
PEGylated Liposomal Methyl Prednisolone Succinate does not Induce Infusion Reactions in Patients: A Correlation Between in Vitro Immunological and in Vivo Clinical Studies

 ,
,  , ,
, ,
Abstract
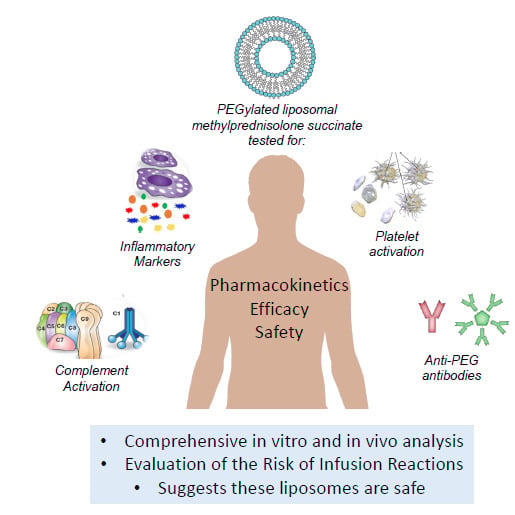
:
1. Introduction
2. Results
2.1. In Vitro Immune-Compatibility of NSSL-MPS
2.2. Clinical Study
2.3. In Vivo Complement Activation and Inflammatory Disease Markers
2.4. Screening of Anti-PEG Antibodies in a Healthy Population
2.5. Anti-PEG Antibodies in the Patient After Administration of NSSL-MPS
2.6. Pharmacokinetics of Repeated Administrations of Liposomal MPS
3. Discussion
4. Material and Methods
4.1. Preparation of the NSSL-MPS
4.2. Physicochemical Characterization of NSSL-MPS
4.3. In Vitro Complement Activation by the NSSL-MPS
4.4. In vitro Cytokines Release
4.5. Platelet Aggregation and Collagen-Induced Platelet Aggregation
4.6. Leucocyte Pro-Coagulant Activity
4.7. In Vitro Evaluation of NSSL-MPS Hemolytic Activity
4.8. Patient’s Treatment with NSSL-MPS
4.9. Blood Sampling
4.10. Anti-PEG Detection Assays in Human Plasma
4.11. In Vivo Quantification of Complement Activation Markers During NSSL-MPS Infusion
4.12. Pharmacokinetic Study
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Unezaki, S.; Maruyama, K.; Hosoda, J.-I.; Nagae, I.; Koyanagi, Y.; Nakata, M.; Ishida, O.; Iwatsuru, M.; Tsuchiya, S. Direct measurement of the extravasation of polyethyleneglycol-coated liposomes into solid tumor tissue by in vivo fluorescence microscopy. Int. J. Pharm. 1996, 144, 11–17. [Google Scholar] [CrossRef]
- Awasthi, V.D.; Goins, B.; Klipper, R.; Phillips, W.T. Accumulation of PEG-liposomes in the inflamed colon of rats: Potential for therapeutic and diagnostic targeting of inflammatory bowel diseases. J. Drug Target. 2002, 10, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Metselaar, J.M.; Wauben, M.H.; Wagenaar-Hilbers, J.P.; Boerman, O.C.; Storm, G. Complete remission of experimental arthritis by joint targeting of glucocorticoids with long-circulating liposomes. Arthritis Rheum. 2003, 48, 2059–2066. [Google Scholar] [CrossRef] [PubMed]
- Azzopardi, E.A.; Ferguson, E.L.; Thomas, D.W. The enhanced permeability retention effect: A new paradigm for drug targeting in infection. J. Antimicrob. Chemother. 2013, 68, 257–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anchordoquy, T.J.; Barenholz, Y.; Boraschi, D.; Chorny, M.; Decuzzi, P.; Dobrovolskaia, M.A.; Farhangrazi, Z.S.; Farrell, D.; Gabizon, A.; Ghandehari, H.; et al. Mechanisms and barriers in cancer nanomedicine: Addressing challenges, looking for solutions. ACS Nano 2017, 11, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Hamad, I.; Hunter, A.C.; Szebeni, J.; Moghimi, S.M. Poly(ethylene glycol)s generate complement activation products in human serum through increased alternative pathway turnover and a MASP-2-dependent process. Mol. Immunol. 2008, 46, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Barenholz, Y. Complement activation, immunogenicity and immune suppression as potential side effects of liposomes. In Harnessing Biomaterials for Nanomedicine: Preparation, Toxicity and Applications; Peer, D., Ed.; Pan Stanford Publishing Pte Ltd.: Singapore, 2012; pp. 309–334. [Google Scholar]
