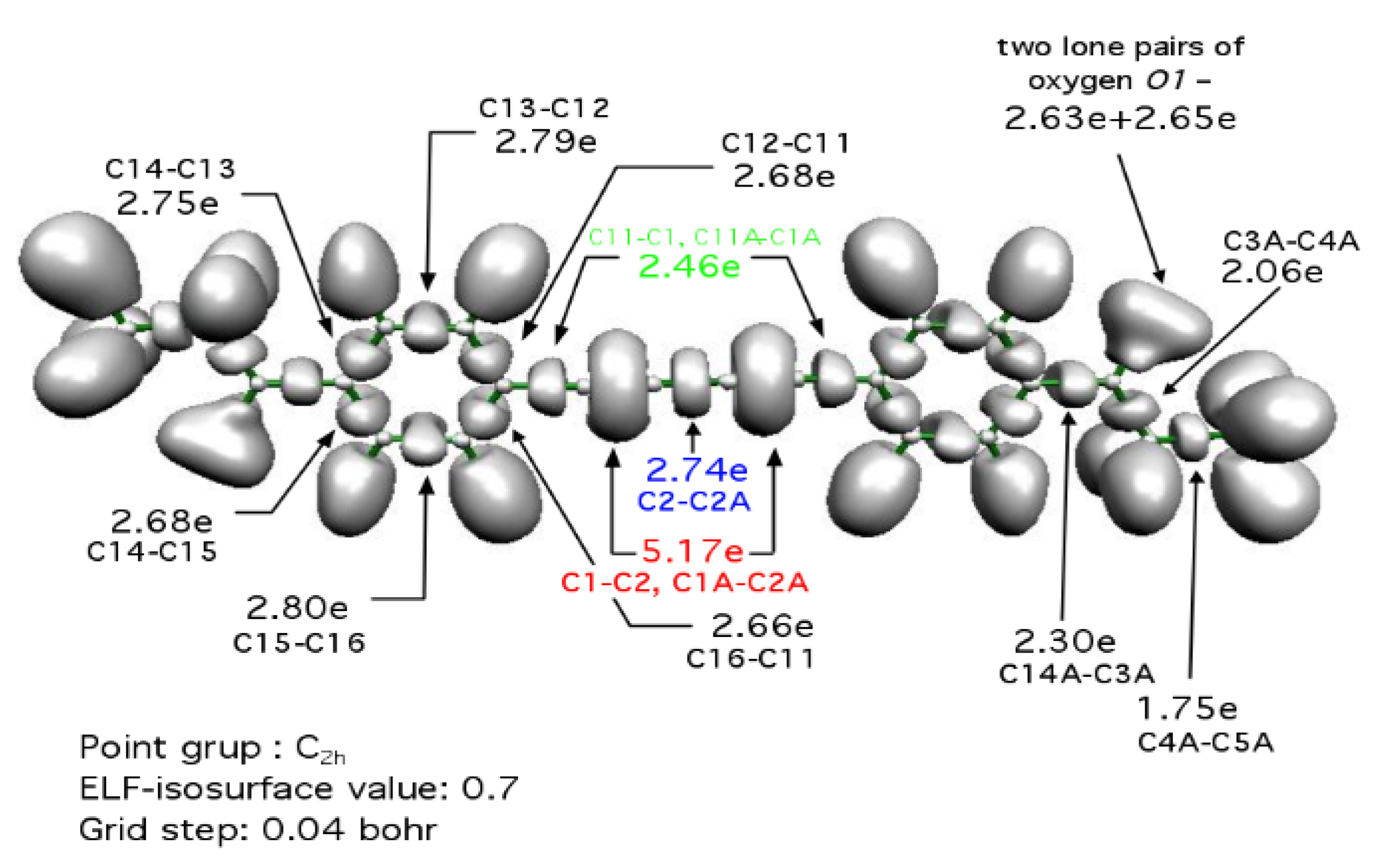
2. Results and Discussion
3. Experimental Section
3.1. Multipole Refinement
3.2. Theoretical Calculations
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cataldo, F. Polyynes. Synthesis, Properties and Applications; Taylor & Francis: Boca Raton, FL, USA, 2006. [Google Scholar]
- Chalifoux, W.A.; Tykwinski, R.R. Synthesis of extended polyynes: Toward carbyne. Comptes Rendus Chim. 2009, 12, 341–358. [Google Scholar] [CrossRef]
- Banhart, F. Chains of carbon atoms: A vision or a new nanomaterial? Beilstein J. Nanotechnol. 2015, 6, 559–569. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Artyukhov, V.I.; Lee, H.; Xu, F.; Yakobson, B.I. Carbyne from First Principles: Chain of C Atoms, a Nanorod or a Nanorope. ACS Nano 2013, 7, 10075–10082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timoshevskii, A.; Kotrechko, S.; Matviychuk, Y. Atomic structure and mechanical properties of carbyne. Phys. Rev. B Condens. Matter 2015, 91, 245434. [Google Scholar] [CrossRef] [Green Version]
- Mohr, W.; Stahl, J.; Hampel, F.; Gladysz, J.A. Synthesis, Structure, and Reactivity of sp Carbon Chains with Bis(phosphine)Pentafluorophenylplatinum Endgroups: Butadiynediyl (C4) through Hexadecaoctaynediyl (C16) Bridges, and Beyond. Chem. Eur. J. 2003, 9, 3324–3340. [Google Scholar] [CrossRef] [PubMed]
- Chalifoux, W.A.; McDonald, R.; Ferguson, M.J.; Tykwinski, R.R. tert-Butyl-End-Capped Polyynes: Crystallographic Evidence of Reduced Bond-Length Alternation. Angew. Chem. Int. Ed. 2009, 48, 7915–7919. [Google Scholar] [CrossRef]
- Eisler, S.; Slepkov, A.D.; Elliot, E.; Luu, T.; McDonald, R.; Hegmann, F.A.; Tykwinski, R.R. Polyynes as a Model for Carbyne: Synthesis, Physical Properties, and Nonlinear Optical Response. J. Am. Chem. Soc. 2005, 127, 2666–2676. [Google Scholar] [CrossRef]
- Gibtner, T.; Hampel, F.; Gisselbrecht, J.-P.; Hirsch, A. End-Cap Stabilized Oligoynes: Model Compounds for the Linear sp Carbon Allotrope-Carbyne. Chem. Eur. J. 2002, 8, 408–432. [Google Scholar] [CrossRef]
- Zheng, Q.; Gladysz, J.A. A Synthetic Breakthrough into an Unanticipated Stability Regime: Readily Isolable Complexes in which C16-C28 Polyynediyl Chains Span Two Platinum Atoms. J. Am. Chem. Soc. 2005, 127, 10508–10509. [Google Scholar] [CrossRef]
- Bruce, M.I.; Zaitseva, N.N.; Nicholson, B.K.; Skelton, B.W.; White, A.H. Syntheses and molecular structures of some compounds containing many-atom chains end-capped by tricobalt carbonyl clusters. J. Organomet. Chem. 2008, 693, 2887–2897. [Google Scholar] [CrossRef]
- Dembinski, R.; Bartik, T.; Bartik, B.; Jaeger, M.; Gladysz, J.A. Toward Metal-Capped One-Dimensional Carbon Allotropes: Wirelike C6-C20 Polyynediyl Chains That Span Two Redox-Active (η5-C5Me5)Re(NO)(PPh3) Endgroups. J. Am. Chem. Soc. 2000, 122, 810–822. [Google Scholar] [CrossRef]
