1. Introduction
Over the last 70 years, organoboron compounds have dramatically changed the landscape of organic chemistry through a wide range of valuable and indispensable synthetic applications, e.g., cross-coupling chemistry, photochemistry, and alkylboration, which has led to new constructions of C–C and C–heteroatom bonds [
1,
2,
3,
4,
5,
6]. Additionally, boron functionality is dispersed in natural products and synthetic drugs. Natural products include the antibiotics aplasmomycin, boromycin, and tartolon B, in which boron functionality appears as a borate complex [
7,
8,
9,
10,
11]. Drugs such as Tavaborole and Bortezomib also incorporate boron functionality [
12,
13,
14]. In terms of these aspects, multiborylated compounds, e.g.,
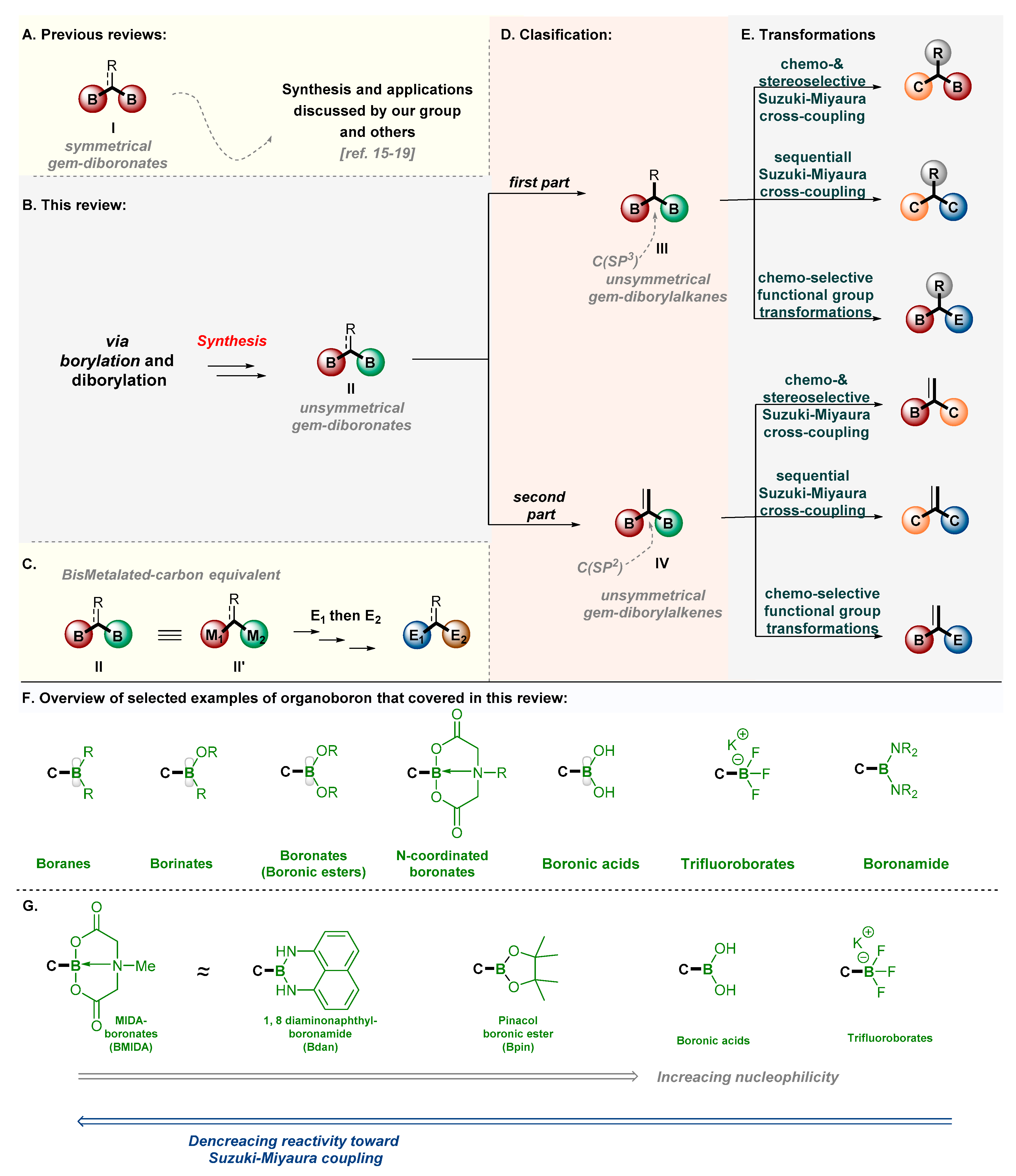
I, are even more attractive due to their synthetic versatility and chemical stability, which allow for the selective synthesis of multifunctionalized molecules (
Scheme 1) [
15,
16,
17,
18,
19,
20]. Recently, our group [
16,
17] and other research groups have mainly reviewed the new class of symmetrical 1,1-diboranes,
I (bis-metallated reagents) (
Scheme 1A,C), their preparation, and their application in organic synthesis for forming organoboranes and bifunctionalized products [
15,
16,
17,
18,
19,
20]. In contrast, the unsymmetrical 1,1-bis(boranes),
II, have rarely been reviewed, despite their importance in the chemo- and stereoselective building of C–C and C–X bonds. These classes of compounds mainly include 1,1-bis(boron) (
III) and alkenyldiboronates (
IV) (
Scheme 1B). These compounds offer two distinct boron substituents for ideal sites as well as stereoselective synthetic strategies to obtain stereo-controlled alkanes [
16,
17] and multisubstituted olefins, which are of major importance in organic synthesis [
21,
22,
23,
24,
25,
26]. Most importantly, 1,1-bis(boryl)alkenes have been utilized in the synthesis of the anticancer agent tamoxifen [
27].