- Szebeni, J.; Muggia, F.; Gabizon, A.; Barenholz, Y. Activation of complement by therapeutic liposomes and other lipid excipient-based therapeutic products: prediction and prevention. Adv. Drug Deliv. Rev. 2011, 63, 1020–1030. [Google Scholar] [CrossRef]
- Szebeni, J. Complement activation-related pseudoallergy: A stress reaction in blood triggered by nanomedicines and biologicals. Mol. Immunol. 2014, 61, 163–173. [Google Scholar] [CrossRef]
- Szebeni, J.; Simberg, D.; Gonzalez-Fernandez, A.; Barenholz, Y.; Dobrovolskaia, M.A. Roadmap and strategy for overcoming infusion reactions to nanomedicines. Nat. Nanotechnol. 2018, 22, 18–273. [Google Scholar] [CrossRef]
- Richter, A.W.; Åkerblom, E. Polyethylene glycol reactive antibodies in man: Titer distribution in allergic patients treated with monomethoxy polyethylene glycol modified allergens or placebo, and in healthy blood donors. Int. Arch. Allergy Immunol. 1984, 74, 36–39. [Google Scholar] [CrossRef]
- Chen, B.-M.; Su, Y.-C.; Chang, C.-J.; Burnouf, P.-A.; Chuang, K.-H.; Chen, C.-H.; Cheng, T.-L.; Chen, Y.-T.; Wu, J.-Y.; Roffler, S.R. Measurement of pre-existing IgG and IgM antibodies against polyethylene glycol in healthy individuals. Anal. Chem. 2016, 88, 10661–10666. [Google Scholar] [CrossRef] [PubMed]
- Ganson, N.J.; Povsic, T.J.; Sullenger, B.A.; Alexander, J.H.; Zelenkofske, S.L.; Sailstad, J.M.; Rusconi, C.P.; Hershfield, M.S. Pre-existing anti–polyethylene glycol antibody linked to first-exposure allergic reactions to pegnivacogin, a PEGylated RNA aptamer. J. Allergy Clin. Immunol. 2016, 137, 1610–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershfield, M.S.; Ganson, N.J.; Kelly, S.J.; Scarlett, E.L.; Jaggers, D.A.; Sundy, J.S. Induced and pre-existing anti-polyethylene glycol antibody in a trial of every 3-week dosing of pegloticase for refractory gout, including in organ transplant recipients. Arthritis Res. Ther. 2014, 16, R63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, J.K.; Hempel, G.; Koling, S.; Chan, L.S.; Fisher, T.; Meiselman, H.J.; Garratty, G. Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer 2007, 110, 103–111. [Google Scholar] [CrossRef]
- Sundy, J.S.; Ganson, N.J.; Kelly, S.J.; Scarlett, E.L.; Rehrig, C.D.; Huang, W.; Hershfield, M.S. Pharmacokinetics and pharmacodynamics of intravenous PEGylated recombinant mammalian urate oxidase in patients with refractory gout. Arthritis Rheum. 2007, 56, 1021–1028. [Google Scholar] [CrossRef]
- Neun, B.; Barenholz, Y.; Szebeni, J.; Dobrovolskaia, M. Understanding the role of anti-PEG antibodies in the complement activation by doxil in vitro. Molecules 2018, 23, 1700. [Google Scholar] [CrossRef] [Green Version]
- Dams, E.T.; Laverman, P.; Oyen, W.J.; Storm, G.; Scherphof, G.L.; van Der Meer, J.W.; Corstens, F.H.; Boerman, O.C. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J. Pharmaco.l Exp. Ther. 2000, 292, 1071–1079. [Google Scholar]