- Chalifoux, W.A.; Tykwinski, R.R. Synthesis of polyynes to model the sp-carbon allotrope carbyne. Nat. Chem. 2010, 2, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Xi, B.; Jodoin, D.S.; Zhang, L.; Cummings, S.P.; Gao, Y.; Tyler, S.F.; Fanwick, P.E.; Crutchley, R.J.; Ren, T. Diruthenium-Polyyn-diyl-Diruthenium Wires: Electronic Coupling in the Long Distance Regime. J. Am. Chem. Soc. 2014, 136, 12174–12183. [Google Scholar] [CrossRef] [PubMed]
- Schermann, G.; Grösser, T.; Hampel, F.; Hirsch, A. Dicyanopolyynes: A Homologuous Series of End-Capped Linear sp Carbon. Chem. Eur. J. 1997, 3, 1105–1112. [Google Scholar] [CrossRef]
- Luu, T.; Elliot, E.; Slepkov, A.D.; Eisler, S.; McDonald, R.; Hegmann, F.A.; Tykwinski, R.R. Synthesis, Structure, and Nonlinear Optical Properties of Diarylpolyynes. Org. Lett. 2005, 7, 51–54. [Google Scholar] [CrossRef]
- Werz, D.B.; Gleiter, R.; Rominger, F. Tellurium-capped carbon rods: Syntheses and electronic and structural properties. Organometallics 2003, 22, 843–849. [Google Scholar] [CrossRef]
- Werz, D.B.; Gleiter, R. Polyalkynes capped by sulfur and selenium. J. Org. Chem. 2003, 68, 9400–9405. [Google Scholar] [CrossRef]
- Coat, F.; Paul, F.; Lapinte, C.; Toupet, L.; Costuas, K.; Halet, J.-F. Chemistry of the 1,3,5,7-octatetraynediyl carbon rod end-capped by two electron-rich (η5-C5Me5)(η2-dppe) Fe groups. J. Organomet. Chem. 2003, 683, 368–378. [Google Scholar] [CrossRef]
- Xu, G.-L.; Zou, G.; Ni, Y.-H.; DeRosa, M.C.; Crutchley, R.J.; Ren, T. Polyyn-diyls capped by diruthenium termini: A new family of carbon-rich organometallic compounds and distance-dependent electronic coupling therein. J. Am. Chem. Soc. 2003, 125, 10057–10065. [Google Scholar] [CrossRef]
- Weisbach, N.; Kuhn, H.; Amini, H.; Ehnbom, A.; Hampel, F.; Reibenspies, J.H.; Hall, M.B.; Gladysz, J.A. Triisopropylsilyl (TIPS) Alkynes as Building Blocks for Syntheses of Platinum Triisopropylsilylpolyynyl and Diplatinum Polyynediyl Complexes. Organometallics 2019, 38, 3294–3310. [Google Scholar] [CrossRef]
- Yam, V.W.-Y.; Wong, K.M.-C.; Zhu, N. Luminescent Platinum(II) Terpyridyl-Capped Carbon-Rich Molecular Rods—An Extension from Molecular- to Nanometer-Scale Dimensions. Angew. Chem. Int. Ed. 2003, 42, 1400–1403. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, A.; Akita, M.; Moro-oka, Y. Synthesis and Characterization of the Dodecahexaynediyldiiron Complex, Fp*−(C≡C)6−Fp* [Fp*= Fe(η5-C5Me5)(CO)2], the Longest Structurally Characterized Polyynediyl Complex. Organometallics 1999, 18, 3241–3244. [Google Scholar] [CrossRef]