However, unsymmetrical 1,1-bis(boryl)alkanes (III) and -alkenes (IV) represent a unique class of boron compounds with a tunable reactivity for boron site-selective functionalization. This review will cover synthetic approaches for forming unsymmetrical 1,1-bis(boron) species as well as for deriving chemo- and stereoselective transformations for the synthesis of complex molecular structures. It is hoped that this review will provide useful knowledge for scientists seeking to discover new things about unsymmetrical 1,1-bis(boryl)alkanes and -alkenes in organic synthesis. The synthesis of these unsymmetrical 1,1-bisboryls includes mainly the hydroboration of alkynes and alkenes. Unsymmetrical 1,1-bis(boryl)alkanes and -alkenes are mainly utilized in chemo- and stereoselective (sequential) cross-coupling reactions and other chemoselective transformations. In order to simplify things for the readers, we classified this review article into two categories based on the hybridization of the carbon attached to the 1,1-bis(boron) functionalities:
- (i)
Unsymmetrical sp3-centered type-III; and
- (ii)
Unsymmetrical sp2-centered type-IV.
In both cases, we discuss the synthesis and utility of the unsymmetrical 1,1-bis(boron) species in organic chemistry.
Before going into the discussion, let us first introduce the characteristics of the C–boron bond in terms of reactivity, on the basis of their substitution pattern. It is well known that the organoboron groups (see
Scheme 1F) [
28] have different reactivities due to the steric and electronic properties of the Lewis acidic boron moiety, which can be easily tuned just by changing the substitution around the organoboron group, which allows for diverse reactivity as a stoichiometric reagent and as a catalyst. For example, boron reagents behave like nucleophilic component is a Suzuki–Miyaura (SM) cross-coupling reaction; therefore, their reactivity depends on their nucleophilicity. Nucleophilicity scale of organoborons (
Scheme 1G) shows that organoboronic acids and organo-trifluoroborates have greater nucleophilicity than do MIDA-boronates (MIDA =
N-methyliminodiacetic acid), (1,8-diaminonaphthyl boronamide, and boronic esters (as described in
Scheme 1G) [
28]; thus, MIDA-boronates have less reactivity toward the SM cross-coupling reaction [
3]. Therefore, the two different organoboron groups in 1,1-bis(boron) species tend to show two different reactivities and perform in a chemoselective manner.