- Im, H.J.; England, C.G.; Feng, L.; Graves, S.A.; Hernandez, R.; Nickles, R.J.; Liu, Z.; Lee, D.S.; Cho, S.Y.; Cai, W. Accelerated blood clearance phenomenon reduces the passive targeting of PEGylated nanoparticles in peripheral arterial disease. ACS Appl. Mater. Interfaces 2016, 8, 17955–17963. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Kiwada, H. Accelerated blood clearance (ABC) phenomenon upon repeated injection of PEGylated liposomes. Int. J. Pharm. 2008, 354, 56–62. [Google Scholar] [CrossRef]
- Suzuki, T.; Ichihara, M.; Hyodo, K.; Yamamoto, E.; Ishida, T.; Kiwada, H.; Ishihara, H.; Kikuchi, H. Accelerated blood clearance of PEGylated liposomes containing doxorubicin upon repeated administration to dogs. Int. J. Pharm. 2012, 436, 636–643. [Google Scholar] [CrossRef]
- Ishida, T.; Ichihara, M.; Wang, X.; Yamamoto, K.; Kimura, J.; Majima, E.; Kiwada, H. Injection of PEGylated liposomes in rats elicits PEG-specific IgM, which is responsible for rapid elimination of a second dose of PEGylated liposomes. J. Control. Release 2006, 112, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Avnir, Y.; Turjeman, K.; Tulchinsky, D.; Sigal, A.; Kizelsztein, P.; Tzemach, D.; Gabizon, A.; Barenholz, Y. Fabrication principles and their contribution to the superior in vivo therapeutic efficacy of nano-liposomes remote loaded with glucocorticoids. PLoS ONE 2011, 6, e25721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avnir, Y.; Ulmansky, R.; Wasserman, V.; Even-Chen, S.; Broyer, M.; Barenholz, Y.; Naparstek, Y. Amphipathic weak acid glucocorticoid prodrugs remote-loaded into sterically stabilized nanoliposomes evaluated in arthritic rats and in a Beagle dog: A novel approach to treating autoimmune arthritis. Arthritis Rheum. 2008, 58, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Moallem, E.; Koren, E.; Ulmansky, R.; Pizov, G.; Barlev, M.; Barenholz, Y.; Naparstek, Y. A liposomal steroid nano-drug for treating systemic lupus erythematosus. Lupus 2016, 25, 1209–1216. [Google Scholar] [CrossRef]
- Turjeman, K.; Bavli, Y.; Kizelsztein, P.; Schilt, Y.; Allon, N.; Katzir, T.B.; Sasson, E.; Raviv, U.; Ovadia, H.; Barenholz, Y. Nano-drugs based on nano sterically stabilized liposomes for the treatment of inflammatory neurodegenerative diseases. PLoS ONE 2015, 10, e0130442. [Google Scholar] [CrossRef] [Green Version]
- Ulmansky, R.; Turjeman, K.; Baru, M.; Katzavian, G.; Harel, M.; Sigal, A.; Naparstek, Y.; Barenholz, Y. Glucocorticoids in nano-liposomes administered intravenously and subcutaneously to adjuvant arthritis rats are superior to the free drugs in suppressing arthritis and inflammatory cytokines. J. Control. Release 2012, 160, 299–305. [Google Scholar] [CrossRef]
- Waknine-Grinberg, J.H.; Even-Chen, S.; Avichzer, J.; Turjeman, K.; Bentura-Marciano, A.; Haynes, R.K.; Weiss, L.; Allon, N.; Ovadia, H.; Golenser, J.; et al. Glucocorticosteroids in nano-sterically stabilized liposomes are efficacious for elimination of the acute symptoms of experimental cerebral malaria. PLoS ONE 2013, 8, e72722. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, K.; Uchida, K.; Koyabu, M.; Miyoshi, H.; Takaoka, M. Recent advances in the concept and diagnosis of autoimmune pancreatitis and IgG4-related disease. J. Gastroenterol. 2011, 46, 277–288. [Google Scholar] [CrossRef]
