
Versatility of the Cyano Group in Intermolecular Interactions


Abstract
:
1. Introduction
2. Results
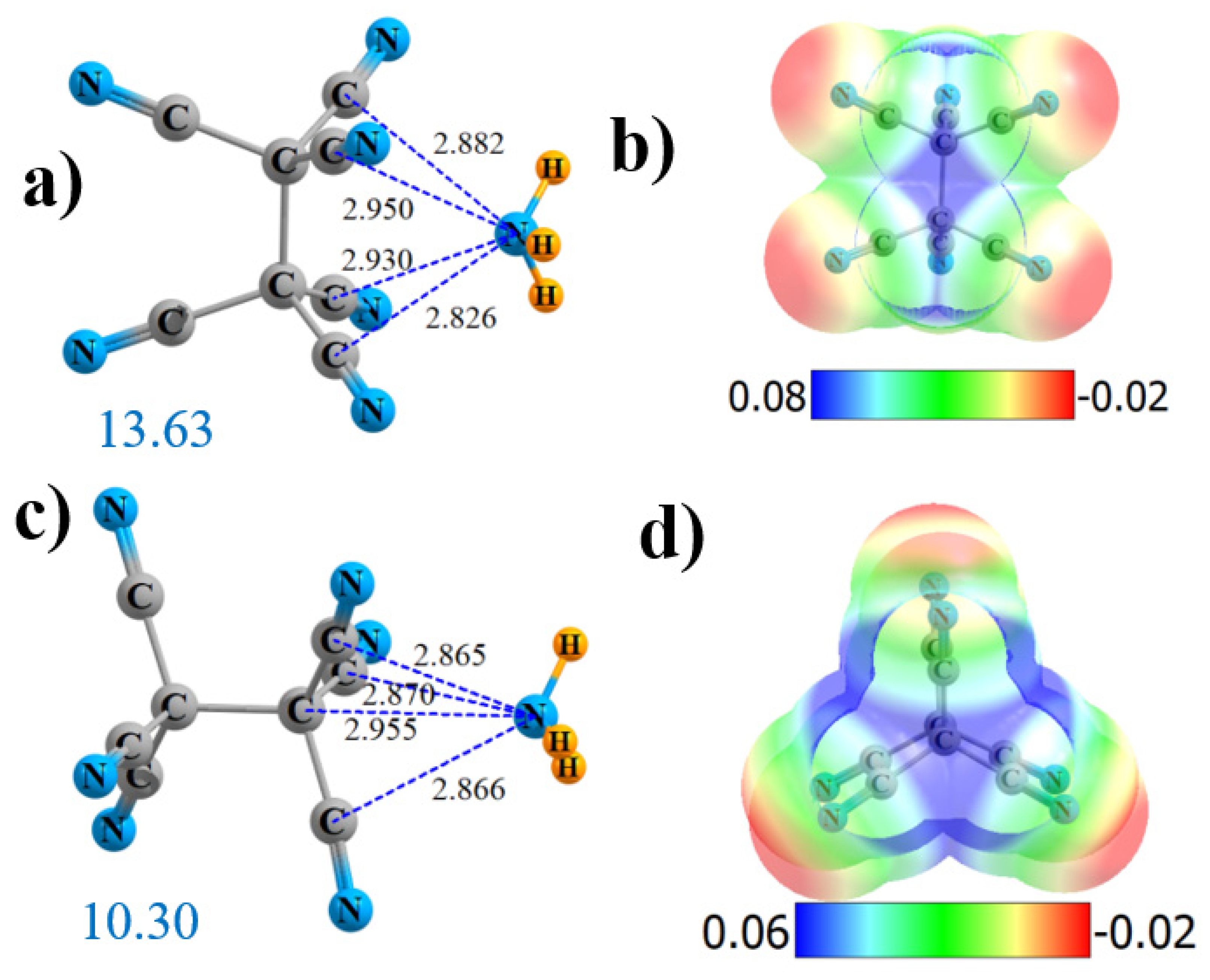
2.1. Hexa-Substituted
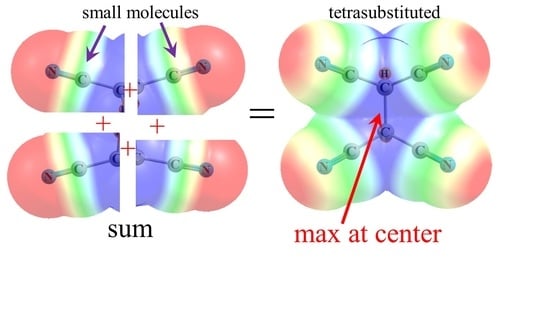
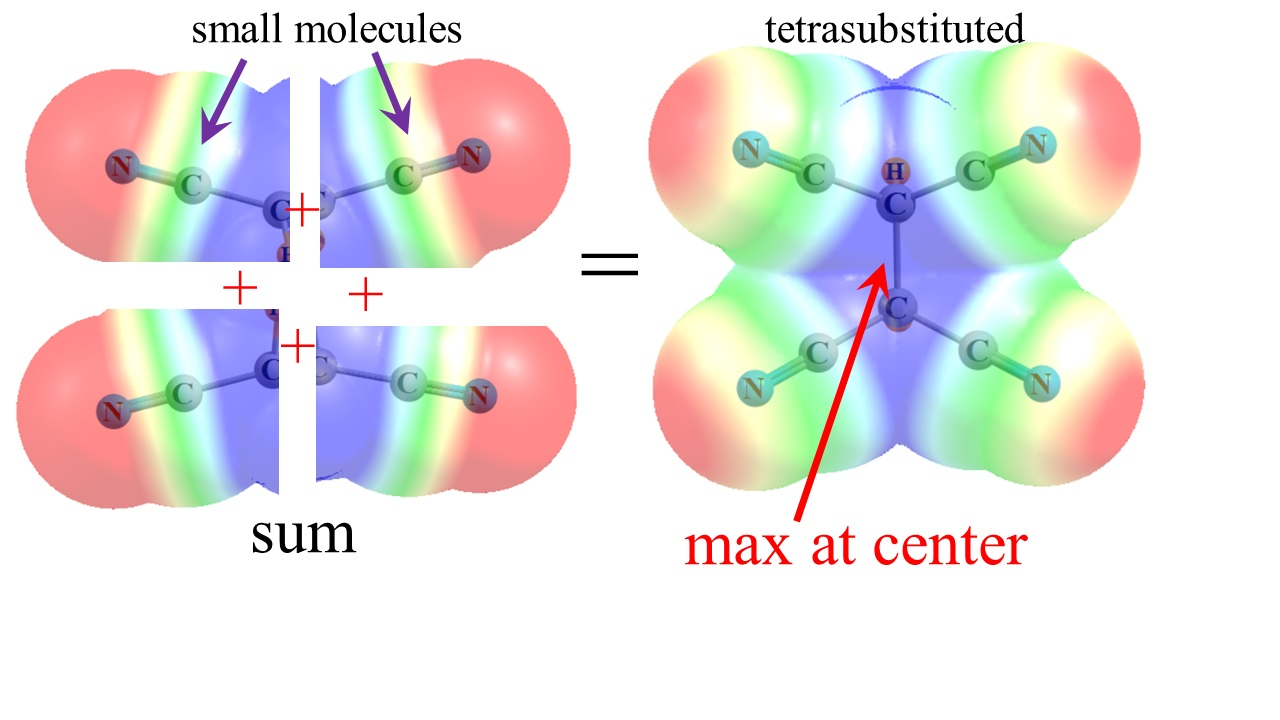
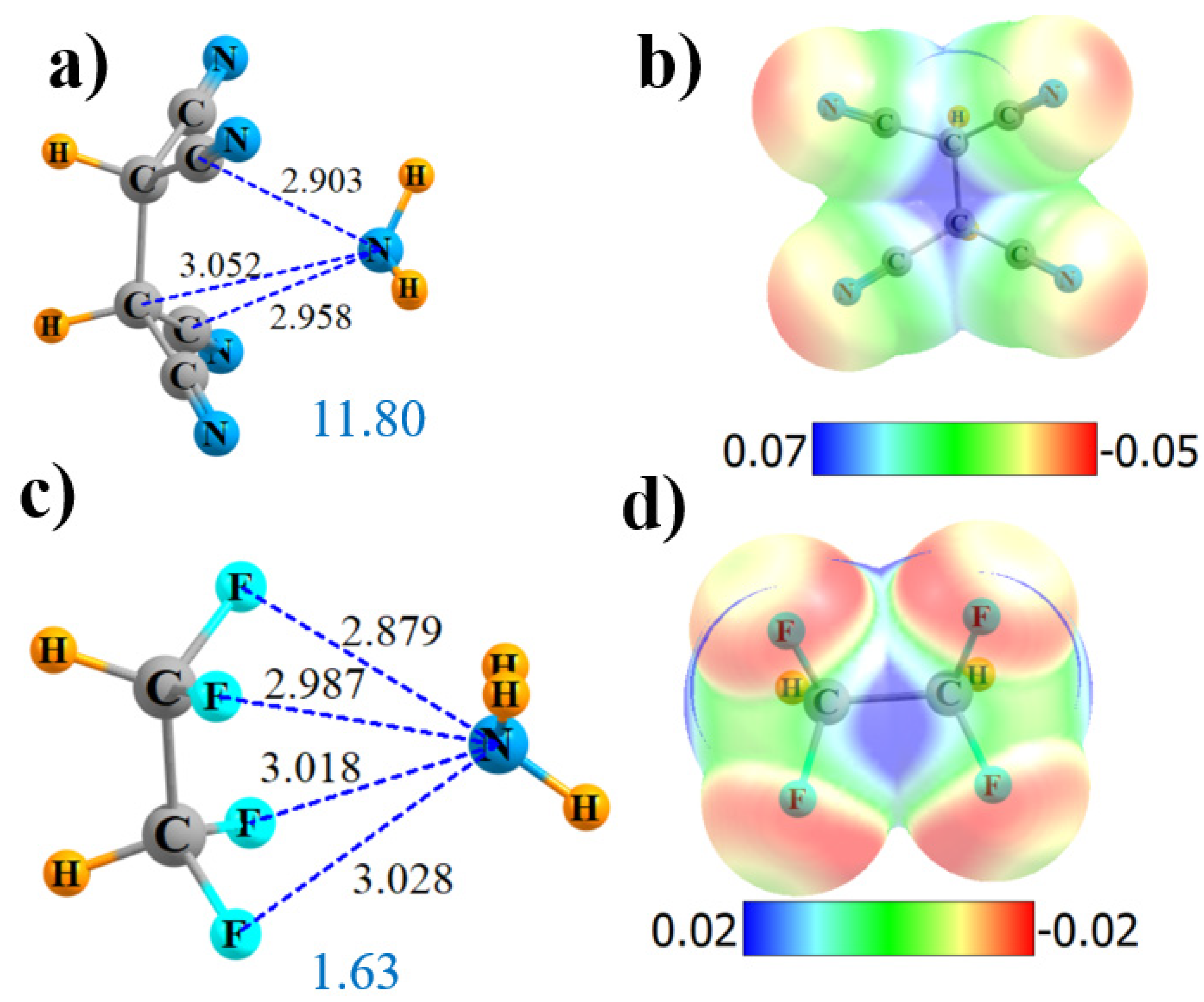
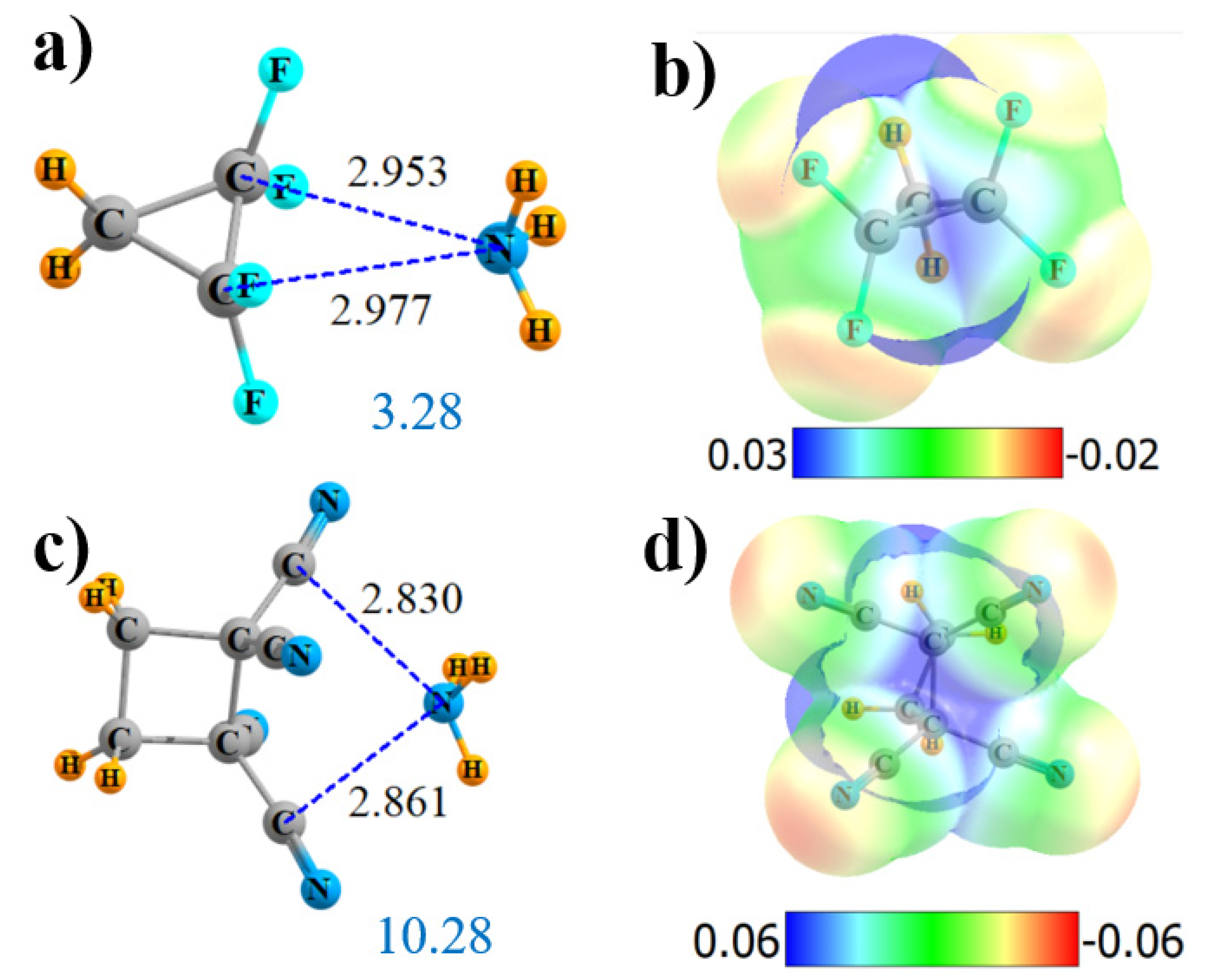
2.2. Tetrasubstituted
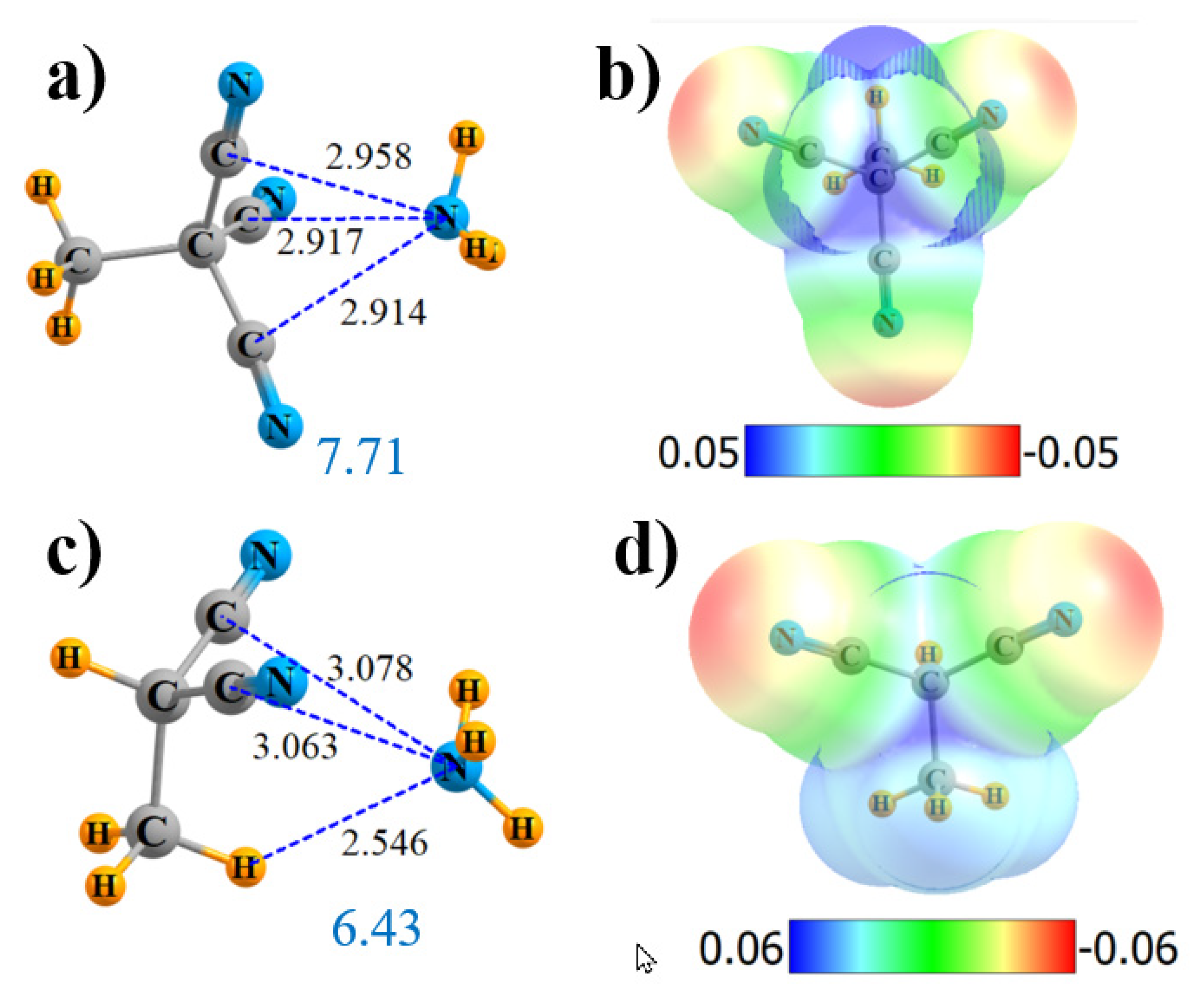
2.3. Less Than Four Substituents
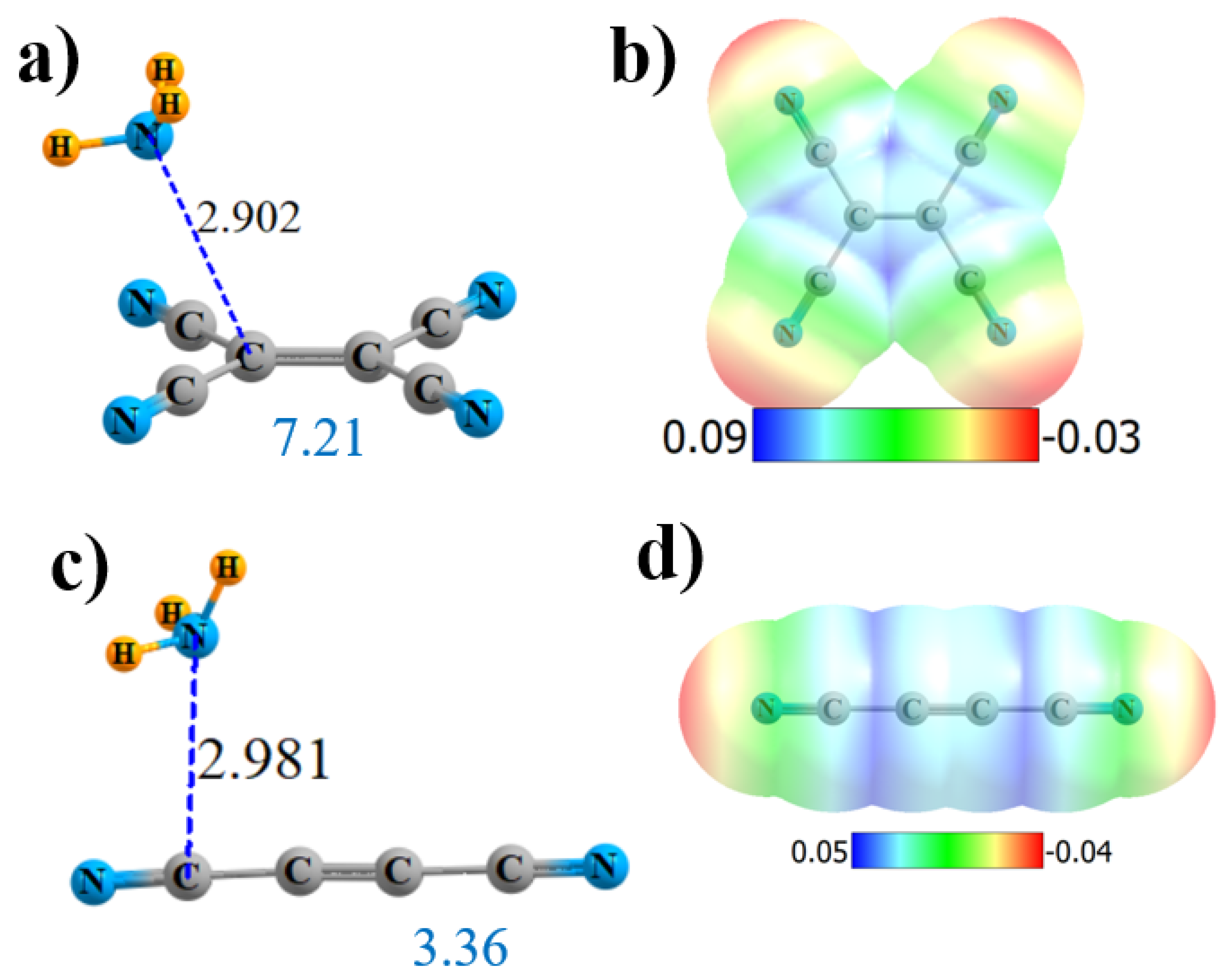
2.4. Substituted Alkene and Alkyne
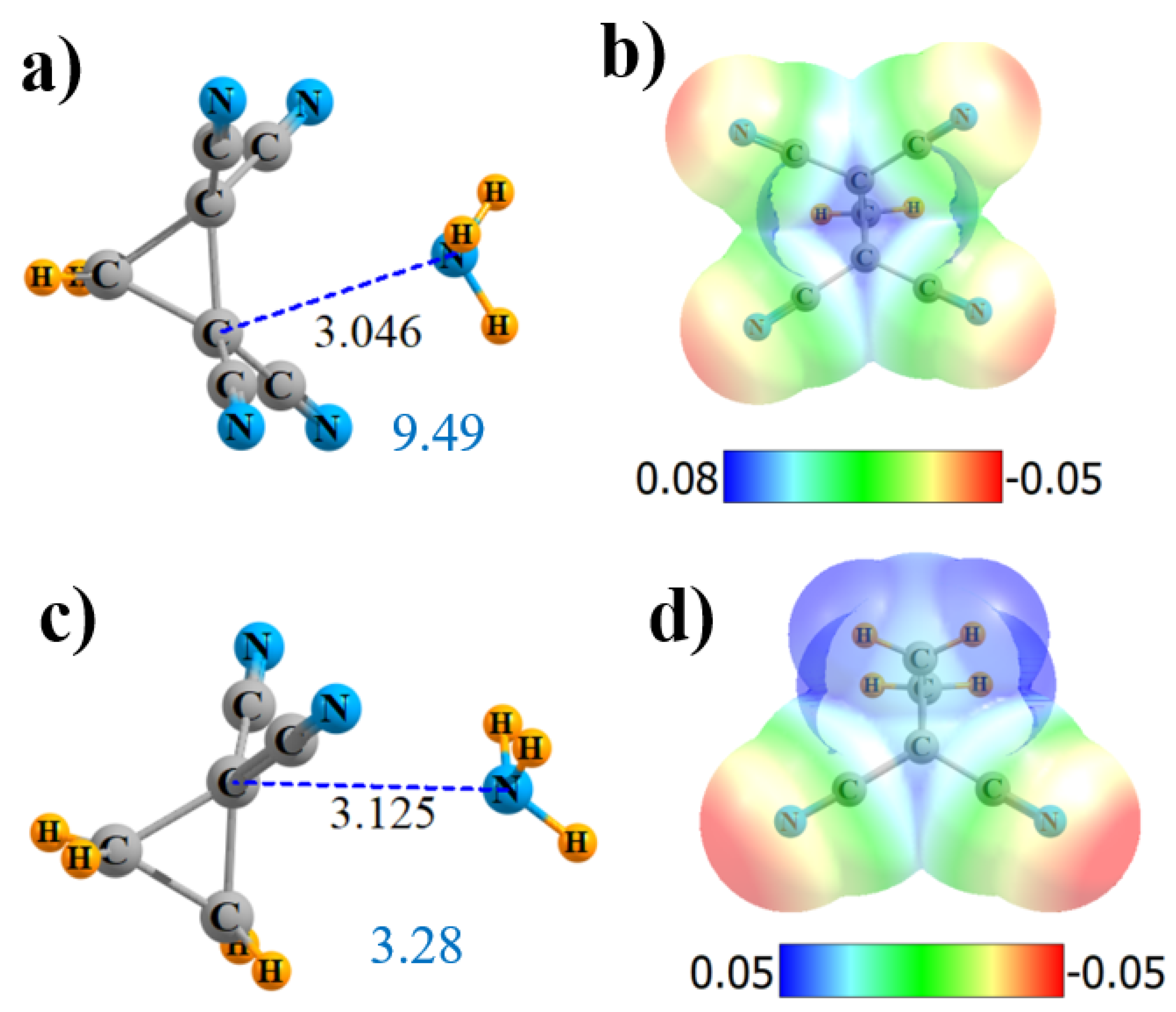
2.5. Cyclic Lewis Acids
2.6. Monomer Perturbations
3. Discussion
4. Conclusions
5. Computational Methods
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond; Freeman: San Francisco, CA, USA, 1960. [Google Scholar]
- Vinogradov, S.N.; Linnell, R.H. Hydrogen Bonding; Van Nostrand-Reinhold: New York, NY, USA, 1971. [Google Scholar]
- Joesten, M.D.; Schaad, L.J. Hydrogen Bonding; Marcel Dekker: New York, NY, USA, 1974; p. 622. [Google Scholar]
- Schuster, P.; Zundel, G.; Sandorfy, C. The Hydrogen Bond. Recent Developments in Theory and Experiments; North-Holland Publishing Co.: Amsterdam, The Netherlands, 1976. [Google Scholar]
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer: Berlin, Germany, 1991. [Google Scholar]
- Scheiner, S. Hydrogen Bonding. A Theoretical Perspective; Oxford University Press: New York, NY, USA, 1997; p. 375. [Google Scholar]
- Gilli, G.; Gilli, P. The Nature of the Hydrogen Bond; Oxford University Press: Oxford, UK, 2009; p. 313. [Google Scholar]
- Gougoula, E.; Medcraft, C.; Alkorta, I.; Walker, N.R.; Legon, A.C. A chalcogen-bonded complex H3N···S=C=S formed by ammonia and carbon disulfide characterised by chirped-pulse, broadband microwave spectroscopy. J. Chem. Phys. 2019, 150, 084307. [Google Scholar] [CrossRef] [Green Version]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Pnicogen bonds in complexes with CO and CS: Differentiating properties. Mol. Phys. 2019, 117, 1117–1127. [Google Scholar] [CrossRef]
- Grabowski, S.J. Pnicogen and tetrel bonds—Tetrahedral Lewis acid centres. Struct. Chem. 2019, 30, 1141–1152. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Grabowski, S.J. Pnicogen and hydrogen bonds: Complexes between PH3X+ and PH2X systems. Phys. Chem. Chem. Phys. 2015, 17, 3261–3272. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, S.J. Halogen bond with the multivalent halogen acting as the Lewis acid center. Chem. Phys. Lett. 2014, 605, 131–136. [Google Scholar] [CrossRef]
- Navarro-García, E.; Galmés, B.; Velasco, M.D.; Frontera, A.; Caballero, A. Anion Recognition by Neutral Chalcogen Bonding Receptors: Experimental and Theoretical Investigations. Chem. Eur. J. 2020, 26, 4706–4713. [Google Scholar] [CrossRef] [PubMed]