2. Unsymmetrical sp3-Centered 1,1-Bis(boryl) Compounds: Synthesis and Applications
In 2011, Hall and coworkers elegantly reported the first preparation of optically enriched unsymmetrical 1,1-bis(boryl) compounds (
2) with excellent enantioselectivity via a copper-catalyzed asymmetric conjugate borylation of
-boronylacrylates (
1) with pinacolatodiborane (
Scheme 2A) [
29]. The obtained enriched unsymmetrical 1,1-bis(boryl) compound (
2) was then treated with KHF
2, forming the corresponding trifluoroborate salt
3. Of note, the Bpin (Bpin = B-pinacolato) group in
2 selectively underwent a trifluorination reaction over Bdan (Bdan = B-1,8-diaminonaphthalene) functionality, most likely due to the higher Lewis acidity available for activation of the p-orbital of the boron in Bpin compared to the lower Lewis acidity of the p-orbital of the boron in Bdan, which is aromatically busy (
Scheme 2) [
30]. An X-ray crystallographic analysis confirmed the conjugate borylation product of
2a (R = Me); this provided us with a better understanding of the physical properties of these compounds.
In addition, the trifluoroborate salt
3 was stereo-specifically cross-coupled with aryl bromide in the presence of palladium catalyst and XPhos (2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl) as a ligand: these salts formed the corresponding arylated product
4 in high yield and with good-to-excellent enantioselectivity (88–99% ee). The coordination of the carbonyl oxygen with the boron atom (see
5) and the stabilization provided by the second boronyl unit in the α-B–Pd(II) complex are thought to facilitate the transmetallation process and the cross-coupling reaction. Interestingly, in this cross-coupling reaction, an inversion of the stereochemistry was observed. The rationale for this inversion was presented through a transmetallation reaction that took place via transition state
5, which was responsible for inverting the stereochemistry (
Scheme 2B) [
29].
In 2015, this group expanded its investigations into palladium (Pd(dba)
2) and the XPhos-catalyzed cross-coupling reactions of optically enriched unsymmetrical 1,1-bisboron compound (
6), using aryl bromides in a chemo- and stereoselective manner for the synthesis of the benzylic secondary alkyl boronates
7a–
h, which had a stereochemistry inversion [
31]. They also noted an important observation: by increasing the Lewis basicity of a carbonyl group from an ester to a Weinreb amide, the cross-coupling reaction was more feasible. The coordination of the carbonyl oxygen with the boron atom, together with the stability of the second boron unit in the α-B-Pd(II) intermediate, helped to overcome a difficulty at the transmetallation step. This reaction showed tolerance to a wide range of aryl coupling partners, with excellent chemoselectivity over the Bdan functionality (
Scheme 3).
These enantioenriched cross-coupled products were subjected to various synthetic transformations, as presented in
Scheme 4. The Bdan motif was treated with acid, affording the hydrolyzed product, which further underwent esterification with pinacol, yielding compound
8. Next,
8 was converted into its corresponding potassium trifluoroboronate salt,
9; finally, it underwent a cross-coupling reaction with aryl bromide, affording compound
10 (with an inversion in its stereochemistry). The sequence of two cross-couplings, from
6 to
10, was followed by double inversions of stereochemistry. Finally, amide
10 was transformed into ketone
11 upon a reaction with ethylmagnesium bromide (
Scheme 4) [
31].
Yun and coworkers reported the use of copper-catalyzed hydroboration of borylalkenes to synthesize unsymmetrical 1,1-bis(boryl)alkanes in a high regioselective and enantioselective version [
32]. Various alkene substitutions, such as aryl, primary, and secondary alkyls, were well tolerated in this reaction and provided excellent regioselectivity (
Scheme 5).
Initially, the reaction mechanism involved the reaction of copper
tert-butoxide with pinacolatoborane, affording
LCu-H species
16, which then underwent a regio- and enantioselective addition reaction into borylalkene
12, yielding the chiral organocopper intermediate
17. Next, this intermediate
17 underwent stereoretentive transmetallation with one more equivalents of pinacolatoborane, finally yielding the desired product
14 and regenerating the catalyst (
LCu-H)
16 for another catalytic cycle (
Scheme 5).