- Classen, J.; Gleiter, R.; Rominger, F. Oligoalkyne-Bridged (η4-Cyclobutadiene)(η5-cyclopentadienyl)cobalt Fragments—Syntheses and Properties. Eur. J. Inorg. Chem. 2002, 2002, 2040–2046. [Google Scholar] [CrossRef]
- Adams, R.D.; Qu, B.; Smith, M.D. Synthesis and characterization of 1,12-Bis (ferrocenyl)-1,3,5,7,9,11-dodecahexayne and its coordination to triosmium and dicobalt carbonyls. Organometallics 2002, 21, 3867–3872. [Google Scholar] [CrossRef]
- Antonova, A.B.; Bruce, M.I.; Ellis, B.G.; Gaudio, M.; Humphrey, P.A.; Jevric, M.; Melino, G.; Nicholson, B.K.; Perkins, G.J.; Skelton, B.W.; et al. A novel methodology for the synthesis of complexes containing long carbon chains linking metal centres: Molecular structures of {Ru(dppe)Cp*}2(μ-C14) and {Co3(μ-dppm)(CO)7}2(μ3:μ3-C16). Chem. Commun. 2004, 8, 960–961. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, T.; Lieb, E.H. Proof of the Peierls Instability in One Dimension. Phys. Rev. Lett. 1987, 59, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S.K.; Pouget, J.P.; Comes, R.; Garito, A.F.; Heeger, A. X-ray studies of 2kF and 4kF anomalies in tetrathiafulvalene-tetracyanoquinodimethane (TTF-TCNQ). Phys. Rev. B 1977, 16, 1468–1479. [Google Scholar] [CrossRef]
- Hunter, J.M.; Fye, J.L.; Roskamp, E.J.; Jarrold, M.F. Annealing carbon cluster ions: A mechanism for fullerene synthesis. J. Phys. Chem. 1994, 98, 1810–1818. [Google Scholar] [CrossRef]
- Lagow, R.J.; Kampa, J.J.; Wei, H.-C.; Battle, S.L.; Genge, J.W.; Laude, D.A.; Harper, C.J.; Bau, R.; Stevens, R.C.; Haw, J.F.; et al. Synthesis of linear acetylenic carbon: The “sp” carbon allotrope. Science 1995, 267, 362–367. [Google Scholar] [CrossRef]
- Homan, K.-H. Fullerenes and soot formation. New pathways to large particles in flame. Angew. Chem. Int. Ed. 1998, 37, 2434–2451. [Google Scholar] [CrossRef]
- Liang, C.; Allen, L.C. Double bond deformation: Cumulenes, cumulenones, and Group IV ethylene analogs. J. Am. Chem. Soc. 1991, 113, 1873–1878. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S. Structure, stability, and fragmentation of small carbon clusters. J. Chem. Phys. 1987, 87, 2191–2197. [Google Scholar] [CrossRef]
- Roeges, N.P.G. A Guide to the Complete Interpretation of Infrared Spectra of Organic Structures; John Wiley & Sons: Chichester, UK, 1994. [Google Scholar]
- Horný, L.; Petraco, N.D.K.; Pak, C.; Schaefer, H.F., III. What is the nature of polyacetylene neutral and anionic chains HC2nH and HC2nH− (n = 6–12) that have recently been observed? J. Am. Chem. Soc. 2002, 124, 5861–5864. [Google Scholar]
- Szafert, S.; Gladysz, J.A. Carbon in one dimension: Structural analysis of the higher conjugated polyynes. Chem. Rev. 2003, 103, 4175–4205. [Google Scholar] [CrossRef]
