3.2.2. General Procedure: CuAAc Reaction
To a solution of 5′-azidouridine 2 (1.0 eq) in t-BuOH:H2O (2:1) (10 mL/0.5 mmol), the corresponding alkyne (1.0 eq), copper sulfate pentahydrate (10 mol%) and (+)-sodium ascorbate (0.6 eq) were added sequentially. Then the reaction mixture was stirred at 40 °C and monitored by TLC. When the starting material was completely converted, the reaction was stopped and co-evaporated twice with MeOH (20 mL/0.5 mmol). The residue was purified by flash chromatography (H2O/IPrOH/EtOAc 1/6/3 or DCM/MeOH 9/1) and then the residue was co-evaporated several times with MeOH (x × 15 mL) to remove residual water. Next, a second flash chromatography purification (acetone/MeOH 9/1 or EtOAc/MeOH 9/1) was used to give a pure product. All analysis are described according to each alkyne used.
3-(1-(((2R,3S,4R,5R)-5-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)-methyl)-1H-1,2,3-triazol-4-yl)propanoic acid (3a). According to the general procedure, copper (II) sulfate pentahydrate (6 mg, 0.02 mmol, 10 mol%) and sodium ascorbate (28 mg, 0.14 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (65 mg, 0.24 mmol, 1.0 eq) and 4-pentynoic acid (24 mg, 0.24 mmol, 1.0 eq) with tBuOH:H2O (4 mL). The reaction mixture was stirred for 2.5 h at 40 °C. The residue was purified by a flash chromatography (H2O/iPrOH/EtOAc 1:6:3) followed by a second flash chromatography (acetone/MeOH 9/1) to give the desired product as white solid (50 mg, 56%). Rf (H2O/iPrOH/EtOAc 1:6:3) = 0.30. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.37 (1H, s, N-H); 7.81 (1H, s, =CHtriazol); 7.49 (1H, d, J = 7.95 Hz, =CH); 5.74 (1H, d, J = 5.03 Hz, CH); 5.63 (1H, d, J = 7.95 Hz, CH); 4.56–4.68 (2H, m, CH2); 4.12 (1H, m, CH); 4.01 (1H, m, CH); 3.94 (1H, m, CH); 2.82 (2H, t, J = 7.79 Hz, CH2), 2.52 (2H, t, J = 7.72 Hz, CH2). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 174.5; 163.5; 151.1; 146.6; 141.5; 123.2; 102.5, 89.1; 82.2; 72.6; 71.0; 51.5; 34.3; 21.4. ESI-HRMS calculated for [C14H18N5O7]+ m/z 368.1200 and found m/z 368.1201. IR (neat, cm−1): 3201.0br; 2928.7m; 1682.4s; 1462.7w; 1384.5m; 1261.4m; 1100.6m; 1056.1m; 813.1w. [α]D20 = +32.7 (C = 0.017 M, MeOH). LC-MS purity 97.2%, tR = 3.23 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
4-(1-(((2R,3S,4R,5R)-5-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)-methyl)-1H-1,2,3-triazol-4-yl)butanoic acid (3b). According to the general procedure, copper(II) sulfate pentahydrate (6 mg, 0.02 mmol, 10 mol%) and sodium ascorbate (28 mg, 0.14 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (65 mg, 0.24 mmol, 1.0 eq) and 5-hexynoic acid (27 mg, 0.24 mmol, 1.0 eq) with tBuOH:H2O (4 mL). The reaction mixture was stirred for 2.5 h at 40 °C. The residue was purified by a flash chromatography (H2O/iPrOH/EtOAc 1:6:3) followed by a second flash chromatography (acetone/MeOH 9/1) to give the desired product as white solid (55 mg, 60%). Rf (H2O/iPrOH/EtOAc 1:6:3) = 0.41. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.38 (1H, s, N-H); 7.85 (1H, s, =CHtriazol); 7.50 (1H, d, J = 7.80 Hz, =CH); 5.74 (1H, d, J = 5.66 Hz, CH); 5.63 (1H, d, J = 7.80 Hz, =CH); 4.63 (2H, m, CH2); 4.13 (1H, m, CH); 4.03 (1H, t, J = 5.37 Hz, CH); 3.96 (1H, t, J = 4.78 Hz, CH); 2.62 (2H, t, J = 7.53 Hz, CH2), 2.25 (2H, t, J = 7.37 Hz, CH2) 1.80 (2H, q, J = 7.58 Hz, CH2). 13C-NMR (100 MHz DMSO-d6) δ (ppm): 174.8; 163.4; 151.1; 146.9; 141.5; 123.2; 102.5, 89.1; 82.2; 72.5; 71.0; 51.5; 33.7; 24.9; 24.8. ESI-HRMS calculated for [C15H20N5O7]+ m/z 382.1357 and found m/z 382.1356. IR (neat, cm−1): 3139.1br; 2925.2m; 1682.3s; 1462.2w; 1385.6m; 1261.9m; 1223.7m; 1100.5m; 1055.7m; 812.8w. [α]D20 = + 86.0 (C = 0.025 M, MeOH). LC-MS purity 95.5%, tR = 3.87 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
5-(1-(((2R,3S,4R,5R)-5-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)-methyl)-1H-1,2,3-triazol-4-yl)pentanoic acid (3c). According to the general procedure, copper(II) sulfate pentahydrate (6 mg, 0.02 mmol, 10 mol%) and sodium ascorbate (28 mg, 0.14 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (65 mg, 0.24 mmol, 1.0 eq) and 6-heptynoic acid (34 mg, 0.24 mmol, 1.0 eq) with tBuOH:H2O (4 mL). The reaction mixture was stirred for 1 h at 40 °C. The residue was purified by a flash chromatography (H2O/iPrOH/EtOAc 1:6:3) followed by a second flash chromatography (acetone/MeOH 9/1) to give the desired product as white solid (60 mg, 63%). Rf (H2O/iPrOH/EtOAc 1:6:3) = 0.49. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.35 (1H, s, N-H); 7.81 (1H, s, =CHtriazol); 7.50 (1H, d, J = 8.0 Hz, =CH); 5.73 (1H, d, J = 5.29 Hz, CH); 5.62 (1H, d, J = 8.0 Hz, CH); 4.62 (2H, m, CH2); 4.12 (1H, m, CH); 4.04 (1H, t, J = 5.28 Hz, CH); 3.95 (1H, t, J = 4.80 Hz, CH); 2.60 (2H, t, J = 6.80 Hz, CH2); 2.22 (2H, t, J = 7.26 Hz, CH2); 1.48-1.63 (4H, m, 2 CH2). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 174.9, 163.4, 151.1, 147.2, 141.5, 123.1, 102.5, 89.2, 82.2, 72.5, 71.0, 51.5, 33.9, 28.9, 25.1, 24.6. ESI-HRMS calculated for [C16H22N5O7]+ m/z 396.1513 and found m/z 396.1512. IR (neat, cm−1): 3142.7br; 2929.3m; 1682.5s; 1461.3w; 1384.1m; 1261.9m; 1222.6m; 1100.1m; 1052.0m; 811.7w. [α]D20 = + 44.5 (C = 0.015 M, MeOH). LC-MS purity 95.2%, tR = 4.51 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
1-((2R,3R,4S,5R)-3,4-Dihydroxy-5-((4-(hydroxymethyl)-1H-1,2,3-triazol-1-yl)methyl)tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione (3d). According to the general procedure, copper(II) sulfate pentahydrate (6 mg, 0.02 mmol, 10 mol%) and sodium ascorbate (28 mg, 0.14 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (65 mg, 0.24 mmol, 1.0 eq) and propargyl alcohol (14 mg, 0.24 mmol, 1.0 eq) with tBuOH:H2O (4 mL). The reaction mixture was stirred for 40 min at 40 °C. The residue was purified by a flash chromatography (H2O/iPrOH/EtOAc 0.1:6.9:3) followed by a second flash chromatography (acetone/MeOH 9/1) to give the desired product as white solid (54 mg, 68%). Rf (H2O/iPrOH/EtOAc 1:6:3) = 0.64. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.36 (1H, s, N-H); 7.92 (1H, s, =CHtriazol); 7.50 (1H, d, J = 8.05 Hz, =CH); 5.74 (1H, d, J = 5.43 Hz, CH); 5.63 (1H, d, J = 7.97 Hz, =CH); 5.50 (1H, m, O-H); 5.39 (1H, m, O-H); 5.18 (1H, m, O-H); 4.66 (2H, m, CH2); 4.50 (2H, m, CH2); 4.14 (1H, m, CH); 4.03 (1H, m, CH); 3.97 (1H, m, CH). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 163.4, 151.3, 148.5, 141.5, 124.0, 102.6, 89.0, 82.3, 72.5, 71.0, 55.4, 51.6. ESI-HRMS calculated for [C12H16N5O6]+ m/z 326.1095 and found m/z 326.1096. IR (neat, cm−1): 3436.5br; 3044.1w; 2922.1w; 1689.7s; 1459.5w; 1419.4m; 1390.6m; 1282.4m; 1081.1m; 1015.3m; 993.0m; 792.2w. [α]D20 = + 47.6 (C = 0.015 M, MeOH). LC-MS purity 95%, tR = 8.36 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