These chiral unsymmetrical 1,1-bis(boryl)alkanes were then transformed into allenylboronate
18 in a homologation reaction with 3-chloro-3-methylbut-1-ynyllithium, with almost no erosion of its enantioselectivity. Similarly, the Suzuki–Miyaura cross-coupling (SMCC) of
19 afforded the corresponding arylated product
20 in a low yield and with little loss of its enantioselectivity (but with retention of its configuration) (
Scheme 6) [
32].
In 2015, Fernandez’s research group reported a transition metal-free approach for the synthesis of unsymmetrical 1,1-bis(boron) compounds from diazo compounds,
23, which were obtained in situ from aldehydes as well as from cyclic and noncyclic ketones (
21) via in situ-generated sodium salts of tosylhydrazones followed by treatment with sodium hydride (
Scheme 7) [
33]. This method also provides a wide range of unsymmetrical 1,1-bis(borane) compounds from aldehydes and ketones, with good isolated yields. However, it is strictly limited to aliphatic carbonyls.
In their work, they provided a rationalized mechanism for this method via transition state
26 (based on DFT-Density Functional Theory calculations). The reaction involved the heterolytic cleavage of the mixed diboron reagent
25 and the formation of geminal C–Bpin and C–Bdan bonds via a concerted–asynchronous mechanism, as shown in
Scheme 7.
To explore the diastereoselective 1,1-diboration of diazo compounds, Fernandez’s group substituted cyclohexanones for in situ-formed diazo-compounds and promoted insertion into the pinB–Bdan bond (
Scheme 8) [
33]. When they carried out a reaction with 4-substituted cyclohexanones (
28a) under optimized reaction conditions, they observed reasonable diastereo-selectivity. An X-ray analysis of
29a showed that the Bdan and CF
3 groups were situated at the 1,4-diequitorial (trans) positions, as shown in
Scheme 8. Under optimized conditions, the 3-Ph-cyclohexanone
28b afforded the 1,3-diequitorial (cis)-substituted isomer as the major diastereomer
29f (64%). Interestingly, under a similar reaction condition, the 2-Me-cyclohexanone
31 afforded excellent diastereo-selectivity (
32a/
32b), favoring the Bdan and Me groups with a 1,2-diequitorial (trans) configuration. This high diastereo-selectivity was expected because the 2-position substituent can directly influence the reaction center.
The origin of diastereo-selectivity was explained through DFT analysis by taking the example of the 4-(trifluoromethyl)cyclohexanone
28a, as shown in
Scheme 9. After the reaction of hydrazide and NaH with compound
28a, Fernandez’s group considered in situ-generating two possible chair conformations (
28aNeq and
28aNax) that were different in terms of their CF
3 substituent arrangement: the equatorial conformer (
28aNeq) was 1.7 kcal/mol lower than the axial conformer (
28aNax). Each confirmation could attack the pinB–Bdan substrate through its two diastereo-faces. When the conformer
28aNeq attacked pinB–Bdan through its two diastereomeric faces, the observed computed free energy barriers for the Bdan equatorial and Bdan axial positions were 33.9 and 38.9 kcal/mol, respectively (
Scheme 9, left side). Thus, the lowest energy path led to a major diastereomer with Bdan and a CF
3 substituent in the 1,4-diequitorial (trans) of each
29a. The high energy (~5.0 kcal/mol) in the Bdan axial approach was due to destabilized interactions between 1,3-diaxial and cyclohexane. Similarly, the conformer
28aNax also exhibited a low energy barrier in the case of the Bdan equatorial approach, as opposed to the Bdan axial approach (
Scheme 9, right side).
The energy difference between the two Bdan equatorial approaches was not too large in the two conformers (
28aNeq and
28aNax), with the expected non-negligible formation of conformer
30a with an observed diastereomeric ratio of 70/30 for
29a/
30a [
33].