- Kamisawa, T.; Zen, Y.; Pillai, S.; Stone, J.H. IgG4-related disease. Lancet 2015, 385, 1460–1471. [Google Scholar] [CrossRef]
- Bozzalla Cassione, E.; Stone, J.H. IgG4-related disease. Curr. Opin. Rheumatol. 2017, 29, 223–227. [Google Scholar] [CrossRef]
- Stone, J.H.; Zen, Y.; Deshpande, V. IgG4-Related Disease. N. Engl. J. Med. 2012, 366, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Umehara, H.; Okazaki, K.; Masaki, Y.; Kawano, M.; Yamamoto, M.; Saeki, T.; Matsui, S.; Sumida, T.; Mimori, T.; Tanaka, Y.; et al. A novel clinical entity, IgG4-related disease (IgG4RD): General concept and details. Mod. Rheumatol. 2012, 22, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Roselló, S.; Blasco, I.; García Fabregat, L.; Cervantes, A.; Jordan, K.; on behalf of the ESMO Guidelines Committee. Management of infusion reactions to systemic anticancer therapy: ESMO Clinical Practice Guidelines†. Ann. Oncol. 2017, 28, iv100–iv118. [Google Scholar] [CrossRef] [PubMed]
- Doxil Monograph. Available online: http://www.janssenlabels.com/package-insert/product-monograph/prescribing-information/DOXIL-pi.pdf (accessed on 28 December 2019).
- Tamez-Pérez, H.E.; Quintanilla-Flores, D.L.; Rodríguez-Gutiérrez, R.; González-González, J.G.; Tamez-Peña, A.L. Steroid hyperglycemia: Prevalence, early detection and therapeutic recommendations: A narrative review. World J. Diabetes 2015, 6, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Hamad, I.; Andresen, T.L.; Jørgensen, K.; Szebeni, J. Methylation of the phosphate oxygen moiety of phospholipid-methoxy(polyethylene glycol) conjugate prevents PEGylated liposome-mediated complement activation and anaphylatoxin production. FASEB J. 2006, 20, 2591–2593. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Spielberg, H.; Cliff, R.O.; Wassef, N.M.; Rudolph, A.S.; Alving, C.R. Complement activation and thromboxane secretion by liposome-encapsulated hemoglobin in rats in vivo: Inhibition by soluble complement receptor type 1. Artif. Cells Blood Substit. Biotechnol. 1997, 25, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Wassef, N.M.; Spielberg, H.; Rudolph, A.S.; Alving, C.R. Complement activation in rats by liposomes and liposome-encapsulated hemoglobin: Evidence for anti-lipid antibodies and alternative pathway activation. Biochem. Biophys. Res. Commun. 1994, 205, 255–263. [Google Scholar] [CrossRef]
- Buyon, J.P.; Tamerius, J.; Belmont, H.M.; Abramson, S.B. Assessment of disease activity and impending flare in patients with systemic lupus erythematosus. Comparison of the use of complement split products and conventional measurements of complement. Arthritis Rheum. 1992, 35, 1028–1037. [Google Scholar] [CrossRef]
- Wenisch, C.; Spitzauer, S.; Florris-Linau, K.; Rumpold, H.; Vannaphan, S.; Parschalk, B.; Graninger, W.; Looareesuwan, S. Complement activation in severeplasmodium falciparummalaria. Clin. Immunol. Immunopathol. 1997, 85, 166–171. [Google Scholar] [CrossRef]
- Ouwendijk, R.J.T.; Zijlstra, F.J.; Wilson, J.H.P.; Bonta, I.L.; Vincent, J.E. Raised plasma thromboxane B2 levels in alcoholic liver disease. Prostaglandins Leukot. Med. 1983, 10, 115–122. [Google Scholar] [CrossRef]