- Frontera, A.; Bauzá, A. Halogen Bonds in Protein Nucleic Acid Recognition. J. Chem. Theory Comput. 2020, 16, 4744–4752. [Google Scholar] [CrossRef] [PubMed]
- Franconetti, A.; Quiñonero, D.; Frontera, A.; Resnati, G. Unexpected chalcogen bonds in tetravalent sulfur compounds. Phys. Chem. Chem. Phys. 2019, 21, 11313–11319. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S.; Wysokiński, R.; Michalczyk, M.; Zierkiewicz, W. Pnicogen Bonds Pairing Anionic Lewis Acid with Neutral and Anionic Bases. J. Phys. Chem. A 2020, 124, 4998–5006. [Google Scholar] [CrossRef]
- Wysokiński, R.; Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. How Many Pnicogen Bonds can be Formed to a Central Atom Simultaneously? J. Phys. Chem. A 2020, 124, 2046–2056. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Wysokiński, R.; Michalczyk, M.; Scheiner, S. Chalcogen bonding of two ligands to hypervalent YF4 (Y = S, Se, Te, Po). Phys. Chem. Chem. Phys. 2019, 21, 20829–20839. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Electrostatics and Polarization in σ- and π-Hole Noncovalent Interactions: An Overview. ChemPhysChem 2020, 21, 579–588. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. σ-holes and π-holes: Similarities and differences. J. Comput. Chem. 2018, 39, 464–471. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, B.G.; Zabardasti, A.; do Rego, D.G.; Pour, M.M. The formation of H···X hydrogen bond, C···X carbon-halide or Si···X tetrel bonds on the silylene-halogen dimers (X = F or Cl): Intermolecular strength, molecular orbital interactions and prediction of covalency. Theor. Chem. Acc. 2020, 139, 131. [Google Scholar] [CrossRef]
- Hou, M.; Liu, Z.; Li, Q. The π-hole tetrel bond between X2TO and CO2: Substituent effects and its potential adsorptivity for CO2. Int. J. Quantum Chem. 2020, 120, e26251. [Google Scholar] [CrossRef]
- Heywood, V.L.; Alford, T.P.J.; Roeleveld, J.J.; Lekanne Deprez, S.J.; Verhoofstad, A.; van der Vlugt, J.I.; Domingos, S.R.; Schnell, M.; Davis, A.P.; Mooibroek, T.J. Observations of tetrel bonding between sp3-carbon and THF. Chem. Sci. 2020, 11, 5289–5293. [Google Scholar] [CrossRef]
- Franconetti, A.; Frontera, A. “Like–like” tetrel bonding interactions between Sn centres: A combined ab initio and CSD study. Dalton Trans. 2019, 48, 11208–11216. [Google Scholar] [CrossRef]
- Zierkiewicz, W.; Michalczyk, M.; Wysokiński, R.; Scheiner, S. Dual Geometry Schemes in Tetrel Bonds: Complexes between TF4 (T = Si, Ge, Sn) and Pyridine Derivatives. Molecules 2019, 24, 376. [Google Scholar] [CrossRef] [Green Version]