1-((2R,3R,4S,5R)-3,4-Dihydroxy-5-((4-(2-hydroxyethyl)-1H-1,2,3-triazol-1-yl)methyl)tetrahydrofuran-2-yl)-pyrimidine-2,4(1H,3H)-dione (3e). According to the general procedure, copper(II) sulfate pentahydrate (6 mg, 0.02 mmol, 10 mol%) and sodium ascorbate (28 mg, 0.14 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (65 mg, 0.24 mmol, 1.0 eq) and 3-butyn-1-ol (17 mg, 0.24 mmol, 1.0 eq) with tBuOH:H2O (4 mL). The reaction mixture was stirred for 35 min at 40 °C. The residue was purified by a flash chromatography (acetone/MeOH 98/2) to give the desired product as white solid (47 mg, 57%). Rf (H2O/iPrOH/EtOAc 1:6:3) = 0.61. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.36 (1H, s, N-H); 7.82 (1H, s, =CHtriazol); 7.47 (1H, d, J = 8.04 Hz, =CH); 5.74 (1H, d, J = 5.23 Hz, CH); 5.63 (1H, d, J = 8.04 Hz, =CH); 4.63 (2H, m, CH2); 4.13 (1H, m, CH); 4.00 (1H, m, CH); 3.96 (1H, m, CH); 3.62 (2H, t, J = 6.82 Hz, CH2); 2.76 (2H, t, J = 6.85 Hz, CH2). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 163.4, 151.1, 145.0, 141.5, 123.7, 102.6, 89.0, 82.2, 72.5, 70.9, 60.8, 51.5, 29.6. ESI-HRMS calculated for [C13H18N5O6]+ m/z 340.1251 and found m/z 340.1254. IR (neat, cm−1): 3424.1br; 2921.2w; 1691.5s; 1461.8w; 1422.2m; 1382.2m; 1279.1m; 1233.6m; 1044.1m; 1018.1m; 995.7m; 809.7w. [α]D20 = + 29.2 (C = 0.03 M, MeOH). LC-MS purity 94.2%, tR = 2.58 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
1-((2R,3R,4S,5R)-3,4-Dihydroxy-5-((4-(3-hydroxypropyl)-1H-1,2,3-triazol-1-yl)methyl)tetrahydrofuran-2-yl)pyrimidine-2,4(1H,3H)-dione (3f). According to the general procedure, copper(II) sulfate pentahydrate (6 mg, 0.02 mmol, 10 mol%) and sodium ascorbate (30 mg, 0.16 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (70 mg, 0.26 mmol, 1.0 eq) and 4-pentyn-1-ol (22 mg, 0.26 mmol, 1.0 eq) with tBuOH:H2O (4 mL). The reaction mixture was stirred for 45 min at 40 °C. The residue was purified by a flash chromatography (H2O/iPrOH/EtOAc 0.1:6.9:3) followed by a second flash chromatography (acetone/MeOH 95/5) to give the desired product as white solid (57 mg, 62%). Rf (H2O/iPrOH/EtOAc 1:6:3) = 0.63. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.36 (1H, s, N-H); 7.81 (1H, s, =CHtriazol); 7.48 (1H, d, J = 8.02 Hz, =CH); 5.74 (1H, d, J = 5.55 Hz, CH); 5.62 (1H, d, J = 8.02 Hz, =CH); 5.49 (1H, m, O-H); 5.36 (1H, m, O-H); 4.62 (2H, m, CH2); 4.47 (1H, m, O-H); 4.13 (1H, m, CH); 4.03 (1H, m, CH); 3.96 (1H, m, CH); 3.42 (2H, m, CH2); 2.63 (2H, t, J = 7.57 Hz, CH2); 1.72 (2H, q, J = 6.77 Hz, CH2). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 163.5, 151.1, 147.3, 141.5, 123.1, 102.5, 89.1, 82.2, 72.5, 71.0, 60.5, 51.5, 32.7, 22.1. ESI-HRMS calculated for [C14H20N5O6]+ m/z 354.1408 and found m/z 354.1408. IR (neat, cm−1): 3367.9br; 2945.1m; 1673.9s; 1462.5m; 1422.9m; 1384.7m; 1263.2m; 1224.0m; 1100.2m; 1051.8m; 811.9w. [α]D20 = + 58.0 (C = 0.026 M, MeOH). LC-MS purity 94.6%, tR = 3.24 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
1-((2R,3R,4S,5R)-5-((4-(Aminomethyl)-1H-1,2,3-triazol-1-yl)methyl)-3,4-dihydroxytetrahydrofuran-2-yl)-pyrimidine-2,4(1H,3H)-dione (3g). According to the general procedure, copper(II) sulfate pentahydrate (18.5 mg, 0.07 mmol, 10 mol%) and sodium ascorbate (88.3 mg, 0.45 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (200 mg, 0.74 mmol, 1.0 eq) and propargyl amine (61 mg, 1.10 mmol, 1.5 eq) with tBuOH:H2O (10 mL). The reaction mixture was stirred for 1 h at 40 °C. The residue was purified by a flash chromatography (CHCl3:MeOH:AcOH 1:1:1) followed by a second flash chromatography (H2O:iPrOH:EtOAc:AcOH 1:6:2:1) to give the desired product as white solid (110 mg, 46%). Rf (CHCl3:MeOH:AcOH 1:1:1) = 0.28. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.88 (1H, s, =CHtriazol); 7.52 (1H, d, J = 7.91 Hz, =CH); 5.74 (1H, d, J = 4.90 Hz, CH); 5.63 (1H, d, J = 7.91 Hz, =CH); 4.64 (2H, m, CH); 4.12 (1H, m, CH); 4.01 (1H, m, CH); 3.94 (1H, m, CH); 3.77 (2H, m, CH2). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 163.5, 151.1, 149.1, 141.6, 123.3, 102.5, 89.3, 82.3, 72.7, 71.1, 51.7, 37.2. ESI-HRMS calculated for [C12H17N6O5]+ m/z 325.1254 and found m/z 325.1254. IR (neat, cm−1): 3150.4br; 1673.9s; 1462.5m; 1385.5m; 1263.1m; 1099.7m; 1050.8m; 812.3w. [α]D20 = + 96.0 (C = 0.04 M, MeOH). LC-MS purity 95.1%, tR = 0.72 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
1-(((2R,3S,4R,5R)-5-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazole-4-carboxamide (3h). According to the general procedure, copper(II) sulfate pentahydrate (18.5 mg, 0.07 mmol, 10 mol%) and sodium ascorbate (88.3 mg, 0.45 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (200 mg, 0.74 mmol, 1.0 eq) and propiolamide (51 mg, 0.74 mmol, 1.0 eq) with tBuOH:H2O (10 mL). The reaction mixture was stirred for 19 h at 40 °C. The residue was purified by a flash chromatography (acetone/MeOH 9/1) followed by a second flash chromatography (EtOAc/MeOH 9/1) to give the desired product as white solid (0.17 g, 68%). Rf (EtOAc:MeOH 9:1) = 0.21. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.37 (1H, s, N-H3); 8.47 (1H, s, =CHtriazol); 7.85 (1H, s, N-H); 7.56 (1H, d, J = 8.16 Hz, =CH); 7.46 (1H, s, N-H); 5.74 (1H, d, J = 5.55 Hz, CH); 5.63 (1H, d, J = 8.16 Hz, CH); 5.52 (1H, d, J = 5.62 Hz, O-H); 5.40 (1H, d, J = 5.34 Hz, O-H); 4.74 (2H, m, CH2); 4.18 (1H, m, CH); 4.11 (1H, m, CH); 3.98 (1H, m, CH); 3.77 (2H, m, CH2). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 163.0, 161.5, 151.6, 142.9, 141.2, 127.2, 102.1, 88.9, 81.6, 72.1, 70.6, 51.5. ESI-HRMS calculated for [C12H15N6O6]+ m/z 339.1047 and found m/z 339.1048. IR (neat, cm−1): 3192.8br; 1690.5s; 1652.1s; 1466.2w; 1393.9w; 1266.7w; 1100.0w; 1045.7w; 819.8w; 779.5w. [α]D20 = + 37.9 (C = 0.04 M, MeOH). LC-MS purity 95.2%, tR = 1.94 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