- Osowska, K.; Lis, T.; Szafert, S. Protection/Deprotection-Free Syntheses and Structural Analysis of (Keto-aryl) diynes. Eur. J. Org. Chem. 2008, 27, 4598–4606. [Google Scholar] [CrossRef]
- Messerschmidt, M.; Scheins, S.; Luger, P. Charge density of (−)-strychnine from 100 to 15 K, a comparison of four data sets. Acta Cryst. 2005, B61, 115–121. [Google Scholar] [CrossRef]
- Munshi, P.; Guru Row, T.N. Topological analysis of charge density distribution in concomitant polymorphs of 3-acetylcoumarin, a case of packing polymorphism. Cryst. Growth Des. 2006, 6, 708–718. [Google Scholar] [CrossRef]
- Reji, T.; Lakshmi, S.; Pati, S.K.; Kulkarni, G.U. Role of Triple Bond in 1,2-Diphenylacetylene Crystal: A Combined Experimental andTheoretical Study. J. Phys. Chem. B 2006, 110, 24674–24677. [Google Scholar]
- Cremer, D.; Kraka, E. Role of triple bond in 1, 2-diphenylacetylene crystal: A combined experimental and theoretical study. Croat. Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Bürgi, H.-B.; Capelli, S.C.; Goeta, A.E.; Howard, J.A.K.; Spackman, M.A.; Yufit, D.S. Electron Distribution and Molecular Motion in Crystalline Benzene: An Accurate Experimental Study Combining CCD X-ray Data on C6H6 with Multitemperature Neutron-Diffraction Results on C6D6. Chem. Eur. J. 2002, 8, 3512–3521. [Google Scholar] [CrossRef]
- Chopra, D.; Cameron, T.S.; Ferrara, J.D.; Guru Row, T.N. Pointers toward the Occurrence of C−F···F−C Interaction: Experimental Charge Density Analysis of 1-(4-Fluorophenyl)-3,6,6-trimethyl-2-phenyl-1,5,6,7-tetrahydro-4H-indol-4-one and 1-(4-Fluorophenyl)-6-methoxy-2-phenyl-1,2,3,4-tetrahydroisoquinoline. J. Phys. Chem. A 2006, 110, 10465–10477. [Google Scholar] [CrossRef] [PubMed]
- Zhurova, E.A.; Matta, C.F.; Wu, N.; Zhurov, V.V.; Pinkerton, A.A. Experimental and theoretical electron density study of estrone. J. Am. Chem. Soc. 2006, 128, 8849–8861. [Google Scholar] [CrossRef] [PubMed]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Density-functional exchange-energy approximation with correct asymptotic behaviour. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauling, L. The nature of the chemical bond. Application of results obtained from the quantum mechanics and from a theory of paramagnetic susceptibility to the structure of molecules. J. Am. Chem. Soc. 1931, 53, 1367–1400. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Mierzwicki, K.; Berski, S.; Latajka, Z. Nature of chemical bonds in MCCH (M = Li, Na, K) based on the topological analysis of electron localisation function (ELF) and electron density. Chem. Phys. Lett. 2000, 331, 538–546. [Google Scholar] [CrossRef]
- xd-a, version 4.10; Computer Program Package for Multipole Refinement and Anlysis of Electron Densities from Diffraction Data; Free University: Berlin, Germany, 1995.