- Mundlapati, V.R.; Sahoo, D.K.; Bhaumik, S.; Jena, S.; Chandrakar, A.; Biswal, H.S. Noncovalent Carbon-Bonding Interactions in Proteins. Angew. Chem. Int. Ed. 2018, 57, 16496–16500. [Google Scholar] [CrossRef]
- Scheiner, S. Differential Binding of Tetrel-Bonding Bipodal Receptors to Monatomic and Polyatomic Anions. Molecules 2019, 24, 227. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Scheiner, S. Effects of Halogen, Chalcogen, Pnicogen, and Tetrel Bonds on IR and NMR Spectra. Molecules 2019, 24, 2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mani, D.; Arunan, E. The X–CY (X = O/F, Y = O/S/F/Cl/Br/N/P) ‘carbon bond’ and hydrophobic interactions. Phys. Chem. Chem. Phys. 2013, 15, 14377–14383. [Google Scholar] [CrossRef] [PubMed]
- Mani, D.; Arunan, E. The X-C···Y Carbon Bond. In Noncovalent Forces; Scheiner, S., Ed.; Springer: Heidelberg, Germany, 2015; pp. 323–356. [Google Scholar]
- Azofra, L.M.; Scheiner, S. Complexation of n SO2 Molecules (n = 1,2,3) with Formaldehyde and Thioformaldehyde. J. Chem. Phys. 2014, 140, 034302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naeem Ahmed, M.; Yasin, K.A.; Aziz, S.; Khan, S.U.; Tahir, M.N.; Gil, D.M.; Frontera, A. Relevant π-hole tetrel bonding interactions in ethyl 2-triazolyl-2-oxoacetate derivatives: Hirshfeld surface analysis and DFT calculations. CrystEngComm 2020, 22, 3567–3578. [Google Scholar] [CrossRef]
- Dong, W.; Wang, Y.; Cheng, J.; Yang, X.; Li, Q. Competition between σ-hole pnicogen bond and π-hole tetrel bond in complexes of CF2=CFZH2 (Z = P, As, and Sb). Mol. Phys. 2019, 117, 251–259. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Nurazar, R. Chalcogen bonds formed through π-holes: SO3 complexes with nitrogen and phosphorus bases. Mol. Phys. 2016, 114, 276–282. [Google Scholar] [CrossRef]
- Azofra, L.M.; Alkorta, I.; Scheiner, S. An exploration of the ozone dimer potential energy surface. J. Chem. Phys. 2014, 140, 244311. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, X.; Zeng, Y.; Meng, L.; Zhang, X. Enhancing effects of π-hole tetrel bonds on the σ-hole interactions in complexes involving F2TO (T = Si, Ge, Sn). Struct. Chem. 2019, 30, 1301–1313. [Google Scholar] [CrossRef]
- Shen, S.; Zeng, Y.; Li, X.; Meng, L.; Zhang, X. Insight into the π-hole···π-electrons tetrel bonds between F2ZO (Z = C, Si, Ge) and unsaturated hydrocarbons. Int. J. Quantum Chem. 2018, 118, e25521. [Google Scholar] [CrossRef]
- Wei, Y.; Li, Q. Comparison for σ-hole and π-hole tetrel-bonded complexes involving cyanoacetaldehyde. Mol. Phys. 2018, 116, 222–230. [Google Scholar] [CrossRef]