- Czock, D.; Keller, F.; Rasche, F.M.; Häussler, U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin. Pharmacokinet. 2005, 44, 61–98. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, A.; Catane, R.; Uziely, B.; Kaufman, B.; Safra, T.; Cohen, R.; Martin, F.; Huang, A.; Barenholz, Y. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994, 54, 987–992. [Google Scholar] [PubMed]
- Gabizon, A.; Shmeeda, H.; Barenholz, Y. Pharmacokinetics of pegylated liposomal doxorubicin: Review of animal and human studies. Clin. Pharmacokinet. 2003, 42, 419–436. [Google Scholar] [CrossRef]
- Working, P.K.; Dayan, A.D. Pharmacological-toxicological expert report. CAELYX. (Stealth liposomal doxorubicin HCl). Hum. Exp. Toxicol. 1996, 15, 751–785. [Google Scholar] [PubMed]
- Al-Habet, S.; Rogers, H. Methylprednisolone pharmacokinetics after intravenous and oral administration. Br. J. Clin. Pharmacol. 1989, 27, 285–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wibroe, P.P.; Moghimi, S.M. Complement sensing of nanoparticles and nanomedicines. In ACS Symposium Series; Hepel, M., Zhong, C.-J., Eds.; ACS Publication: Washington, DC, USA, 2012; pp. 365–382. [Google Scholar]
- Chanan-Khan, A.; Szebeni, J.; Savay, S.; Liebes, L.; Rafique, N.M.; Alving, C.R.; Muggia, F.M. Complement activation following first exposure to pegylated liposomal doxorubicin (Doxil): Possible role in hypersensitivity reactions. Ann. Oncol. 2003, 14, 1430–1437. [Google Scholar] [CrossRef] [PubMed]
- Kanhai, K.M.S.; Zuiker, R.G.J.A.; Stavrakaki, I.; Gladdines, W.; Gaillard, P.J.; Klaassen, E.S.; Groeneveld, G.J. Glutathione-PEGylated liposomal methylprednisolone in comparison to free methylprednisolone: Slow release characteristics and prolonged lymphocyte depression in a first-in-human study. Br. J. Clin. Pharmacol. 2018, 84, 1020–1028. [Google Scholar] [CrossRef]
- Masaki, Y.; Dong, L.; Kurose, N.; Kitagawa, K.; Morikawa, Y.; Yamamoto, M.; Takahashi, H.; Shinomura, Y.; Imai, K.; Saeki, T.; et al. Proposal for a new clinical entity, IgG4-positive multiorgan lymphoproliferative syndrome: analysis of 64 cases of IgG4-related disorders. Ann. Rheum. Dis. 2009, 68, 1310–1315. [Google Scholar] [CrossRef]
- Fukui, S.; Fujita, Y.; Origuchi, T.; Maeda, T.; Kawakami, A. Serum complement factor C5a in IgG 4 -related disease. Ann. Rheum. Dis. 2019, 78, e65. [Google Scholar] [CrossRef] [Green Version]
- Wallace, Z.S.; Deshpande, V.; Mattoo, H.; Mahajan, V.S.; Kulikova, M.; Pillai, S.; Stone, J.H. IgG4-related disease: Clinical and laboratory features in one hundred twenty-five patients. Arthritis Rheumatol. 2015, 67, 2466–2475. [Google Scholar] [CrossRef] [Green Version]
- Hirano, T. Interleukin 6 in autoimmune and inflammatory diseases: A personal memoir. Proc. Japan Acad. Ser. B 2010, 86, 717–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Kasashima, S.; Kawashima, A.; Kasashima, F.; Endo, M.; Matsumoto, Y.; Kawakami, K. Inflammatory features, including symptoms, increased serum interleukin-6, and C-reactive protein, in IgG4-related vascular diseases. Heart Vessels 2018, 33, 1471–1481. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Takahashi, H.; Hasebe, K.; Suzuki, C.; Naishiro, Y.; Hayashi, T.; Yamamoto, H.; Ishida, T.; Shinomura, Y. The analysis of interleukin-6 in patients with systemic IgG4-related plasmacytic syndrome--expansion of SIPS to the territory of Castleman’s disease. Rheumatology 2009, 48, 860–862. [Google Scholar] [CrossRef] [Green Version]