- Hansen, N.K.; Coppens, P. Testing aspherical atom refinements on small-molecule data sets. Acta Crystallogr. 1978, A34, 909–921. [Google Scholar] [CrossRef]
- Bader, R.F. Atoms in Molecules—A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- CRYSTAL, version 2003; User’s Manual; University of Torino: Torino, Italy, 2003.
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theoret. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- DGrid, version 4.2, Basin 4.2; Max Planck Institute for Chemical Physics of Solids: Dresden, Germany, 2007.
- The numerical grid value equaled 0.04 bohr was used. The “closed-shell” formula of ELF has been used.
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Gaussian, version 03, Revision D.02; Gaussian, Inc.: Wallingford, CT, USA, 2004.
- Molekel, version 4.1; Swiss National Supercomputing Centre: Manno, Switzerland, 2000–2001.
Sample Availability: Sample of the compound 1 is available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
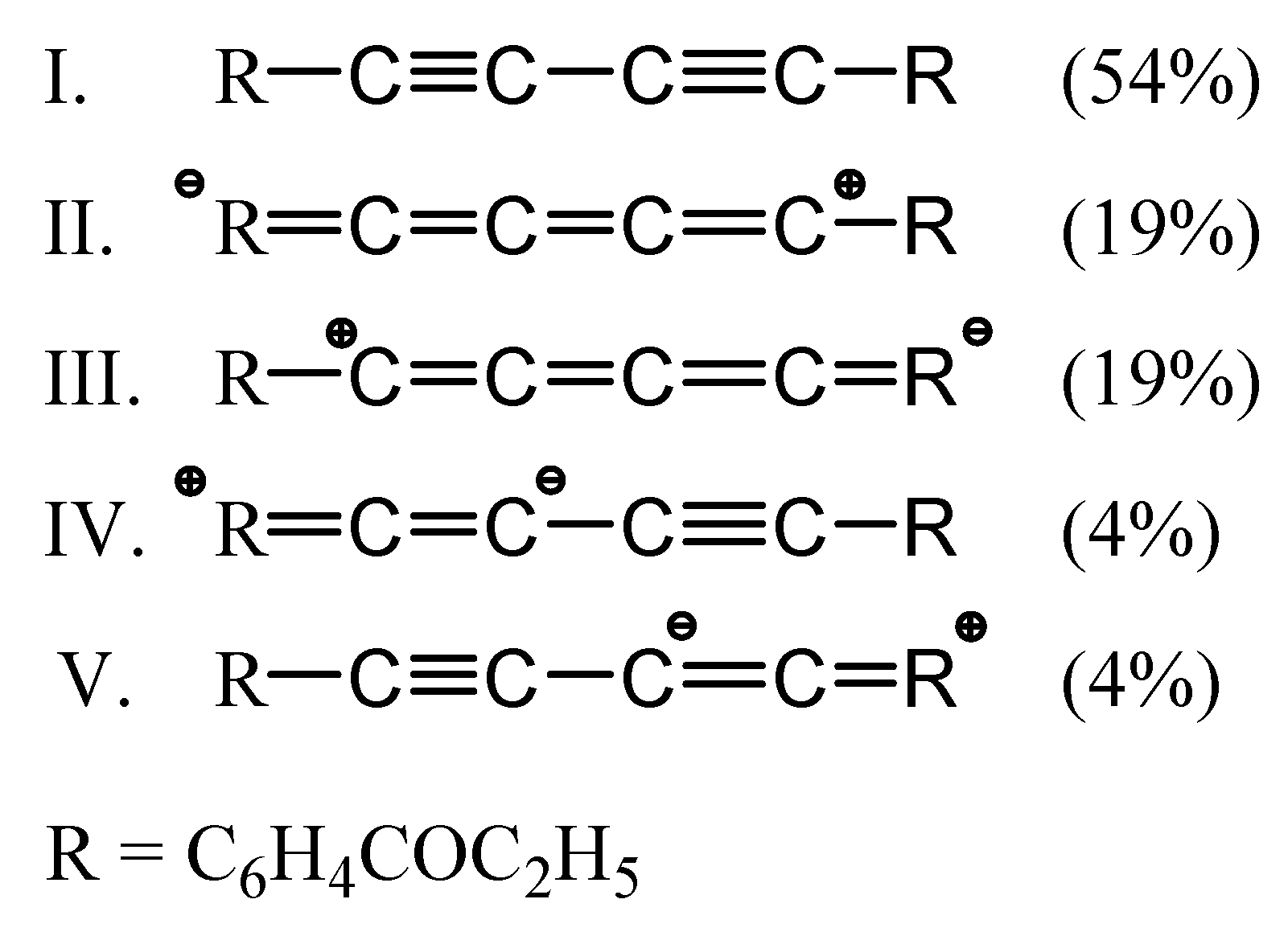{kind=link}
| CR | MP | CR | MP | ||
|---|---|---|---|---|---|
| O(1)-C(3) | 1.2231(8) | 1.2248(4) | C(11)-C(12) | 1.4077(8) | 1.4080(5) |
| C(1)-C(2) | 1.2120(8) | 1.2169(4) | C(11)-C(16) | 1.4055(8) | 1.4060(5) |
| C(1)-C(11) | 1.4300(9) | 1.4259(4) | C(12)-C(13) | 1.3933(9) | 1.3934(4) |
| C(2)-C(2A) | 1.3670(11) | 1.3636(6) | C(13)-C(14) | 1.3981(8) | 1.3987(4) |
| C(3)-C(4) | 1.5141(8) | 1.5109(4) | C(14)-C(15) | 1.4039(8) | 1.4039(4) |
| C(3)-C(14) | 1.5016(9) | 1.5008(4) | C(15)-C(16) | 1.3908(9) | 1.3924(4) |
| C(4)-C(5) | 1.5202(9) | 1.5237(5) |
| Atom | Charge | Atom | Charge | Atom | Charge |
|---|---|---|---|---|---|
| O(1) | −0.59(5) | C(12) | 0.10(4) | H(51) | 0.12(9) |
| C(1) | −0.30(4) | C(13) | 0.04(4) | H(52) | −0.28(9) |
| C(2) | −0.25(4) | C(14) | 0.02(3) | H(53) | 0.09(8) |