- Galmés, B.; Martínez, D.; Infante-Carrió, M.F.; Franconetti, A.; Frontera, A. Theoretical ab Initio Study on Cooperativity Effects between Nitro π-hole and Halogen Bonding Interactions. ChemPhysChem 2019, 20, 1135–1144. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. π-Hole Bonds: Boron and Aluminum Lewis Acid Centers. ChemPhysChem 2015, 16, 1470–1479. [Google Scholar] [CrossRef] [PubMed]
- Jabłoński, M. Hydride-Triel Bonds. J. Comput. Chem. 2018, 39, 1177–1191. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Lei, J.; Feng, G.; Grabow, J.-U.; Gou, Q. The rotational spectrum of acetophenone-CO2: Preferred non-covalent interactions. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 238, 118424. [Google Scholar] [CrossRef]
- Lu, T.; Zhang, J.; Gou, Q.; Feng, G. Structure and C⋯N tetrel-bonding of the isopropylamine–CO2 complex studied by microwave spectroscopy and theoretical calculations. Phys. Chem. Chem. Phys. 2020, 22, 8467–8475. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Potential Energy Surfaces of HN(CH)SX:CO2 for X = F, Cl, NC, CN, CCH, and H: N···C Tetrel Bonds and O···S Chalcogen Bonds. J. Phys. Chem. A 2019, 123, 7270–7277. [Google Scholar] [CrossRef]
- Azofra, L.M.; Scheiner, S. Complexes Containing CO2 and SO2. Mixed Dimers, Trimers and Tetramers. Phys. Chem. Chem. Phys. 2014, 16, 5142–5149. [Google Scholar] [CrossRef] [Green Version]
- Azofra, L.M.; Scheiner, S. Tetrel, chalcogen, and CH··O hydrogen bonds in complexes pairing carbonyl-containing molecules with 1, 2, and 3 molecules of CO2. J. Chem. Phys. 2015, 142, 034307. [Google Scholar] [CrossRef]
- Del Bene, J.; Elguero, J.; Alkorta, I. Complexes of CO2 with the Azoles: Tetrel Bonds, Hydrogen Bonds and Other Secondary Interactions. Molecules 2018, 23, 906. [Google Scholar] [CrossRef] [Green Version]
- Dang Cam-Tu, P.; Thi Ngan, V.; Tien Trung, N. General trends in structure, stability and role of interactions in the complexes of acetone and thioacetone with carbon dioxide and water. Chem. Phys. 2020, 530, 110580. [Google Scholar] [CrossRef]
- Bhattarai, S.; Sutradhar, D.; Chandra, A.K.; Zeegers-Huyskens, T. Nature of the Interaction of Pyridines with OCS. A Theoretical Investigation. Molecules 2020, 25, 416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Exploring N···C tetrel and O···S chalcogen bonds in HN(CH)SX:OCS systems, for X = F, NC, Cl, CN, CCH, and H. Chem. Phys. Lett. 2019, 730, 466–471. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Del Bene, J.E. Complexes of O=C=S with Nitrogen Bases: Chalcogen Bonds, Tetrel Bonds, and Other Secondary Interactions. ChemPhysChem 2018, 19, 1886–1894. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Legon, A. An Ab Initio Investigation of the Geometries and Binding Strengths of Tetrel-, Pnictogen-, and Chalcogen-Bonded Complexes of CO2, N2O, and CS2 with Simple Lewis Bases: Some Generalizations. Molecules 2018, 23, 2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zierkiewicz, W.; Fanfrlík, J.; Michalczyk, M.; Michalska, D.; Hobza, P. S···N chalcogen bonded complexes of carbon disulfide with diazines. Theoretical study. Chem. Phys. 2018, 500, 37–44. [Google Scholar] [CrossRef]