Interestingly, 1,1-bis(boryl)alkanes can behave as catalysts, too. Piers’s borane
V [HB(C
6H
5)
2] precatalyzed the hydroboration of terminal and internal alkynes for the synthesis of
E-alkenyl pinacol boronic ester
36, and excellent selectivities were reported by Stephan and coworkers in 2016 [
34]. In this hydroboration, they found that unsymmetrical 1,1-bis(borane)
34 catalyzed the reaction. An independent reaction of Piers borane
V [HB(C
6H
5)
2] with phenylacetylene afforded the corresponding
E-alkenyl boronic ester
33a, which, upon additional treatment with pinacol boranes, yielded the regioselective stereogenic unsymmetrical 1,1-bis(borane)
34 (
Scheme 10). Actually, 1,1-bis(borane)
34 acted as an electrophilic catalyst for the hydroboration of alkyne into alkene in the presence of HBpin (pinacolborane). To support this, the electrophilic boron center of
34 was confirmed through an X-ray diffraction analysis of the
tert-butylisonitrile adduct
35.
The proposed mechanism for the unsymmetrical 1,1-bis(borane)
34 catalyzing the hydroboration of alkynes for the synthesis of
E-alkenyl boronic ester
36 is shown in
Scheme 10. In this mechanism, unsymmetrical 1,1-bisboranes
34 acts as a Lewis acid catalyst, which activates alkyne to form complex
VI: then, HBpin reacts with complex
VI in a concerted
syn-1,2-hydroboration manner (complex
VII) to afford the hydroborylated product
36 and regenerate the unsymmetrical 1,1-bis(borane)
34 [
34].
In 2019, Sharma research group reported a novel and concise method for the synthesis of a wide range of MIDA (MIDA =
N-methyliminodiacetic acid) acylboronates (
41) via the chemoselective oxidation of 1,1-bisboranes (
39) (
Scheme 11) [
35]. MIDA acylboronates are synthetically challenging; however, they can be utilized as powerful building blocks for bioorthogonal amide formation and protein ligation.
First, Sharma prepared unsymmetrical 1,1-bis(borane) products (
39) from symmetrical diboranes (
38) by heating MIDA and triethylorthoformate (
Scheme 11). The mechanism underlying this selective desymmetrization is still unclear. However, steric considerations could be among the reasons for this selectivity.
Next, Sharma chemoselectively oxidized pinacolate boran functionality over the MIDA boronate of unsymmetrical 1,1-bis(borane) products (
39a–
b) to obtain α-hydroxymethyl MIDA boronates (
40) with good yields (
Scheme 12). Thereafter, these α-hydroxymethyl MIDA boronates (
40) were successfully oxidized into MIDA acylboronates (
41) using Dess–Martin periodinane (DMP), with moderate to good yields (
Scheme 12).
Then, Sharma successfully applied a similar strategy for the synthesis of unique α, β-unsaturated MIDA acyl boronates (
44) with an acceptable yield (
Scheme 13).
Additionally, Sharma applied the same strategy to the unsymmetrical diborylmethane
39c to obtain hydroxymethyl MIDA
45. Interestingly, the DMP oxidation of hydroxymethyl MIDA afforded acetoxy MIDA boronate
46 instead of formyl MIDA boronate (
Scheme 14) [
35].
In the same year, the Masarwa research group reported a late-stage desymmetrization of symmetrical 1,1-bis(boryl)alkanes via nucleophilic trifluorination while constructing unsymmetrical 1,1-bis(boryl)alkanes bearing trifluoroborate salts (
Scheme 15) [
36]. This method was tolerable within a wide range of substrate scopes, with good to excellent yields. Most interestingly, this method did not need any column purification, as the product was obtained upon crystallization. Our group proposed a mechanism to account for this desymmetrization methodology: First, nucleophilic fluoride attacks the vacant
p-orbital of one of the boron centers in the symmetrical diborane 38 and generates monofluorinated compound I (step 1), which is more electrophilic than the parental symmetrical diborane 38. Hence, it forces the second fluorination (step 2) and then the third fluorination (step 3) at the boron center shown in
Scheme 15. Consequently, the newly generated BF
3 moiety develops a partial negative charge on the fluoride, and the fluoride may stabilize the flanking Bpin group through a fluoride bridge, as shown in 47′, which helps to prevent a subsequent attack by fluoride.