N-((1-(((2R,3S,4R,5R)-5-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)hexanamide (3i). According to the general procedure, copper(II) sulfate pentahydrate (6 mg, 0.02 mmol, 10 mol%) and sodium ascorbate (28 mg, 0.14 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (65 mg, 0.24 mmol, 1.0 eq) and N-(prop-2-yn-1-yl)hexanamide (37 mg, 0.24 mmol, 1.0 eq) with tBuOH:H2O (3 mL). The reaction mixture was stirred for 2 h at 40 °C. The residue was purified by a flash chromatography (H2O/iPrOH/EtOAc 0.1:6.9:3) to give the desired product as white solid (99 mg, 99%). Rf (H2O/iPrOH/EtOAc 1:6:3) = 0.78. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.38 (1H, brs, N-H); 8.27 (1H, t, J = 5.68 Hz, N-H); 7.85 (1H, s, =CHtriazol); 7.51 (1H, d, J = 8.09 Hz, =CH); 5.75 (1H, d, J = 5.24 Hz, CH); 5.65 (1H, d, = 8.09 Hz, =CH); 5.51 (1H, d, J = 5.70 Hz, O-H); 5.39 (1H, d, J = 5.22 Hz, O-H); 4.66 (2H, m, CH2); 4.27 (2H, d, J = 5.70 Hz, CH2); 4.13 (1H, m, CH); 4.05 (1H, m, CH); 3.96 (1H, m, CH); 2.07 (2H, t, J = 7.41 Hz, CH2); 1.49 (2H, m, CH2), 1.16-1.30 (4H, m, 2 CH2); 0.84 (3H, t, J = 6.78 Hz, CH3). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 172.6, 163.4, 151.1, 145.6, 141.5, 124.0, 102.6, 89.0, 82.2, 72.5, 71.0, 51.6, 35.6, 34.5, 29.1, 26.0, 22.3, 14.3. ESI-HRMS calculated for [C18H27N6O6]+ m/z 423.1986 and found m/z 423.1985. IR (neat, cm−1): 3295.5br; 2930.2m; 2871.3m; 1682.4s; 1538.6m; 1462.0m; 1424.0m; 1381.3m; 1258.8m; 1101.1m; 1051.7m; 813.0w. [α]D20 = + 42.1 (C = 0.02 M, MeOH).LC-MS purity 95%, tR = 5.71 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
N-((1-(((2R,3S,4R,5R)-5-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)octanamide (3j). According to the general procedure, copper(II) sulfate pentahydrate (6 mg, 0.02 mmol, 10 mol%) and sodium ascorbate (28 mg, 0.14 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (65 mg, 0.24 mmol, 1.0 eq) and N-(prop-2-yn-1-yl)octanamide (44 mg, 0.24 mmol, 1.0 eq) with tBuOH:H2O (3 mL). The reaction mixture was stirred for 2 h at 40 °C. The residue was purified by a flash chromatography (H2O/iPrOH/EtOAc 0.1:6.9:3) to give the desired product as white solid (85 mg, 77%). Rf (H2O/iPrOH/EtOAc 1:6:3) = 0.81. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.38 (1H, brs, N-H); 8.27 (1H, t, J = 5.56 Hz, N-H); 7.85 (1H, s, =CHtriazol); 7.51 (1H, d, J = 8.06 Hz, =CH); 5.75 (1H, d, J = 5.24 Hz, CH); 5.65 (1H, d, J = 8.06 Hz, =CH); 5.51 (1H, d, J = 5.67 Hz, O-H); 5.38 (1H, d, J = 5.24 Hz, O-H); 4.66 (2H, m, CH2); 4.27 (2H, d, J = 5.46 Hz, CH2); 4.13 (1H, m, CH); 4.05 (1H, m, CH); 3.96 (1H, m, CH); 2.07 (2H, t, J = 7.34 Hz, CH2); 1.48 (2H, m, CH2), 1.23 (8H, m, 4 CH2); 0.85 (3H, t, J = 6.31 Hz, CH3). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 172.6, 163.4, 151.1, 145.6, 141.4, 124.0, 102.6, 89.1, 82.2, 72.6, 71.0, 51.6, 35.7, 34.5, 31.6, 29.1, 28.9, 25.7, 22.5, 14.4. ESI-HRMS calculated for [C20H31N6O6]+ m/z 451.2299 and found m/z 451.2299. IR (neat, cm−1): 3293.4br; 2927.2m; 2855.8m; 1682.3s; 1538.7m; 1462.1m; 1381.7m; 1263.1m; 1101.0m; 1052.4m; 812.0w. [α]D20 = + 61.4 (C = 0.02 M, MeOH). LC-MS purity 98.3%, tR = 7.63 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
Dimethyl 5-(1-(((2R,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)isophthalate (3k). According to the general procedure, copper(II) sulfate pentahydrate (29 mg, 0.11 mmol, 10 mol%) and sodium ascorbate (136 mg, 0.69 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxy-uridine 2 (310 mg, 1.14 mmol, 1.0 eq) and dimethyl-5-ethynylisophathalate (250 mg, 1.14 mmol, 1.0 eq) with tBuOH:H2O (10 mL). The reaction mixture was stirred for 5 h 30 min at 40 °C. The residue was purified by a flash chromatography (DCM/MeOH 93/7 to 90/10) followed by a second flash chromatography (acetone/MeOH 99/1) to give the desired product as white solid (0.40 g, 71%). Rf (DCM/MeOH 9:1) = 0.45. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.36 (1H, s, N-H); 8.91 (1H, s, =CHtriazol); 8.63 (2H, s, Ar-H); 8.39 (1H, m, Ar-H); 7.56 (1H, d, J = 8.09 Hz, =CH); 5.77 (1H, d, J = 5.06 Hz, CH); 5.59 (1H, d, J = 8.09 Hz, =CH); 5.52 (1H, d, J = 5.58 Hz, O-H); 5.41 (1H, d, J = 5.46 Hz, O-H); 4.76 (2H, m, CH2); 4.25 (1H, m, CH); 4.11 (1H, m, CH); 4.03 (1H, m, CH); 3.92 (6H, s, 2 CH3). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 165.2, 163.0, 150.6, 144.6, 141.2, 132.0, 131.0, 129.7, 128.6, 123.4, 102.1, 89.0, 82.5, 72.1, 70.5, 52.6, 51.6. ESI-HRMS calculated for [C21H22N5O9]+ m/z 488.1412 and found m/z 488.1411. IR (neat, cm−1): 3136.9br; 2955.2m; 1704.9s; 1467.1m; 1432.7m; 1302.5m; 1242.4s; 1204.8m; 1093.5m; 1057.2m; 996.4w; 819.5w; 753.7w. [α]D20 = + 23.5 (C = 0.03 M, MeOH). LC-MS purity 98.4%, tR = 7.21 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
1-((2R,3R,4S,5R)-5-((4-(3,5-Dimethoxyphenyl)-1H-1,2,3-triazol-1-yl)methyl)-3,4-dihydroxytetrahydro-furan-2-yl)pyrimidine-2,4(1H,3H)-dione (3l). According to the general procedure, copper(II) sulfate pentahydrate (18.5 mg, 0.07 mmol, 10 mol%) and sodium ascorbate (88 mg, 0.46 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (200 mg, 0.74 mmol, 1.0 eq) and 3,5-dimethoxyphenyl-1-ethynyl (120 mg, 0.74 mmol, 1.0 eq) with tBuOH:H2O (10 mL). The reaction mixture was stirred for 17 h at 40 °C. The residue was purified by a flash chromatography (DCM/MeOH 9/1) followed by a second flash chromatography (EtOAc/MeOH 95/5) to give the desired product as white solid (0.25 g, 78%). Rf (DCM/MeOH 9:1) = 0.33. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.36 (1H, d, J = 2.07 Hz, N-H); 8.60 (1H, s, =CHtriazol); 7.53 (1H, d, J = 8.13 Hz, =CH); 7.02 (2H, d, J = 2.24 Hz, 2 Ar-H); 6.47 (1H, t, J = 2.24 Hz, Ar-H); 5.76 (1H, d, J = 5.32 Hz, =CH); 5.58 (1H, dd, J = 2.07 Hz, J = 8.13 Hz, =CH); 5.51 (1H, d, J = 5.56 Hz, O-H); 5.40 (1H, d, J = 5.45 Hz, O-H); 4.73 (2H, m, CH2); 4.21 (1H, m, CH); 4.09 (1H, m, CH); 4.02 (1H, m, CH); 3.79 (6H, s, 2 CH3). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 163.0, 160.9, 150.6, 146.3, 141.2, 132.5, 122.5, 103.1, 102.0, 100.0, 88.9, 81.6, 72.1, 70.5, 55.3, 51.4. ESI-HRMS calculated for [C19H22N5O7]+ m/z 432.1513 and found m/z 432.1512. IR (neat, cm−1): 3233.9br; 2943.7w; 2840.0w; 1682.2s; 1596.2s; 1556.9w; 1456.1m; 1423.2m; 1370.5m; 1265.4m; 1203.5m; 1153.1m; 1100.2m; 1043.8m; 925.4w; 809.6w; 685.8w. [α]D20 = + 74.7 (C = 0.04 M, MeOH). LC-MS purity 99.5%, tR = 6.94 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