- Legon, A.C. Tetrel, pnictogen and chalcogen bonds identified in the gas phase before they had names: A systematic look at non-covalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 14884–14896. [Google Scholar] [CrossRef]
- Previtali, V.; Sánchez-Sanz, G.; Trujillo, C. Theoretical Investigation of Cyano-Chalcogen Dimers and Their Importance in Molecular Recognition. ChemPhysChem 2019, 20, 3186–3194. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, L.; Xuan, X. The C=N···C-X s-hole interaction acts as a conformational lock. New J. Chem. 2017, 41, 42–46. [Google Scholar] [CrossRef]
- Berrueta Martínez, Y.; Rodríguez Pirani, L.S.; Erben, M.F.; Boese, R.; Reuter, C.G.; Vishnevskiy, Y.V.; Mitzel, N.W.; Della Védova, C.O. Structures of Trichloromethyl Thiocyanate, CCl3SCN, in Gaseous and Crystalline State. ChemPhysChem 2016, 17, 1463–1467. [Google Scholar] [CrossRef]
- Michalczyk, M.; Zierkiewicz, W.; Wysokiński, R.; Scheiner, S. Hexacoordinated Tetrel-Bonded Complexes between TF4 (T=Si, Ge, Sn, Pb) and NCH: Competition between σ- and π-Holes. ChemPhysChem 2019, 20, 959–966. [Google Scholar] [CrossRef] [Green Version]
- Zierkiewicz, W.; Michalczyk, M.; Scheiner, S. Regium bonds between Mn clusters (M = Cu, Ag, Au and n = 2–6) and nucleophiles NH3 and HCN. Phys. Chem. Chem. Phys. 2018, 20, 22498–22509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikherdov, A.S.; Novikov, A.S.; Boyarskiy, V.P.; Kukushkin, V.Y. The halogen bond with isocyano carbon reduces isocyanide odor. Nat. Commun. 2020, 11, 2921. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok van der Werve, A.; van Dijk, Y.R.; Mooibroek, T.J. p-Hole/n to p* interactions with acetonitrile in crystal structures. Chem. Commun. 2018, 54, 10742–10745. [Google Scholar] [CrossRef] [PubMed]
- Nziko, V.d.P.N.; Scheiner, S. Comparison of p-hole tetrel bonding with s-hole halogen bonds in complexes of XCN (X = F, Cl, Br, I) and NH3. Phys. Chem. Chem. Phys. 2016, 18, 3581–3590. [Google Scholar] [CrossRef] [Green Version]
- Bauzá, A.; Frontera, A.; Mooibroek, T.J. 1,1,2,2-Tetracyanocyclopropane (TCCP) as supramolecular synthon. Phys. Chem. Chem. Phys. 2016, 18, 1693–1698. [Google Scholar] [CrossRef] [Green Version]