- Mckay, L.I.; Cidlowski, J.A. Physiologic and pharmacologic effects of corticosteroids. In Holland-Frei Cancer Medicine; Kufe, D.W., Pollock, R.E., Weichselbaum, R.R., Bast, R.C., Gansler, T.S., Holland, J.F., Frei, E., Eds.; BC Decker: Hamilton, ON, USA, 2003. [Google Scholar]
- Shoenfeld, Y.; Gurewich, Y.; Gallant, L.A.; Pinkhas, J. Prednisone-induced leukocytosis. Am. J. Med. 1981, 71, 773–778. [Google Scholar] [CrossRef]
- Chung, C.H. Managing premedications and the risk for reactions to infusional monoclonal antibody therapy. Oncologist 2008, 13, 725–732. [Google Scholar] [CrossRef]
- Shmeeda, H.; Even-Chen, S.; Honen, R.; Cohen, R.; Weintraub, C.; Barenholz, Y. Enzymatic assays for quality control and pharmacokinetics of liposome formulations: Comparison with nonenzymatic conventional methodologies. In Methods Enzymol; Academic Press: Cambridge, MA, USA, 2003; Volume 367, pp. 272–292. [Google Scholar]
- Wei, X.; Shamrakov, D.; Nudelman, S.; Peretz-Damari, S.; Nativ-Roth, E.; Regev, O.; Barenholz, Y. Cardinal role of intraliposome doxorubicin-sulfate nanorod crystal in doxil properties and performance. ACS Omega 2018, 3, 2508–2517. [Google Scholar] [CrossRef] [Green Version]
- Neun, B.W.; Ilinskaya, A.N.; Dobrovolskaia, M.A. Analysis of complement activation by nanoparticles. In Methods in Molecular Biology; McNeil, S.E., Ed.; Springer: New York, NY, USA; Dordrecht, The Netherlands; Heidelberg, Germany; London, UK, 2018; pp. 149–160. [Google Scholar]
- Potter, T.M.; Neun, B.W.; Rodriguez, J.C.; Ilinskaya, A.N.; Dobrovolskaia, M.A. Analysis of pro-inflammatory cytokine and type II interferon induction by nanoparticles. In Methods in Molecular Biology; McNeil, S.E., Ed.; Springer: New York, NY, USA; Dordrecht, The Netherlands; Heidelberg, Germany; London, UK, 2018; pp. 173–187. [Google Scholar]
- Potter, T.M.; Rodriguez, J.C.; Neun, B.W.; Ilinskaya, A.N.; Cedrone, E.; Dobrovolskaia, M.A. In vitro assessment of nanoparticle effects on blood coagulation. In Methods in Molecular Biology; McNeil, S.E., Ed.; Springer: New York, NY, USA; Dordrecht, The Netherlands; Heidelberg, Germany; London, UK, 2018; pp. 103–124. [Google Scholar]
- Dobrovolskaia, M.A.; Clogston, J.D.; Neun, B.W.; Hall, J.B.; Patri, A.K.; McNeil, S.E. Method for analysis of nanoparticle hemolytic properties in vitro. Nano. Lett. 2008, 8, 2180–2187. [Google Scholar] [CrossRef]
- Neun, B.W.; Ilinskaya, A.N.; Dobrovolskaia, M.A. Updated method for in vitro analysis of nanoparticle hemolytic properties. In Methods in Molecular Biology; McNeil, S.E., Ed.; Springer: New York, NY, USA; Dordrecht, The Netherlands; Heidelberg, Germany; London, UK, 2018; pp. 91–102. [Google Scholar]
- Onpattro dosing and preparation information. Available online: https://www.onpattrohcp.com/sites/default/files/pdfs/ONPATTRO-Dosing-and-Preparation-Guide.pdf (accessed on 28 December 2019).