1-((2R,3R,4S,5R)-3,4-Dihydroxy-5-((4-(4-nitrophenyl)-1H-1,2,3-triazol-1-yl)methyl)tetrahydrofuran-2-yl)-pyrimidine-2,4(1H,3H)-dione (3m). According to the general procedure, copper(II) sulfate pentahydrate (18.5 mg, 0.07 mmol, 10 mol%) and sodium ascorbate (88 mg, 0.46 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (200 mg, 0.74 mmol, 1.0 eq) and 1-ethynyl-4-nitrophenyl (110 mg, 0.74 mmol, 1.0 eq) with tBuOH:H2O (10 mL). The reaction mixture was stirred for 17 h at 40 °C. The residue was purified by a flash chromatography (DCM/MeOH 92/8 to 90/10) followed by a second flash chromatography (EtOAc/MeOH 9/1) to give the desired product as white solid (0.15 g, 48%). Rf (DCM/MeOH 9:1) = 0.26. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 10.46 (1H, s, N-H); 9.11 (1H, s, =CHtriazol); 8.77 (2H, d, J = 8.46 Hz, 2 Ar-H); 8.63 (2H, d, J = 8.46 Hz, 2 Ar-H); 7.88 (1H, d, J = 8.08 Hz, =CH); 6.27 (1H, m, CH); 5.97 (1H, d, J = 8.08 Hz, =CH); 5.35 (2H, m, CH2); 5.23 (1H, d, J = 4.28 Hz, O-H); 5.08 (1H, d, J = 5.73 Hz, O-H); 4.71-4.85 (3H, m, 3 CH). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 162.9, 150.8, 147.3, 145.6, 141.6, 138.1, 126.6, 124.6, 124.2, 102.6, 92.1, 82.1, 73.4, 71.4, 51.9. ESI-HRMS calculated for [C17H17N6O7]+ m/z 417.1153 and found m/z 417.1153. IR (neat, cm−1): 3104.2br; 2938.7w; 1674.4s; 1606.1m; 1514.7m; 1461.2m; 1378.7m; 1335.0s; 1238.2m; 1107.0m; 1043.4m; 853.4w; 813.0w; 756.0w. [α]D20 = + 57.7 (C = 0.02 M, MeOH). LC-MS purity 99.5%, tR = 6.49 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
1-((2R,3R,4S,5R)-3,4-Dihydroxy-5-((4-(pyridin-2-yl)-1H-1,2,3-triazol-1-yl)methyl)tetrahydrofuran-2-yl)-pyrimidine-2,4(1H,3H)-dione (3n). According to the general procedure, copper(II) sulfate pentahydrate (18.5 mg, 0.07 mmol, 10 mol%) and sodium ascorbate (88 mg, 0.46 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (200 mg, 0.74 mmol, 1.0 eq) and 2-ethynylpyridine (0.11 mL, 1.11 mmol, 1.5 eq) with tBuOH:H2O (10 mL). The reaction mixture was stirred for 19 h at 40 °C. The residue was purified by a flash chromatography (DCM/MeOH 9/1) followed by a second flash chromatography (EtOAc/MeOH 95/5 to 90/10) to give the desired product as white solid (0.14 g, 50%). Rf (DCM/MeOH 9:1) = 0.15. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.36 (1H, d, J = 2.05 Hz, N-H); 8.58 (1H, d, J = 4.71 Hz, Ar-H); 8.56 (1H, s, =CHtriazol); 8.03 (1H, d, J = 7.77 Hz, Ar-H); 7.89 (1H, td, J = 7.67 Hz, J = 1.75 Hz, Ar-H); 7.60 (1H, d, J = 8.06 Hz, =CH); 7.34 (1H, dd, J = 1.15 Hz, J = 4.86 Hz, Ar-H); 5.76 (1H, d, J = 5.57 Hz, CH); 5.60 (1H, dd, J = 2.05 Hz, J = 8.06 Hz, =CH); 5.51 (1H, d, J = 5.67 Hz, O-H); 5.40 (1H, d, J = 5.30 Hz, O-H); 4.77 (2H, m, CH2); 4.23 (1H, q, J = 4.39 Hz, CH); 4.11 (1H, q, J = 5.52 Hz, CH); 4.02 (1H, q, J = 5.06 Hz, CH). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 162.9, 150.6, 149.8, 149.6, 147.3, 141.1, 137.2, 124.0, 123.0, 119.4, 102.0, 88.7, 81.7, 72.0, 70.6, 51.4. ESI-HRMS calculated for [C16H17N6O5]+ m/z 373.1254 and found m/z 373.1254. IR (neat, cm−1): 3527.6w; 3433.8m; 3139.4w; 3067.3w; 2929.8w; 1764.0w; 1710.2s; 1686.9s; 1613.8s; 1555.9w; 1488.4w; 1464.3m; 1423.5w; 1379.6w; 1350.0w; 1325.4w; 1279.6w; 1250.8m; 1211.3w; 1191.3w; 1104.3w; 1062.1m; 1045.7w; 1027.9w; 943.4w; 898.5w; 859.7w; 814.5w; 788.3w; 717.7w; 628.5w. [α]D20 = + 46.4 (C = 0.04 M, MeOH). LC-MS purity 95.8%, tR = 4.33 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
1-((2R,3R,4S,5R)-3,4-Dihydroxy-5-((4-(thiophen-2-yl)-1H-1,2,3-triazol-1-yl)methyl)tetrahydrofuran-2-yl)-pyrimidine-2,4(1H,3H)-dione (3o). According to the general procedure, copper(II) sulfate pentahydrate (18.5 mg, 0.07 mmol, 10 mol%) and sodium ascorbate (88 mg, 0.46 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxy-uridine 2 (200 mg, 0.74 mmol, 1.0 eq) and 2-ethynylthiophene (0.18 mL, 1.86 mmol, 2.5 eq) with tBuOH:H2O (10 mL). The reaction mixture was stirred for 19 h at 40 °C. The residue was purified by a flash chromatography (DCM/MeOH 9/1) followed by a second flash chromatography (EtOAc/MeOH 95/5) to give the desired product as white solid (0.20 g, 71%). Rf (DCM/MeOH 9:1) = 0.26. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.36 (1H, d, J = 2.20 Hz, N-H); 8.46 (1H, s, =CHtriazol); 7.54 (1H, d, J = 8.15 Hz, =CH); 7.53 (1H, dd, J = 1.17 Hz, J = 5.15 Hz, Ar-H); 7.43 (1H, dd, J = 3.55 Hz, J = 1.18 Hz, Ar-H); 7.12 (1H, dd, J = 3.56 Hz, J = 5.05 Hz, Ar-H); 5.76 (1H, d, J = 5.33 Hz, CH); 5.59 (1H, dd, J = 2.20 Hz, J = 8.15 Hz, =CH); 5.51 (1H, d, J = 5.65 Hz, O-H); 5.39 (1H, d, J = 5.46 Hz, O-H); 4.72 (2H, m, CH2); 4.20 (1H, q, J = 4.42 Hz, CH); 4.10 (1H, q, J = 5.45 Hz, CH); 4.00 (1H, q, J = 5.10 Hz, CH). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 162.9, 150.6, 141.8, 141.1, 132.8, 127.9, 125.4, 124.2, 121.4, 102.0, 88.9, 81.6, 72.1, 70.5, 51.4. ESI-HRMS calculated for [C15H16N5O5S]+ m/z 378.0866 and found m/z 378.0866. IR (neat, cm−1): 3280.9br; 3108.2m; 2951.7w; 2900.9w; 1675.3s; 1634.1m; 1461.2m; 1418.2w; 1391.3w; 1356.5w; 1275.5m; 1226.6w; 1210.1w; 1138.1w; 1102.6m; 1064.0m; 1040.9w; 996.7w; 935.7w; 823.2w; 685.7w. [α]D20 = −24.3 (C = 0.04 M, MeOH). LC-MS purity 99.5%, tR = 5.84 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