- Escudero-Adán, E.C.; Bauzá, A.; Frontera, A.; Ballester, P. Nature of Noncovalent Carbon-Bonding Interactions Derived from Experimental Charge-Density Analysis. ChemPhysChem 2015, 16, 2530–2533. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Small Cycloalkane (CN)2C-C(CN)2 Structures Are Highly Directional Non-covalent Carbon-Bond Donors. Chem. Eur. J. 2014, 20, 10245–10248. [Google Scholar] [CrossRef]
- Roeleveld, J.J.; Lekanne Deprez, S.J.; Verhoofstad, A.; Frontera, A.; van der Vlugt, J.I.; Mooibroek, T.J. Engineering Crystals Using sp3-C Centred Tetrel Bonding Interactions. Chem. Eur. J. 2020, 26, 10126–10132. [Google Scholar] [CrossRef]
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Jabłoński, M. Bond paths between distant atoms do not necessarily indicate dominant interactions. J. Comput. Chem. 2018, 39, 2183–2195. [Google Scholar] [CrossRef]
- Wick, C.R.; Clark, T. On bond-critical points in QTAIM and weak interactions. J. Mol. Model. 2018, 24, 142. [Google Scholar] [CrossRef] [PubMed]
- Jabłoński, M. On the Uselessness of Bond Paths Linking Distant Atoms and on the Violation of the Concept of Privileged Exchange Channels. ChemistryOpen 2019, 8, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Jabłoński, M. Counterintuitive bond paths: An intriguing case of the C(NO2)3- ion. Chem. Phys. Lett. 2020, 759, 137946. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Keith, T.A. AIMALL; TK Gristmill Software: Overland Park, KS, USA, 2013. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Ditchfield, R. GIAO studies of magnetic shielding in FHF− and HF. Chem. Phys. Lett. 1976, 40, 53–56. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. Ab initio (GIAO) calculations of absolute nuclear shieldings for representative compounds containing 1(2)H, 6(7)Li, 11B, 13C, 14(15)N, 17O, 19F, 29Si, 31P, 33S, and 35Cl nuclei. Struct. Chem. 1998, 9, 187–202. [Google Scholar] [CrossRef]
- Pankratyev, E.Y.; Tulyabaev, A.R.; Khalilov, L.M. How reliable are GIAO calculations of 1H and 13C NMR chemical shifts? A statistical analysis and empirical corrections at DFT (PBE/3z) level. J. Comput. Chem. 2011, 32, 1993–1997. [Google Scholar] [CrossRef]
- Szalewicz, K.; Jeziorski, B.; Rybak, S. Perturbation theory calculations of intermolecular interaction energies. Int. J. Quantum Chem. 1991, 40, 23–36. [Google Scholar] [CrossRef]
- Szalewicz, K.; Jeziorski, B. Symmetry-adapted perturbation theory of intermolecular interactions. In Molecular Interactions. From van der Waals to Strongly Bound Complexes; Scheiner, S., Ed.; Wiley: New York, NY, USA, 1997; pp. 3–43. [Google Scholar]
- Werner, H.-J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M.; Celani, P.; Korona, T.; Lindh, R.; Mitrushenkov, A.; Rauhut, G.; et al. MOLPRO, Version 2010.1, a Package of ab Initio Programs. Available online: http://www.molpro.net (accessed on 20 August 2020).