- Cheng, T.-L.L.; Cheng, C.-M.M.; Chen, B.-M.M.; Tsao, D.-A.A.; Chuang, K.-H.H.; Hsiao, S.-W.W.; Lin, Y.-H.H.; Roffler, S.R. Monoclonal antibody-based quantitation of poly(ethylene glycol)-derivatized proteins, liposomes, and nanoparticles. Bioconjugate Chem. 2005, 16, 1225–1231. [Google Scholar] [CrossRef]
- Su, Y.C.; Chen, B.M.; Chuang, K.H.; Cheng, T.L.; Roffler, S.R. Sensitive quantification of PEGylated compounds by second-generation anti-poly(ethylene glycol) monoclonal antibodies. Bioconjugate Chem. 2010, 21, 1264–1270. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds NSSL-MPS are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treat-ment # | Dose Liposomal MPS | R2 | Terminal Half-Life | Cmax | Cmax/Dose | AUClast | AUClast/Dose | Clobs | MRTlast |
|---|---|---|---|---|---|---|---|---|---|
| mg | h | µg/mL | µg/mL/mg | h*µg/mL | h*µg/mL/mg | mL/h | h | ||
| 1 | 50 | 0.98 | 14 | 35 | 0.7 | 1262 | 25 | 38 | 25 |
| 2 | 100 | 0.98 | 17 | 68 | 0.7 | 2163 | 22 | 46 | 27 |
| 3 | 150 | 0.998 | 19 | 108 | 0.7 | 3922 | 26 | 35 | 27 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bavli, Y.; Chen, B.-M.; Roffler, S.R.; Dobrovolskaia, M.A.; Elnekave, E.; Ash, S.; Barenholz, Y.; Turjeman, K. PEGylated Liposomal Methyl Prednisolone Succinate does not Induce Infusion Reactions in Patients: A Correlation Between in Vitro Immunological and in Vivo Clinical Studies. Molecules 2020, 25, 558. https://doi.org/10.3390/molecules25030558
Bavli Y, Chen B-M, Roffler SR, Dobrovolskaia MA, Elnekave E, Ash S, Barenholz Y, Turjeman K. PEGylated Liposomal Methyl Prednisolone Succinate does not Induce Infusion Reactions in Patients: A Correlation Between in Vitro Immunological and in Vivo Clinical Studies. Molecules. 2020; 25(3):558. https://doi.org/10.3390/molecules25030558
Chicago/Turabian StyleBavli, Yaelle, Bing-Mae Chen, Steve R. Roffler, Marina A. Dobrovolskaia, Eldad Elnekave, Shifra Ash, Yechezkel Barenholz, and Keren Turjeman. 2020. "PEGylated Liposomal Methyl Prednisolone Succinate does not Induce Infusion Reactions in Patients: A Correlation Between in Vitro Immunological and in Vivo Clinical Studies" Molecules 25, no. 3: 558. https://doi.org/10.3390/molecules25030558
APA StyleBavli, Y., Chen, B. -M., Roffler, S. R., Dobrovolskaia, M. A., Elnekave, E., Ash, S., Barenholz, Y., & Turjeman, K. (2020). PEGylated Liposomal Methyl Prednisolone Succinate does not Induce Infusion Reactions in Patients: A Correlation Between in Vitro Immunological and in Vivo Clinical Studies. Molecules, 25(3), 558. https://doi.org/10.3390/molecules25030558