(2R,3R,4S,5R,6R)-2-(Acetoxymethyl)-6-((1-(((2R,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)tetrahydro-2H-pyran-3,4,5-triyl triacetate (3p). According to the general procedure, copper(II) sulfate pentahydrate (28 mg, 0.11 mmol, 10 mol%) and sodium ascorbate (132 mg, 0.67 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (300 mg, 1.11 mmol, 1.0 eq) and 2-propynyl-tetra-O-acetyl-β-D-glucopyronoside (430 mg, 1.11 mmol, 1.0 eq) with tBuOH:H2O (15 mL). The reaction mixture was stirred for 6 h at 25 °C. The residue was purified by a flash chromatography (DCM/MeOH 93/7 to 90/10) followed by a second flash chromatography (acetone/MeOH 99/1) to give the desired product as white solid (0.44 g, 60%). Rf (DCM/MeOH 9:1) = 0.41. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.35 (1H, s, N-H); 8.03 (1H, s, =CHtriazol); 7.54 (1H, d, J = 8.13 Hz, =CH); 5.74 (1H, d, J = 5.32 Hz, CH); 5.64 (1H, d, J = 8.08 Hz, =CH); 5.50 (1H, d, J = 5.47 Hz, O-H); 5.38 (1H, d, J = 5.28 Hz, O-H); 5.25 (1H, t, J = 9.49 Hz, CH); 4.87-4.95 (2H, m, 2 CH); 4.60-4.83 (5H, m, 2 CH2, 1 CH); 3.95-4.23 (6H, m, 1 CH2, 4 CH); 2.03 (3H, s, CH3); 1.98 (3H, s, CH3); 1.92 (3H, s, CH3); 1.90 (3H, s, CH3). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 170.1, 169.5, 169.3, 169.0, 163.0, 150.6, 143.1, 141.2, 124.9, 102.1, 98.7, 88.9, 81.7, 72.0, 70.9, 70.7, 70.6, 68.1, 61.9, 61.7, 51.3, 20.5, 20.4, 20.3. ESI-HRMS calculated for [C26H34N5O15]+ m/z 656.2045 and found m/z 656.2044. IR (neat, cm−1): 3271.1br; 2954.3w; 1744.8s; 1688.5s; 1458.2w; 1429.5w; 1368.8m; 1217.4s; 1168.6w; 1034.9s; 905.3w; 813.4w. [α]D20 = + 4.5 (C = 0.02 M, MeOH). LC-MS purity 95.3%, tR = 5.96 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
(5-(1-(((2R,3S,4R,5R)-5-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)-methyl)-1H-1,2,3-triazol-4-yl)pentanoyl)-L-alanine (4). According to the general procedure, copper(II) sulfate pentahydrate (23 mg, 0.09 mmol, 10 mol%) and sodium ascorbate (112 mg, 0.56 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (254 mg, 0.94 mmol, 1.0 eq) and 6-heptynoic acid (132 mg, 0.94 mmol, 1.0 eq) with tBuOH:H2O (8 mL). The reaction mixture was stirred for 1 h at 40 °C. The residue was purified by a flash chromatography (H2O/iPrOH/EtOAc 1:6:3) to give the desired product as white solid. The pure compound was dried under reduced pressure to remove residual water. Then, in a flame-dried round-bottom flask, the corresponding carboxylic acid derivative 3c (373 mg, 0.94 mmol, 1.0 eq) was stirred with anhydrous DMF (10 mL) under argon atmosphere at 0 °C. Then, BOP reagent (417 mg, 0.94 mmol, 1.0 eq), HOBt (127 mg, 0.94 mmol, 1.0 eq), DIPEA (0.43 mL, 2.46 mmol, 3.0 eq) and molecular sieves 4 Å (one spatula) were successively added and stirred for 10 min at 0 °C. Thus, L-alanine methyl ester hydrochloride (107 mg, 1.04 eq, 1.1 eq) was added to the reaction mixture and stirred at 25 °C for 16 h. The reaction was stopped and concentrated to dryness in vacuo with several co-evaporation with n-heptane (3 × 15 mL) to remove residual DMF. The residue was purified by flash chromatography (H2O/IPrOH/EA 0.1/6.9/3) followed by a second flash chromatography (acetone/MeOH 96/4 to 90/10) to give a white solid. Then, the desired compound was directly deprotected without further characterization. This peptidic compound (40 mg, 0.08 mmol, 1.0 eq) was stirred with THF:H2O 3:1 and lithium hydroxide monohydrate (7 mg, 0.17 mmol, 2.0 eq) was added to the reaction which was stirred for 18 h at 25 °C. Then, the reaction was quenched with 1M HCl aqueous solution to pH 2. The reaction mixture was concentrated to dryness in vacuo. Then, the residue was purified by a flash chromatography SiO2-C18 (H2O 100%) to give the desired product as white solid (27 mg, 29% in three steps). Rf (SiO2-C18-H2O 100%) = 0.18. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.34 (1H, d, J = 1.70 Hz, N-H); 8.14 (1H, d, J = 7.25 Hz, N-H); 7.83 (1H, s, =CHtriazol); 7.55 (1H, d, J = 8.08 Hz, =CH); 5.73 (1H, d, J = 5.36 Hz, CH); 5.60 (1H, dd, J = 1.70 Hz, J = 8.08 Hz, =CH); 4.64 (2H, m, CH2); 4.10-4.22 (2H, m, 2 CH); 4.06 (1H, t, J = 5.38 Hz, CH); 3.96 (1H, t, J = 4.92 Hz, CH); 2.59 (2H, t, J = 6.84 Hz, CH2); 2.12 (2H, t, J = 7.06 Hz, CH2); 1.45-1.61 (4H, m, 2 CH2); 1.23 (3H, d, J = 7.31 Hz, CH3). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 174.2, 171.9, 163.0, 150.6, 146.8, 141.2, 122.6, 102.0, 88.7, 81.8, 72.1, 70.5, 51.1, 47.4, 34.7, 28.5, 24.8, 24.7, 17.2. ESI-HRMS calculated for [C19H27N6O8]+ m/z 467.1884 and found m/z 467.1882. IR (neat, cm−1): 3366.2br; 3079.2w; 2942.8w; 2867.3w; 1686.6m; 1551.7w; 1460.3w; 1415.8w; 1384.4w; 1261.4w; 1217.1w; 1129.9w; 1102.5w; 1050.0s; 1022.8s; 998.9s; 825.5m; 764.6m; 630.7w. [α]D20 = + 20.6 (C = 0.01 M, MeOH). LC-MS purity 94.9%, tR = 4.37 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
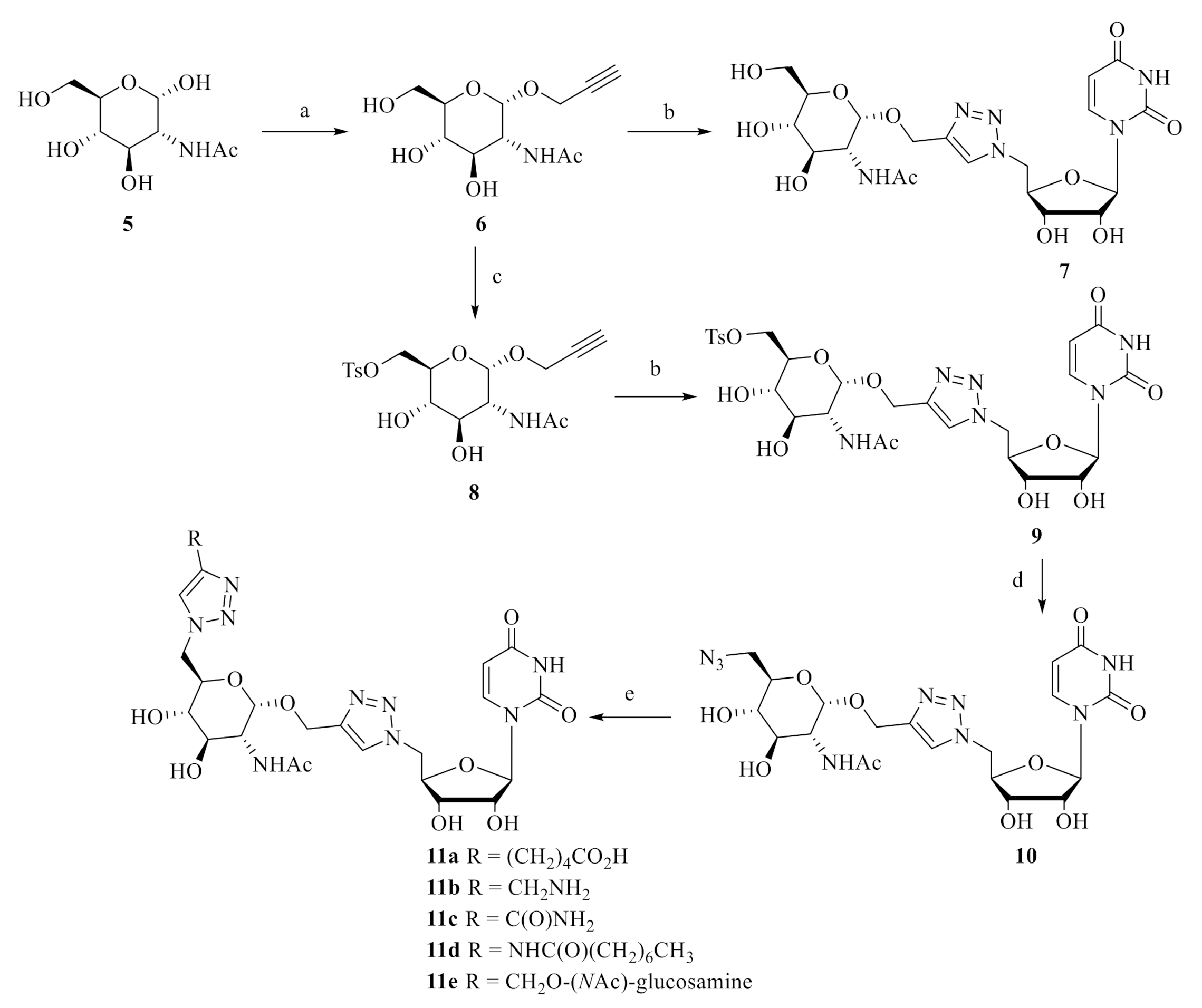
N-((2S,3R,4R,5S,6R)-4,5-Dihydroxy-6-(hydroxymethyl)-2-(prop-2-yn-1-yloxy)tetrahydro-2H-pyran-3-yl)acetamide (6, CAS: 1000593-95-2). To a suspension of commercially N-acetyl-D-glucosamine 5 (1.0 g, 4.52 mmol, 1.0 eq) in propargylic alcohol (10 mL), 4 Å molecular sieves (one spatula) was added under argon atmosphere. At 0 °C, trimetylsilyl triflate (0.82 mL, 4.52 mmol, 1.0 eq) was added drop by drop and the mixture was stirred at 0 °C for an additional 5 min (until the fully solubilization of starting material). Then, the solution was stirred at 80 °C for 3 h. Thus, the reaction was cooled at 0 °C and quenched with addition of solid NaHCO3 (3 g). The suspension was filtered through a Celite pad and this was washed with methanol several times. The filtrate was concentrated to dryness in vacuo and the resulting residue was directly purified by a flash chromatography (DCM/MeOH 9/1) to give a brown solid. Then, the purified residue was dispersed with acetone (10 mL) and this was stirred at 50 °C for 1 h. After, the suspension was cooled at 0 °C for 1 h and this was filtrated and washed with cold acetone (twice) to give a clean white solid (0.61 g, 52%). Rf (DCM/MeOH 9:1) = 0.17. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.75 (1H, d, J = 8.25 Hz, N-H); 5.01 (1H, d, J = 5.62 Hz, O-H); 4.81 (1H, d, J = 3.54 Hz, CH); 4.73 (1H, d, J = 5.74 Hz, O-H); 4.51 (1H, t, J = 6.02 Hz, O-H); 4.18 (2H, dd, J = 2.44 Hz, J = 15.95 Hz, CH2); 3.60-3.73 (2H, m, 2 CH); 3.43-3.50 (2H, m, 2 CH); 3.42 (1H, t, J = 2.39 Hz, CHalkyne); 3.32-3.37 (1H, m, CH); 3.11-3.18 (1H, m, CH); 1.83 (3H, s, CH3). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 169.4, 95.2, 79.7, 77.3, 73.2, 70.7, 70.5, 60.6, 53.4, 22.6. ESI-HRMS calculated for [C11H18NO6]+ m/z 260.1128 and found m/z 260.1126. IR (neat, cm−1): 3585.7w; 3284.0s; 3079.5w; 2921.1w; 1632.5s; 1546.7s; 1412.9w; 1378.4w; 1320.1w; 1248.6w; 1113.0m; 1094.9m; 1029.0s; 1000.8s; 950.0w; 886.8w; 845.7w; 663.6w. [α]D20 = + 186.6 (C = 0.06 M, MeOH).LC-MS purity 96.6%, tR = 2.99 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