| C(3) | 0.05(4) | C(15) | 0.14(5) | H(12) | 0.06(6) |
| C(4) | 0.10(4) | C(16) | 0.24(6) | H(13) | 0.06(6) |
| C(5) | 0.30(6) | H(41) | 0.07(6) | H(15) | 0.06(6) |
| C(11) | −0.05(3) | H(42) | 0.16(5) | H(16) | −0.14(8) |
| Bond | ρcp | Δρcp | Rij | d1 | d2 | Hessian Eigenvalues | ε | ||
|---|---|---|---|---|---|---|---|---|---|
| λ1 | λ2 | λ3 | |||||||
| O(1)-C(3) | 2.78 | −6.3 | 1.2249 | 0.7944 | 0.4305 | −23.95 | −22.93 | 40.59 | 0.04 |
| C(1)-C(2) | 2.92 | −28.0 | 1.2170 | 0.6431 | 0.5739 | −18.45 | −18.14 | 8.54 | 0.02 |
| C(1)-C(11) | 2.01 | −14.9 | 1.4260 | 0.7231 | 0.7029 | −13.43 | −12.81 | 11.33 | 0.05 |
| C(2)-C(2′) | 2.19 | −15.3 | 1.3636 | 0.6818 | 0.6818 | −13.95 | −13.35 | 11.98 | 0.04 |
| C(3)-C(4) | 1.77 | −12.6 | 1.5110 | 0.8174 | 0.6937 | −11.47 | −11.05 | 9.89 | 0.04 |
| C(3)-C(14) | 1.85 | −14.8 | 1.5008 | 0.7521 | 0.7487 | −12.81 | −11.81 | 9.78 | 0.08 |
| C(4)-C(5) | 1.66 | −10.2 | 1.5238 | 0.7655 | 0.7584 | −10.22 | −9.98 | 9.98 | 0.02 |
| C(4)-H(41) | 1.97 | −19.7 | 1.0857 | 0.6240 | 0.4617 | −16.78 | −15.33 | 12.37 | 0.09 |
| C(4)-H(42) | 1.76 | −15.2 | 1.0840 | 0.6834 | 0.4006 | −15.43 | −14.50 | 14.73 | 0.06 |
| C(5)-H(51) | 2.08 | −22.7 | 1.0858 | 0.6276 | 0.4583 | −17.76 | −16.64 | 11.73 | 0.07 |
| C(5)-H(52) | 1.97 | −15.5 | 1.0851 | 0.5402 | 0.5449 | −15.04 | −13.98 | 13.49 | 0.08 |
| C(5)-H(53) | 1.94 | −19.1 | 1.0892 | 0.6296 | 0.4596 | −16.48 | −15.05 | 12.45 | 0.10 |
| C(11)-C(12) | 2.18 | −19.5 | 1.4081 | 0.7208 | 0.6873 | −15.57 | −13.61 | 9.68 | 0.14 |
| C(11)-C(16) | 2.12 | −19.0 | 1.4061 | 0.6881 | 0.7180 | −15.36 | −13.13 | 9.52 | 0.17 |
| C(12)-C(13) | 2.21 | −19.5 | 1.3936 | 0.6888 | 0.7048 | −15.48 | −13.80 | 9.78 | 0.12 |
| C(12)-H(12) | 2.06 | −21.8 | 1.0835 | 0.6186 | 0.4649 | −17.36 | −16.87 | 12.43 | 0.03 |
| C(13)-C(14) | 2.14 | −18.8 | 1.3988 | 0.6811 | 0.7177 | −14.92 | −13.66 | 9.73 | 0.09 |