Sample Availability: Calculated data are available from the author. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
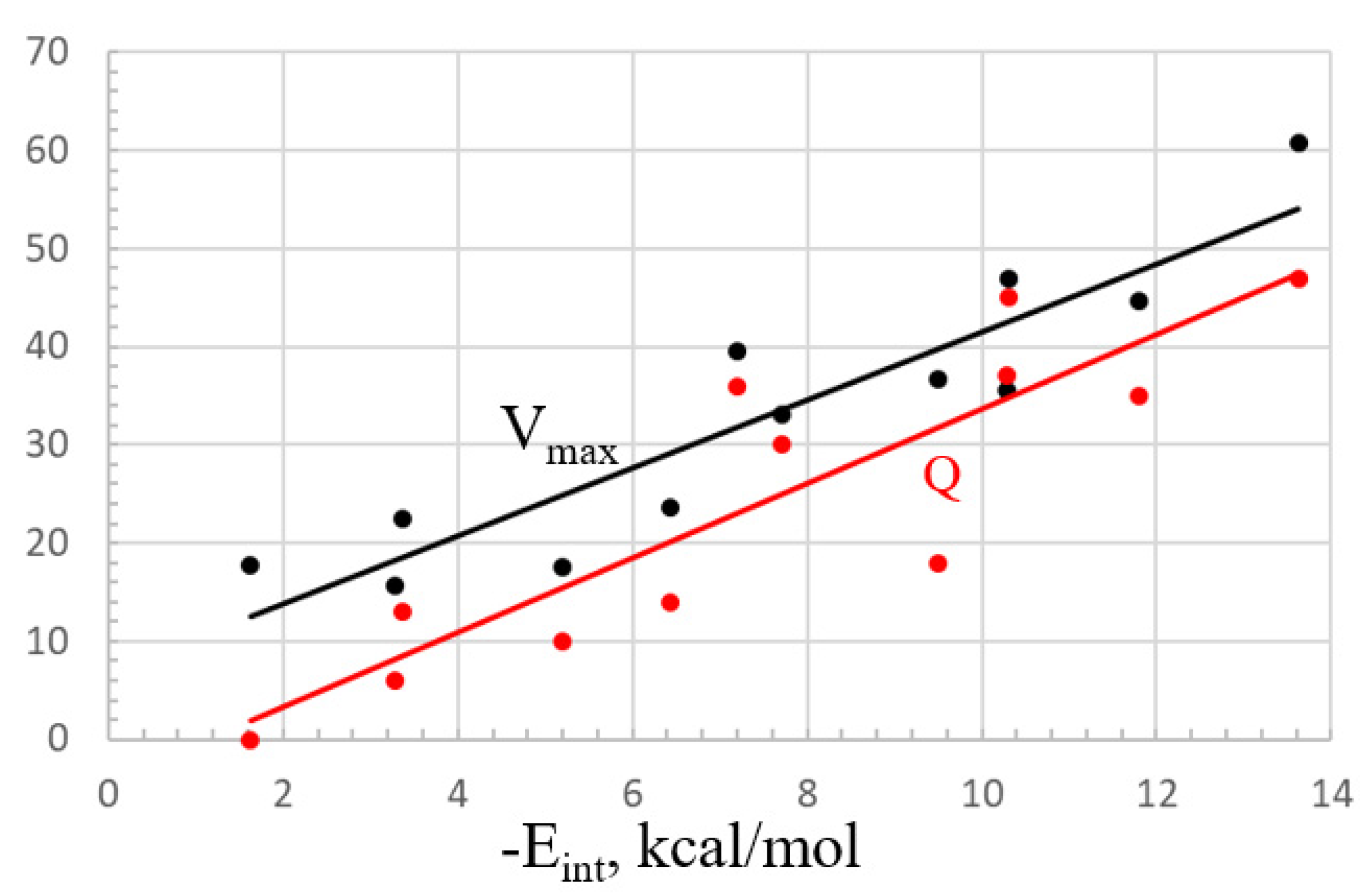
| Lewis Acid | N a | −Eint, kcal/mol | Vmax, kcal/mol | ρBCP × 104, au b | E(2), kcal/mol c | Q d, e |
|---|---|---|---|---|---|---|
| noncyclic | ||||||
| (CN)3C-C(CN)3 side | 4 | 13.63 | 60.7 | 4 × CN 130 | 4 × C≡N 1.4 2 × C-CN 0.7 | 0.047 |
| (CN)3C-C(CN)3 end | 3 | 10.30 | 46.9 | 3 × CN 135 C 131 | 3 × C≡N 2.3 C-C 0.9 | 0.045 |
| (CN)2HC-CH(CN)2 | 4 | 11.80 | 44.7 | 2 × CN 118 C 111 | 4 × C≡N 1.4 2 × CH 0.6 | 0.035 |
| F2HC-CHF2 | 4 | 1.63 | 17.7 | 4 × F 90 | 4 × CF 0.2 2 × CH 0.2 | 0.000 |
| H3C-C(CN)3 end | 3 | 7.71 | 33.1 | 3 × CN 120 | 3 × C≡N 1.8 C-C 0.5 | 0.030 |
| (CN)H2C-CH2(CN) | 2 | CH--N | - | |||
| H3C-CH(CN)2 | 2 | 6.43 | 23.7 | CH··N 112 2 × CN 86 | CH··N 2.7 2 C≡N 0.5 | 0.014 |
| H3C-CH2(CN) | 1 | CH--N | - | |||
| (CN)2C=C(CN)2 | 2 | 7.21 | 39.6 | C 133 | C=C 2.2 2 × C≡N 0.9 | 0.036 |
| (CN)C≡C(CN) | 1 | 3.36 | 22.4 | CN 100 | C≡C 0.7 C≡N 0.9 | 0.013 |
| cyclic | ||||||
| CH2{C(CN)2}2 | 4 | 9.49 | 36.7 | C 115 | 2 × CC 0.5 4 × C≡N 0.5 | 0.018 |
| CH2CH2C(CN)2 | 2 | 5.19 | 17.5 | C 89 | CC 0.4 2 × C≡N 0.5 | 0.010 |
| CH2{CF2}2 | 4 | 3.28 | 15.6 | C 126 | 2 × CC 1.0 | 0.006 |
| CH2CH2{C(CN)2}2 | 4 | 10.28 | 35.6 | 2 × CN 132 | 2 × CC 0.6 4 × C≡N 1.1 | 0.037 |
| Lewis Acid | C-C | C-CN | CN≡N |
|---|---|---|---|
| noncyclic | |||
| (CN)3C-C(CN)3 side | −0.0098 | 0.0002 | 0.0001 |
| (CN)3C-C(CN)3 end | −0.0451 | −0.0005 | 0.0001 |
| (CN)2HC-CH(CN)2 | −0.0075 | −0.0004 | 0.0002 |
| H3C-C(CN)3 end | 0.0011 | −0.0010 | 0.0003 |
| H3C-CH(CN)2 | 0.0005 | 0.0074 | −0.0003 |
| (CN)2C=C(CN)2 | −0.0024 | 0.0000 | −0.0002 |
| (CN)C≡C(CN) | −0.0002 | 0.0007 | 0.0000 |
| cyclic | |||
| CH2{C(CN)2}2 | −0.0123 | −0.0006 | 0.0002 |
| CH2CH2C(CN)2 | −0.0072 | −0.0010 | 0.0003 |
| CH2CH2{C(CN)2}2 | −0.0019 | −0.0024 | 0.0003 |
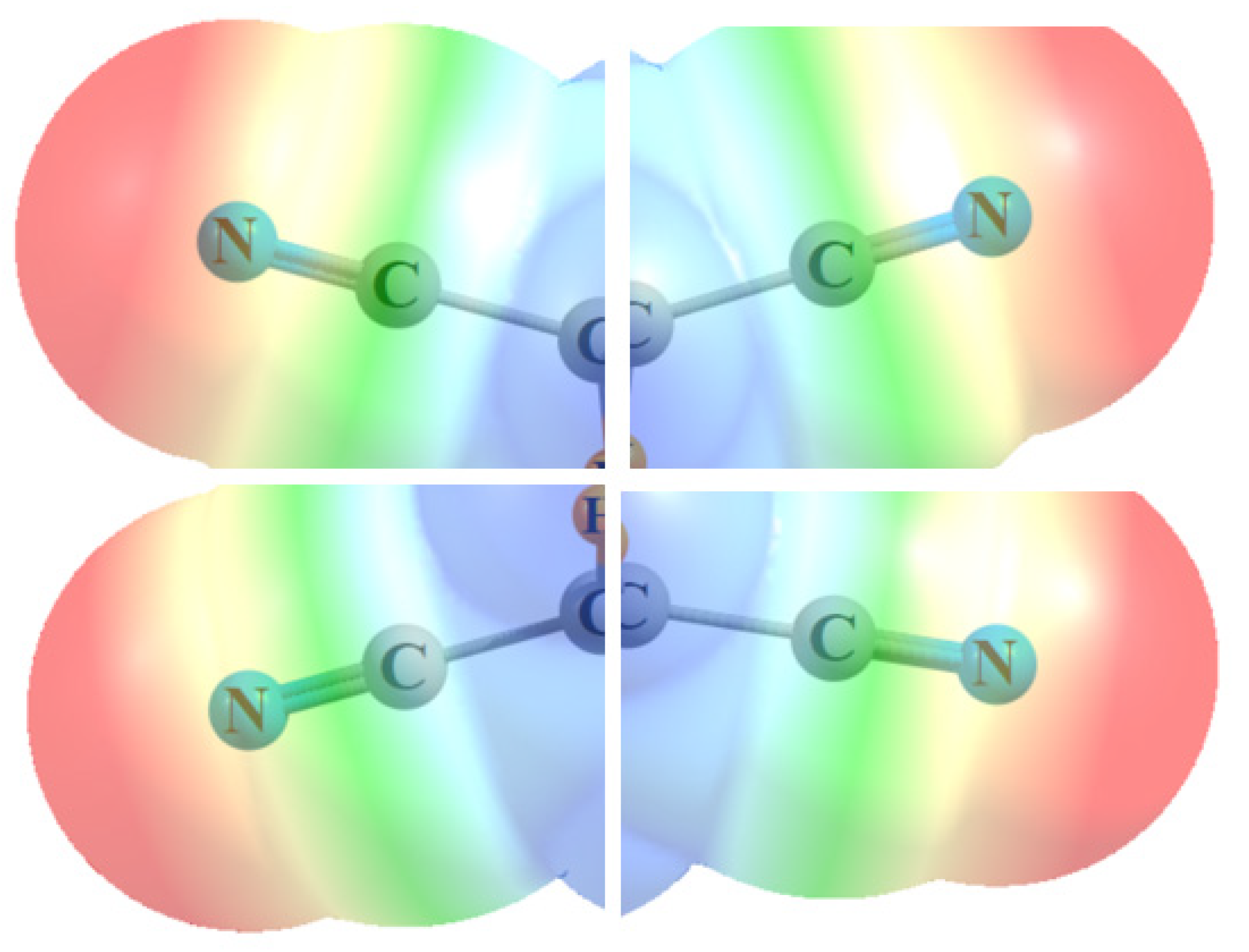
| Lewis Acid | Min | Max | Center |
|---|---|---|---|
| (CN)3C-C(CN)3 side | 0.4 | 9.2 | 4.8 |
| (CN)3C-C(CN)3 end | 3.0 | 5.0 | 4.0 |
| (CN)2HC-CH(CN)2 | −5.2 | 11.0 | 2.9 |
| H3C-C(CN)3 end | −6.7 | 3.5 | −1.6 |