N-((2S,3R,4R,5S,6R)-2-((1-(((2R,3S,4R,5R)-5-(2,4-Dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxy-tetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)-4,5-dihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-3-yl)acetamide (7, CAS: 1152423-06-7). According to the general procedure, copper(II) sulfate pentahydrate (19.3 mg, 0.08 mmol, 10 mol%) and sodium ascorbate (91.7 mg, 0.46 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (208 mg, 0.77 mmol, 1.0 eq) and 2-propynyl-N-acetyl-α-D-glucosamine 6 (200 mg, 0.77 mmol, 1.0 eq) with tBuOH:H2O (15 mL). The reaction mixture was stirred for 4 h at 40 °C. The residue was purified by a flash chromatography (acetone/H2O 95/5) followed by a second flash chromatography (H2O/iPrOH/EtOAc 1/6/3) to give the desired product as white solid (0.24 g, 59%). Rf (H2O/iPrOH/EtOAc 1/6/3) = 0.28. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.36 (1H, s, N-H); 8.08 (1H, s, =CHtosyl); 7.68 (1H, d, J = 8.17 Hz, N-H); 7.58 (1H, d, J = 8.09 Hz, =CH); 5.74 (1H, d, J = 5.38 Hz, CH); 5.65 (1H, d, J = 8.09 Hz, =CH); 5.51 (1H, d, J = 5.64 Hz, O-H); 5.40 (1H, d, J = 5.38 Hz, O-H); 4.99 (1H, d, JH19′ = 5.34 Hz, O-H); 4.75 (1H, d, J = 3.57 Hz, CH); 4.60–4.74 (4H, m, O-H/CH2/CH); 4.46-4.54 (2H, m, O-H/CH); 4.06-4.17 (2H, m, 2 CH); 3.97 (1H, m, CH); 3.61-3.69 (2H, m, 2 CH); 3.41-3.53 (3H, m, 3 CH); 3.14 (1H, m, CH); 1.78 (3H, s, CH3). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 169.4, 163.0, 150.6, 143.7, 141.2, 124.6, 102.1, 96.2, 88.9, 81.7, 73.0, 72.0, 70.8, 70.5, 60.8, 59.8, 53.7, 51.3, 22.5. ESI-HRMS calculated for [C20H29N6O11]+ m/z 529.1888 and found m/z 529.1883. IR (neat, cm−1): 3307.1br; 3099.6m; 2929.3m; 2833.5w; 1674.1s; 1548.6w; 1462.6w; 1423.5w; 1380.9w; 1320.9w; 1263.0w; 1101.1m; 1019.8s; 951.2w; 897.4w; 814.1w; 765.3w; 708.8w. [α]D20 = + 152.0 (C = 0.03 M, H2O).LC-MS purity 95.5%, tR = 3.39 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
((2R,3S,4R,5R,6S)-5-Acetamido-3,4-dihydroxy-6-(prop-2-yn-1-yloxy)tetrahydro-2H-pyran-2-yl)methyl4-methylbenzenesulfonate (8, CAS: 1895096-35-1). To a solution of 2-propynyl-N-acetyl-α-D-glucosamine 6 (1.0 g, 3.86 mmol, 1.0 eq) in adry pyridine (15 mL) at 0 °C under argon atmosphere, Tosyl chloride (0.81 g, 4.24 mmol, 1.1 eq) was added and the reaction was stirred at 25 °C for 16 h. After total conversion of starting material, the reaction mixture was stopped and concentrated to dryness in vacuo with co-evaporation once with toluene (50 mL) and once with cyclohexane (50 mL). The resulting residue was directly purified by a flash chromatography (DCM/MeOH 95/5) followed by a second chromatography (EtOAc 100%) to give a white solid (0.79 g, 50%). Rf (DCM/MeOH 9:1) = 0.38. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 7.78 (2H, d, J = 8.32 Hz, 2 Ar-H); 7.77 (1H, m, N-H); 7.48 (2H, d, J = 8.32 Hz, 2 Ar-H); 5.31 (1H, d, J = 5.93 Hz, O-H); 4.86 (1H, d, J = 6.06 Hz, O-H); 4.74 (1H, d, J = 3.55 Hz, CH); 4.18 (2H, dd, J = 1.92 Hz, J = 10.75 Hz, CH2); 4.11 (2H, Dd, J = 2.44 Hz, J = 16.11 Hz, CH2); 3.65 (1H, m, CH); 3.55 (1H, m, CH); 3.44 (1H, t, J = 2.43 Hz, C-Halkyne); 3.41 (1H, m, CH); 3.11 (1H, m, CH); 2.43 (3H, s, CH3); 1.82 (3H, s, CH3). 13C-NMR (100 MHz, DMSO-d6) δ (ppm):169.4, 144.9, 132.4, 130.1, 127.6, 95.4, 79.4, 77.5, 70.1, 70.0, 69.9, 53.9, 53.2, 22.5, 21.1. ESI-HRMS calculated for [C18H24NO8S]+ m/z 414.1217 and found m/z 414.1219. IR (neat, cm−1): 3282.0br; 2921.7w; 1651.6m; 1597.1w; 1538.1m; 1441.4w; 1353.7m; 1293.0w; 1189.4w; 1173.7s; 1122.2w; 1095.9w; 1032.0m; 996.2w; 969.2w; 929.4w; 891.5w; 813.4w; 664.8w. [α]D20 = + 122.2 (C = 0.07 M, MeOH).LC-MS purity 94.3%, tR = 6.88 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
((2R,3S,4R,5R,6S)-5-Acetamido-6-((1-(((2R,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)-3,4-dihydroxytetrahydro-2H-pyran-2-yl)methyl 4-methylbenzenesulfonate (9). According to the general procedure, copper(II) sulfate pentahydrate (47 mg, 0.19 mmol, 10 mol%) and sodium ascorbate (220 mg, 1.12 mmol, 0.6 eq) were added to a solution of 5′-azido-5′-deoxyuridine 2 (500 mg, 1.86 mmol, 1.0 eq) and 6-tosylate-2-propynyl-N-acetyl-α-D-glucosamine 8 (770 mg, 1.86 mmol, 1.0 eq) with tBuOH:H2O (25 mL). The reaction mixture was stirred for 2 h at 40 °C. The residue was purified by a flash chromatography (acetone/H2O 98/2) followed by a second flash chromatography (H2O/iPrOH/EtOAc 0.5/6.5/3) to give the desired product as colorless oil (1.04 g, 82%). Rf (H2O/iPrOH/EtOAc 1/6/3) = 0.68. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.36 (1H, s, N-H); 8.06 (1H, s, =CHtriazol); 7.79 (2H, d, J = 8.35 Hz, 2 Ar-H); 7.70 (1H, d, J = 8.12 Hz, N-H); 7.58 (1H, d, J = 8.12 Hz, =CH); 7.47 (2H, d, J = 8.35 Hz, 2 Ar-H); 5.74 (1H, d, J = 5.31 Hz, CH); 5.64 (1H, dd, J = 1.38 Hz, J = 8.12 Hz, =CH); 5.51 (1H, d, J = 5.63 Hz, O-H); 5.40 (1H, d, J = 5.44 Hz, O-H); 5.30 (1H, d, J = 5.84 Hz, O-H); 4.83 (1H, d, J = 6.01 Hz, O-H); 4.70 (1H, d, J = 3.39 Hz, CH); 4.60–4.74 (2H, m, CH2); 4.40–4.60 (2H, m, CH2); 4.24 (1H, m, CH); 4.10–4.17 (3H, m, 3 CH); 3.98 (1H, m, CH); 3.57–3.67 (2H, m, 2 CH); 3.36–3.44 (1H, m, CH); 3.06–3.14 (1H, m, CH); 2.41 (3H, s, CH3); 1.76 (3H, s, CH3). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 169.5, 163.0, 150.6, 144.9, 143.5, 141.2, 132.4, 130.1, 127.6, 124.6, 102.1, 96.2, 88.9, 81.8, 72.0, 70.6, 70.2, 70.0, 69.8, 60.1, 53.4, 51.4, 22.5, 21.1. ESI-HRMS calculated for [C27H35N6O13S]+ m/z 683.1977 and found m/z 683.1963. IR (neat, cm−1): 3303.8br; 2929.2m; 2832.7w; 1682.4s; 1598.0w; 1548.6w; 1461.8w; 1427.3w; 1379.5w; 1351.8m; 1265.7w; 1189.5w; 1174.0s; 1123.3m; 1096.8m; 1020.2m; 998.7m; 969.6w; 931.0w; 813.2w; 768.8w; 666.1w. [α]D20 = + 91.7 (C = 0.02 M, MeOH).LC-MS purity 98.3%, tR = 5.55 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