| C(13)-H(13) | 2.08 | −22.7 | 1.0836 | 0.6383 | 0.4453 | −18.12 | −17.59 | 13.04 | 0.03 |
| C(14)-C(15) | 2.21 | −20.2 | 1.4041 | 0.6904 | 0.7137 | −15.73 | −14.06 | 9.61 | 0.12 |
| C(15)-C(16) | 2.21 | −20.1 | 1.3924 | 0.6783 | 0.7141 | −15.81 | −13.66 | 9.38 | 0.16 |
| C(15)-H(15) | 2.10 | −23.4 | 1.0834 | 0.6068 | 0.4766 | −17.76 | −17.44 | 11.78 | 0.02 |
| C(16)-H(16) | 2.20 | −23.4 | 1.0843 | 0.5232 | 0.5610 | −18.18 | −17.41 | 12.21 | 0.04 |
| Energy | SVWN | PWGGA | B3LYP | B3PW |
|---|---|---|---|---|
| Ecryst | −100.7 | −81.4 | −54.9 | −28.2 |
| Ecryst + BSSE | −78.3 | −58.1 | −32.3 | −8.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Starynowicz, P.; Berski, S.; Gulia, N.; Osowska, K.; Lis, T.; Szafert, S. Is It Conjugated or Not? The Theoretical and Experimental Electron Density Map of Bonding in p-CH3CH2COC6H4-C≡C-C≡C-p-C6H4COCH3CH2. Molecules 2020, 25, 4388. https://doi.org/10.3390/molecules25194388
Starynowicz P, Berski S, Gulia N, Osowska K, Lis T, Szafert S. Is It Conjugated or Not? The Theoretical and Experimental Electron Density Map of Bonding in p-CH3CH2COC6H4-C≡C-C≡C-p-C6H4COCH3CH2. Molecules. 2020; 25(19):4388. https://doi.org/10.3390/molecules25194388
Chicago/Turabian StyleStarynowicz, Przemysław, Sławomir Berski, Nurbey Gulia, Karolina Osowska, Tadeusz Lis, and Sławomir Szafert. 2020. "Is It Conjugated or Not? The Theoretical and Experimental Electron Density Map of Bonding in p-CH3CH2COC6H4-C≡C-C≡C-p-C6H4COCH3CH2" Molecules 25, no. 19: 4388. https://doi.org/10.3390/molecules25194388
APA StyleStarynowicz, P., Berski, S., Gulia, N., Osowska, K., Lis, T., & Szafert, S. (2020). Is It Conjugated or Not? The Theoretical and Experimental Electron Density Map of Bonding in p-CH3CH2COC6H4-C≡C-C≡C-p-C6H4COCH3CH2. Molecules, 25(19), 4388. https://doi.org/10.3390/molecules25194388