| H3C-CH(CN)2 | −4.3 | 6.5 | 1.1 |
| (CN)2C=C(CN)2 | −0.7 | −2.2 | −1.4 |
| (CN)C≡C(CN) | 7.9 | 2.3 | 5.1 |
| Lewis Acid | C | CN | N |
|---|---|---|---|
| noncyclic | |||
| (CN)3C-C(CN)3 side | −1.72 | −1.97 | 10.66 |
| (CN)3C-C(CN)3 end | −9.67 | −1.83 | 12.19 |
| (CN)2HC-CH(CN)2 | −1.67 | −2.10 | 9.11 |
| H3C-C(CN)3 end | −5.20 | −2.88 | 6.33 |
| H3C-CH(CN)2 | −0.41 | −2.86 | 4.04 |
| (CN)2C=C(CN)2 | −7.42 | −1.82 | 11.05 |
| (CN)C≡C(CN) | −2.63 | −1.99 | 5.90 |
| cyclic | |||
| CH2{C(CN)2}2 | −5.00 | −2.76 | 6.03 |
| CH2CH2C(CN)2 | −5.95 | −2.90 | 5.14 |
| CH2CH2{C(CN)2}2 | −0.81 | −2.82 | 5.66 |
| Lewis Acid | kcal/mol | Percentage | |||||
|---|---|---|---|---|---|---|---|
| n | ES | IND | DISP | ES | IND | DISP | |
| (CN)3C-C(CN)3 side | 4 | 22.37 | 16.17 | 10.37 | 45.7 | 33.1 | 21.2 |
| (CN)2HC-CH(CN)2 | 4 | 18.61 | 12.83 | 9.01 | 46.0 | 31.7 | 22.3 |
| cyclic CH2{C(CN)2}2 | 4 | 13.49 | 8.41 | 6.89 | 46.9 | 29.2 | 23.9 |
| (CN)3C-C(CN)3 end | 3 | 17.65 | 12.69 | 7.54 | 46.6 | 33.5 | 19.9 |
| cyclic CH2CH2C(CN)2 | 2 | 7.40 | 3.86 | 4.71 | 46.3 | 24.2 | 29.5 |
| F2HC-CHF2 | 4 | 4.98 | 5.45 | 4.25 | 33.9 | 37.1 | 29.0 |
| cyclic CH2{CF2}2 | 4 | 7.53 | 7.17 | 4.60 | 39.0 | 37.1 | 23.8 |
| aug-cc-pVDZ | aug-cc-pVTZ | |
|---|---|---|
| Δν(C≡N), cm−1 | ||
| min | −6.7 | −5.2 |
| max | +3.5 | +0.9 |
| center | −1.6 | −2.1 |
| Δσ, ppm | ||
| C | −5.2 | −7.9 (−6.3) a |
| CN | −2.9 | −2.4 (−2.7) a |
| N | 6.3 | 7.1 (7.2) a |
| −Eint, kcal/mol | 7.71 | 7.77 (5.60) b |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheiner, S. Versatility of the Cyano Group in Intermolecular Interactions. Molecules 2020, 25, 4495. https://doi.org/10.3390/molecules25194495
Scheiner S. Versatility of the Cyano Group in Intermolecular Interactions. Molecules. 2020; 25(19):4495. https://doi.org/10.3390/molecules25194495
Chicago/Turabian StyleScheiner, Steve. 2020. "Versatility of the Cyano Group in Intermolecular Interactions" Molecules 25, no. 19: 4495. https://doi.org/10.3390/molecules25194495
APA StyleScheiner, S. (2020). Versatility of the Cyano Group in Intermolecular Interactions. Molecules, 25(19), 4495. https://doi.org/10.3390/molecules25194495