N-((2S,3R,4R,5S,6R)-6-(Azidomethyl)-2-((1-(((2R,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)-4,5-dihydroxytetrahydro-2H-pyran-3-yl)acetamide (10). To flame dried round-bottom flask, compound 9 (0.91 g, 1.33 mmol, 1.0 eq) was stirred with dry DMF (10 mL) at 25 °C under argon atmosphere. Then, sodium azide (0.26 g, 3.99 mmol, 3.0 eq) was added to the solution and this was stirred for 72 h. Thus, the reaction mixture was concentrated to dryness in vacuo with co-evaporation with n-heptane to remove residual DMF. The residue was purified by a flash chromatography (acetonitrile/H2O 9/1) to give the desired product as white solid (0.47 g, 64%). Rf (acetonitrile/H2O 9/1) = 0.36. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.36 (1H, s, N-H); 8.08 (1H, s, =CHtriazol); 7.74 (1H, d, J = 8.12 Hz, N-H); 7.59 (1H, d, J = 8.09 Hz, =CH); 5.74 (1H, d, J = 5.37 Hz, CH); 5.65 (1H, d, J = 8.09 Hz, =CH); 5.52 (1H, d, J = 5.63 Hz, O-H); 5.40 (1H, d, J = 5.33 Hz, O-H); 5.28 (1H, m, O-H); 4.82 (1H, d, J = 5.97 Hz, O-H); 4.80 (1H, d, J = 3.54 Hz, CH); 4.61–4.75 (2H, m, CH2); 4.51–4.70 (2H, m, CH); 4.09–4.18 (2H, m, 2 CH); 3.98 (1H, m, CH); 3.61–3.72 (2H, m, 2 CH); 3.37–3.50 (3H, m, CH2/CH); 3.15 (1H, m, CH); 1.78 (3H, s, CH3). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 169.5, 163.0, 150.6, 143.5, 141.2, 124.6, 102.1, 96.5, 88.9, 81.7, 72.0, 71.5, 71.4, 70.6, 70.2, 60.2, 53.5, 51.3, 51.1, 22.5. ESI-HRMS calculated for [C20H28N9O10]+ m/z 554.1953 and found m/z 554.1949. IR (neat, cm−1): 3289.0br; 3258.4m; 3193.2m; 3151.7m; 3077.0m; 2929.4br; 2722.4w; 2595.2w; 2101.1s; 1682.3s; 1577.6w; 1529.9w; 1546.7w; 1463.2w; 1421.5w; 1381.5w; 1318.7w; 1267.5w; 1234.2w; 1121.0m; 1098.4m; 1053.8s; 897.3w; 839.9w; 815.0w; 767.8w. [α]D20 = + 72.8 (C = 0.01 M, MeOH).LC-MS purity 94.0%, tR = 5.78 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
5-(1-(((2R,3S,4R,5R,6S)-5-Acetamido-6-((1-(((2R,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)-3,4-dihydroxytetrahydro-2H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)pentanoic acid (11a). According to the general procedure, copper(II) sulfate pentahydrate (3.9 mg, 0.02 mmol, 10 mol%) and sodium ascorbate (18.5 mg, 0.09 mmol, 0.6 eq) were added to a solution of compound 10 (86 mg, 0.16 mmol, 1.0 eq) and 6-heptynoic acid (26.1 mg, 0.19 mmol, 1.2 eq) with tBuOH:H2O (10 mL). The reaction mixture was stirred for 5 h at 40 °C. The reaction was concentrated to dryness in vacuo with co-evaporation of methanol several times. The residue was purified by a flash chromatography (H2O/iPrOH/EtOAc 1/6/3) to give the desired product as white solid (87 mg, 82%). Rf (H2O/iPrOH/EtOAc 1/6/3) = 0.23. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.35 (1H, s, N-H); 7.93 (1H, s, =CHtriazol); 7.84 (1H, s, =CHtriazol); 7.77 (1H, d, J = 8.17 Hz, N-H); 7.59 (1H, d, J = 8.09 Hz, =CH); 5.73 (1H, d, J = 5.43 Hz, CH); 5.64 (1H, d, J = 8.09 Hz, =CH); 4.71 (1H, d, J = 3.42 Hz, CH); 4.58–4.73 (3H, m, CH2/CH); 4.37–4.45 (1H, m, CH); 4.18–4.28 (2H, m, CH2); 4.15 (1H, m, CH); 4.06 (1H, t, J = 5.38 Hz, CH); 3.93 (1H, m, CH); 3.82 (1H, m, CH); 3.66 (1H, m, CH); 3.47 (1H, m, CH); 3.07 (1H, m, CH); 2.58 (2H, t, = 7.01 Hz, CH2); 2.05 (2H, t, J = 7.05 Hz, CH2); 1.76 (3H, s, CH3); 1.41–1.61 (4H, m, 2 CH2). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 175.7, 169.5, 163.5, 150.6, 146.8, 143.0, 141.2, 124.6, 122.6, 102.1, 95.9, 89.0, 81.8, 72.1, 70.8, 70.6, 70.3, 59.4, 53.5, 51.3, 50.6, 35.0, 28.8, 24.9, 24.7, 22.5,. ESI-HRMS calculated for [C27H38N9O12]+ m/z 680.2634 and found m/z 680.2627. IR (neat, cm−1): 3294.2br; 3149.3m; 2927.8br; 2858.9m; 1689.3s; 1549.6m; 1461.5m; 1423.2m; 1380.5m; 1262.9m; 1224.5m; 1101.7m; 1053.1m; 1037.5m; 896.2w; 813.3w; 774.4w; 720.7w. [α]D20 = + 44.2 (C = 0.01 M, MeOH).LC-MS purity 96.7%, tR = 2.58 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
N-((2S,3R,4R,5S,6R)-6-((4-(Aminomethyl)-1H-1,2,3-triazol-1-yl)methyl)-2-((1-(((2R,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)-4,5-dihydroxytetrahydro-2H-pyran-3-yl)acetamide (11b). According to the general procedure, copper(II) sulfate pentahydrate (3.9 mg, 0.02 mmol, 10 mol%) and sodium ascorbate (18.5 mg, 0.09 mmol, 0.6 eq) were added to a solution of compound 10 (86 mg, 0.16 mmol, 1.0 eq) and propargylamine (12.8 mg, 0.23 mmol, 1.5 eq) with tBuOH:H2O (10 mL). The reaction mixture was stirred for 5 h at 40 °C. The reaction was concentrated to dryness in vacuo with co-evaporation of methanol several times. The residue was purified by a flash chromatography (Acetone/H2O 95/5) to give the desired product as pale yellow solid (18 mg, 19%). Rf (acetone/H2O 95/5) = 0.25. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.34 (1H, s, N-H); 8.79 (1H, s, =CHtriazol); 8.01 (1H, s, =CHtriazol); 7.74 (1H, d, J = 8.13 Hz, N-H); 7.58 (1H, d, J = 8.10 Hz, =CH); 5.73 (1H, d, J = 5.24 Hz, CH); 5.63 (1H, d, J = 8.07 Hz, CH); 5.57 (1H, d, J = 5.13 Hz, O-H); 5.52 (1H, d, J = 5.33 Hz, O-H); 5.42 (1H, d, J = 4.92 Hz, O-H); 4.92 (1H, d, J = 5.74 Hz, O-H); 4.73 (1H, d, J = 3.43 Hz, CH); 4.57–4.82 (3H, m, CH2/CH); 4.37–4.51 (1H, m, CH); 4.22–4.36 (2H, m, CH2); 4.05–4.18 (4H, m, N-H2/CH/CH); 3.97 (1H, m, CH); 3.88 (1H, m, CH); 3.62 (1H, m, CH); 3.48 (1H, m, CH); 3.17 (1H, d, J = 5.03 Hz, CH); 3.05 (1H, m, CH); 1.76 (3H, s, CH3). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 169.5, 163.0, 150.6, 146.7, 144.1 141.2, 129.3, 124.5, 102.1, 96.4, 89.0, 81.7, 72.0, 71.9, 70.6, 70.1, 59.9, 55.8, 51.3, 48.6, 22.4. ESI-HRMS calculated for [C23H30N9O11]+ m/z 608.2059 and found m/z 608.2053. IR (neat, cm−1): 3275.5br; 2953.8m; 2922.9s; 2852.8m; 1688.6s; 1537.8w; 1463.0w; 1428.3w; 1379.6w; 1260.0w; 1231.2w; 1187.8w; 1126.1w; 1102.0m; 1083.0m; 1050.1s; 816.0w; 776.8w; 719.3w. [α]D20 = + 43.3 (C = 0.03 M, H2O).LC-MS purity 95.0%, tR = 2.22 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
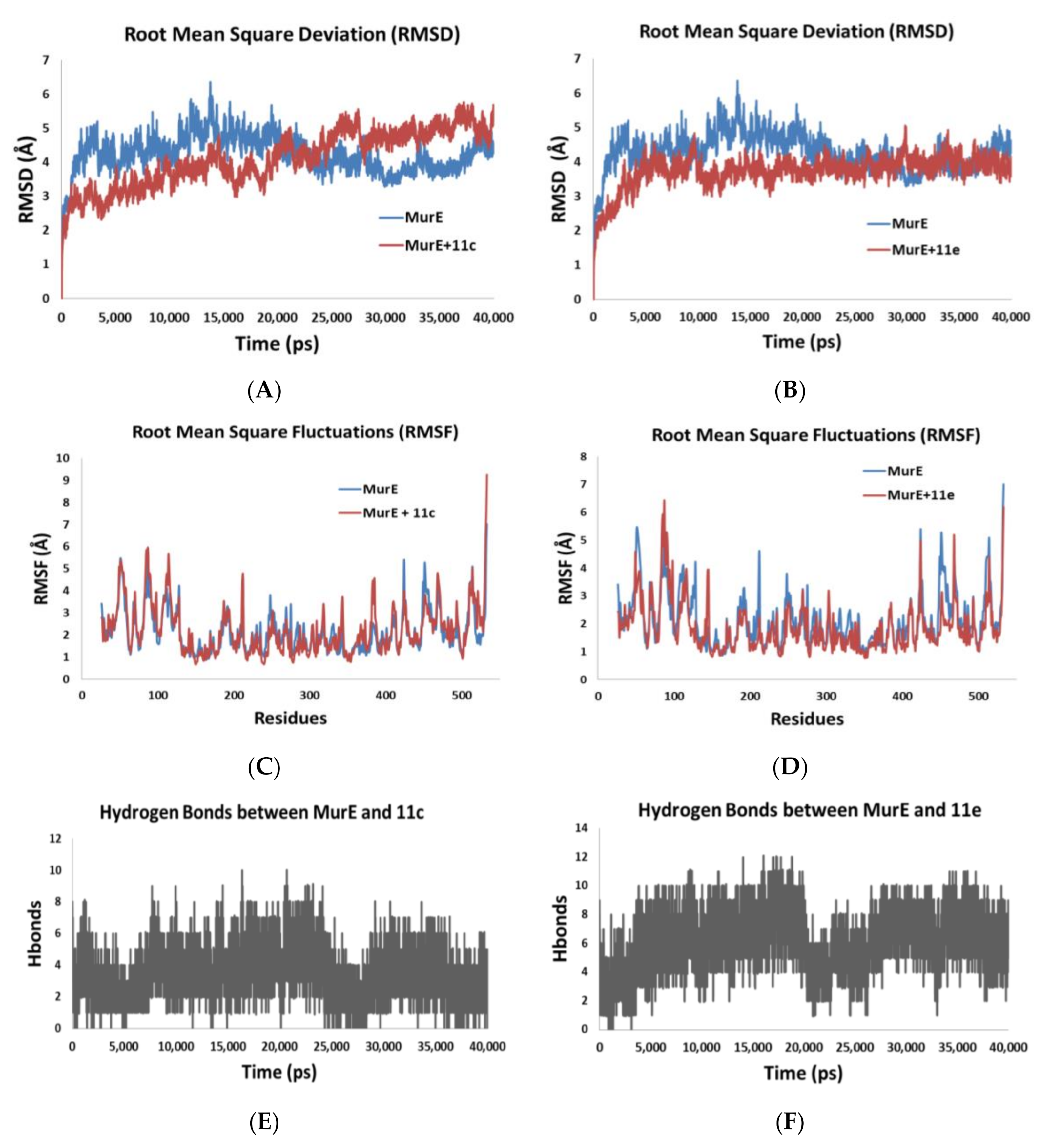
1-(((2R,3S,4R,5R,6S)-5-Acetamido-6-((1-(((2R,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)-3,4-dihydroxytetrahydro-2H-pyran-2-yl)methyl)-1H-1,2,3-triazole-4-carboxamide (11c). According to the general procedure, copper(II) sulfate pentahydrate (3.9 mg, 0.02 mmol, 10 mol%) and sodium ascorbate (18.5 mg, 0.09 mmol, 0.6 eq) were added to a solution of compound 10 (86 mg, 0.16 mmol, 1.0 eq) and propiolamide (12 mg, 0.17 mmol, 1.1 eq) with tBuOH:H2O (10 mL). The reaction mixture was stirred for 5 h at 40 °C. The reaction was concentrated to dryness in vacuo with co-evaporation of methanol several times. The residue was purified by a flash chromatography (H2O/iPrOH/EtOAc 1/6/3) to give the desired product as white solid (87 mg, 90%). Rf (Acetone/H2O 95/5) = 0.31. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.36 (1H, s, N-H); 8.45 (1H, s, =CHtriazol); 8.02 (1H, s, =CHtriazol); 7.84 (1H, s, N-H); 7.75 (1H, d, J = 8.06 Hz, N-H); 7.59 (1H, d, J = 8.06 Hz, =CH); 7.43 (1H, s, N-H); 5.74 (1H, d, J = 5.32 Hz, CH); 5.65 (1H, d, J = 8.04 Hz, =CH); 4.74 (1H, d, J = 3.38 Hz, CH); 4.53–4.78 (4H, m, CH/CH2); 4.24–4.37 (2H, m, CH2); 4.10–4.18 (2H, m, 2 CH); 3.98 (1H, m, CH); 3.89 (1H, m, CH); 3.63 (1H, m, CH); 3.49 (1H, m, CH); 3.04 (1H, t, J = 9.22 Hz, CH); 1.76 (3H, s, CH3). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 169.5, 163.0, 161.6, 150.6, 143.5, 142.7, 141.3, 127.6, 124.5, 102.1, 96.3, 89.0, 81.8, 72.0, 71.9, 70.6, 70.2, 59.9, 53.5, 51.4, 50.9, 22.5. ESI-HRMS calculated for [C23H31N10O11]+ m/z 623.2168 and found m/z 623.2164. IR (neat, cm−1): 3304.7br; 2922.8m; 2851.5w; 1667.0s; 1556.8m; 1463.0m; 1423.1m; 1382.1m; 1322.8w; 1263.2m; 1232.2m; 1101.2m; 1049.8s; 1019.6s; 896.1w; 814.8w; 773.3w. [α]D20 = + 62.8 (C = 0.02 M, MeOH).LC-MS purity 96.1%, tR = 2.88 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
N-((1-(((2R,3S,4R,5R,6S)-5-Acetamido-6-((1-(((2R,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)-3,4-dihydroxytetrahydro-2H-pyran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methyl)octanamide (11d). According to the general procedure, copper(II) sulfate pentahydrate (3.9 mg, 0.02 mmol, 10 mol%) and sodium ascorbate (18.5 mg, 0.09 mmol, 0.6 eq) were added to a solution of compound 10 (86 mg, 0.16 mmol, 1.0 eq) and N-(prop-2-yn-1-yl)octanamide (31 mg, 0.17 mmol, 1.1 eq) with tBuOH:H2O (10 mL). The reaction mixture was stirred for 5 h at 40 °C. The reaction was concentrated to dryness in vacuo with co-evaporation of methanol several times. The residue was purified by a flash chromatography (acetone/H2O 95/5) to give the desired product as white solid (88 mg, 78%). Rf (acetone/H2O 95/5) = 0.38. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.35 (1H, d, J = 1.30 Hz, N-H); 8.23 (1H, t, J = 5.64 Hz, N-H); 7.99 (1H, s, =CHtriazol); 7.84 (1H, s, =CHtriazol); 7.72 (1H, d, J = 8.16 Hz, N-H); 7.59 (1H, d, J = 8.07 Hz, =CH); 5.74 (1H, d, J = 5.35 Hz, CH); 5.64 (1H, dd, J = 1.30 Hz, J = 8.07 Hz, =CH); 5.51 (1H, d, J = 5.63 Hz, O-H); 5.47 (1H, d, J = 5.79 Hz, O-H); 5.40 (1H, d, J = 5.43 Hz, O-H); 4.88 (1H, d, J = 5.79 Hz, O-H); 4.71 (1H, d, J = 3.57 Hz, CH); 4.60–4.74 (3H, m, CH/CH2); 4.44–4.51 (1H, m, CH); 4.22–4.37 (4H, m, 2 CH2); 4.07–4.18 (2H, m, 2 CH); 3.98 (1H, m, CH); 3.83 (1H, m, CH); 3.63 (1H, m, CH); 3.47 (1H, m, CH); 3.05 (1H, m, CH); 2.03 (3H, t, J = 7.35 Hz, CH3); 1.76 (3H, s, H29′); 1.44 (2H, m, CH2); 1.15–1.28 (8H, m, 4 CH2); 0.84 (3H, t, J = 6.70 Hz, CH3). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 172.1, 169.5, 163.0 150.6, 144.9, 143.2, 141.2, 124,6, 123.7, 102.1, 96.1, 88.9, 81.7, 72.0, 71.9, 70.6, 70.5, 70.2, 59.8, 53.4, 51.4, 50.5, 35.2, 34.1, 31.1, 28.6, 28.4, 25.2, 22.5, 22.0, 13.9. ESI-HRMS calculated for [C31H47N10O11]+ m/z 735.3420 and found m/z 735.3421. IR (neat, cm−1): 3291.1br; 2928.4s; 2855.8m; 1682.8s; 1540.6m; 1461.6m; 1427.5m; 1379.1m; 1264.0m; 1227.8m; 1102.8m; 1052.8s; 814.2w; 776.8w; 720.7w. [α]D20 = + 79.0 (C = 0.02 M, MeOH).LC-MS purity 95.2%, tR = 3.42 min water/methanol (70:30 vol/vol, 0.1% of formic acid).
N-((2S,3R,4R,5S,6R)-6-((4-((((2S,3R,4R,5S,6R)-3-Acetamido-4,5-dihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl)oxy)methyl)-1H-1,2,3-triazol-1-yl)methyl)-2-((1-(((2R,3S,4R,5R)-5-(2,4-dioxo-3,4-dihydro-pyrimidin-1(2H)-yl)-3,4-dihydroxytetrahydrofuran-2-yl)methyl)-1H-1,2,3-triazol-4-yl)methoxy)-4,5-dihydroxytetrahydro-2H-pyran-3-yl)acetamide (11e). According to the general procedure, copper(II) sulfate pentahydrate (3.9 mg, 0.02 mmol, 10 mol%) and sodium ascorbate (18.5 mg, 0.09 mmol, 0.6 eq) were added to a solution of compound 10 (86 mg, 0.16 mmol, 1.0 eq) and 2-propynyl-N-acetyl-α-D-glucosamine 6 (44.3 mg, 0.17 mmol, 1.1 eq) with tBuOH:H2O (10 mL). The reaction mixture was stirred for 5 h at 40 °C. The reaction was concentrated to dryness in vacuo with co-evaporation of methanol several times. The residue was purified by a flash chromatography (acetone/H2O 9/1) to give the desired product as white solid (97 mg, 77%). Rf (acetone/H2O 9/1) = 0.26. 1H-NMR (400 MHz, DMSO-d6) δ (ppm): 11.35 (1H, s, N-H); 8.04 (1H, s, =CHtriazol); 8.00 (1H, s, =CHtriazol); 7.74 (1H, d, J = 8.21 Hz, N-H); 7.70 (1H, d, J = 8.24 Hz, N-H); 7.59 (1H, d, J = 8.11 Hz, =CH); 5.74 (1H, d, J = 5.37 Hz, CH); 5.65 (1H, d, J = 8.11 Hz, =CH); 5.53 (1H, m, O-H); 5.49 (1H, m, O-H); 5.42 (1H, m, O-H); 4.99 (1H, m, O-H); 4.90 (1H, m, O-H); 4.74 (1H, d, J = 3.46 Hz, CH); 4.72 (1H, d, J = 3.39 Hz, CH); 4.59–4.74 (4H, m, 3 CH); 4.46–4.57 (3H, m, 3 CH); 4.25–4.35 (2H, m, CH2); 4.10–4.18 (2H, m, 2 CH); 4.09 (1H, m, O-H); 3.98 (1H, m, CH); 3.86 (1H, m, CH); 3.60–3.70 (3H, m, 3 CH); 3.40–3.53 (4H, m, 4 CH); 3.15 (1H, t, J = 10.28 Hz, CH); 3.05 (1H, t, J = 9.08 Hz, CH); 1.80 (3H, s, CH3); 1.76 (3H, s, CH3). 13C-NMR (100 MHz, DMSO-d6) δ (ppm): 169.5, 163.0, 150.6, 143.5, 143.4, 141.2, 125.1, 124.4, 102.1, 96.4, 96.1, 88.9, 81.8, 73.0, 72.0, 71.9, 70.8, 70.6, 70.5, 70.4, 70.3, 60.8, 60.0, 59.8, 53.6, 53.5, 51.4, 50.6, 22.6, 22.5. ESI-HRMS calculated for [C31H45N10O16]+ m/z 813.3009 and found m/z 813.3006. IR (neat, cm−1): 3306.8br; 2923.4br; 1682.6s; 1548.2m; 1461.6w; 1428.0w; 1378.4w; 1318.5w; 1263.6w; 1229.9w; 1103.2m; 1022.6s; 949.6w; 841.3w; 811.3w; 759.8w. [α]D20 = + 113.5 (C = 0.01 M, MeOH).LC-MS purity 97.1%, tR = 2.48 min water/methanol (70:30 vol/vol, 0.1% of formic acid).



 and
and 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
