Interestingly, this method exhibited excellent diastereo-selectivity when the reaction was performed on the 1,1-bis(boryl)substituted cyclopropanes
38a–c (
Scheme 16). This diastereo-selectivity has been confirmed using 2D-NMR spectroscopic methods. From these results, it can be seen that the diastereo-selectivity mechanism underlying the reaction involves the selective nucleophilic trifluorination of boron, which occupies the less hindered side of cyclopropane.
These unsymmetrical 1,1-bis(boryl)alkanes, bearing a trifluoroborate group, were utilized for various selective functionalizations. For example, the simple hydrolysis of the trifluoroborate group afforded the 1,1-bis(borane) products
48a–c, which has a boronic acid moiety (
Scheme 17A). Additionally, compound
47 treated with diamines and diols as coupling partners, yielded the unsymmetrical 1,1-bis(borane) products
49a–c (
Scheme 17B).
Finally, trifluoroborylated unsymmetrical 1,1-bis(boryl)alkanes were also utilized for cross-coupling reactions. Intermolecular palladium(II) catalyzed a cross-coupling reaction with aryl bromide, which afforded arylated product
50a, which had excellent yield (
Scheme 18A). Similarly, an intramolecular cross-coupling reaction of 2-bromo-substituted phenyl substrates under the same reaction conditions yielded the cyclic product
50b, with a 90% yield (
Scheme 18B) [
36].
In 2017, Erker and coworkers reported that Lewis acid induced a cyclopropyl acetylene rearrangement for the synthesis of unsymmetrical 1,1-bis(borane) [
37]. In this synthesis, when cyclopropyl acetylene
51 was treated with two equivalents of Piers’s borane
V [HB(C
6F
5)
2], it resulted in the formation of substituted α-boryl-tetrahydroborole
53a-cis and
53b-trans in a 7:1 ratio (
Scheme 19A): this was confirmed by in situ NMR spectroscopic studies. Furthermore, they confirmed these structures using X-ray diffraction and found that the pair of substituents at C1 and C4 (in terms of stereochemistry) differed structurally from the
cis and
trans isomers.
The mechanism underlying this reaction involves the sequential hydroboration of cyclopropyl acetylene
51 with 2.0 eq. of Piers’s borane
V, affording 1,1-bis(boryl) compound
VIII. Then, one of the frustrating borons of product
VIII acts as a Lewis acid and induces cyclopropyl ring-opening and the hydride sequence. Then, aryl 1,2 shifts to form borole
53 (
Scheme 19B). The stereochemical outcome of the reaction largely depends on the least sterically hindered pathway, leading to the major diastereomer
cis-borole.
The cyclic 1,1-bis(boryl) contains two Lewis acidic boron sites: they can coordinate only as a frustrated Lewis pair (FLP) or can serve as a dihydrogen splitting reagent. Treatment of a 1:1 ratio of
cis-borole compound and tri-
tert-butylphosphine (as a Lewis base with 2.0 bar of hydrogen in pentane solution) resulted in the precipitation of product
cis-
54a. Similarly, upon treatment with carbon dioxide instead of hydrogen, a new six-membered cyclic ring was formed, in addition to the borole ring
cis-
54b (
Scheme 20).
Interestingly, Erker and coworkers interconverted
cis-borole
53a into
trans-isomer
53b by treating it with a catalytic amount of TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl). This reaction followed reversible H-abstraction at the activated C
1 position of the heterocycle. After they subjected
trans-borole to a similar FLP reaction, they obtained products
54c and
54d in good yields (
Scheme 21) [
37].
In the same year, Erker and coworkers reported that cyclic 1,1-bis(borane)
53 catalyzed the hydroboration of
N-methylindole. Here, 1,1-bis(borane) compounds were utilized as effective catalysts for C–H bond activating the borylation of
N-methylindole with catechol borane. This reaction afforded 3-boryl-
N-methylindole with a 59% yield and
N-methylindoline with a Lewis pair adduct (with HBcat). Furthermore, this adduct, upon treatment with the same catalyst for several hours, yielded a 5-boryl-
N-methylindoline product through the evolution of molecular hydrogen (
Scheme 22) [
38].
3. Unsymmetrical sp2-Centered 1,1-bis(boron) Compounds: Synthesis and Applications
In 2007, Chirik and coworkers developed a cobalt that catalyzed the 1,1-diboration of readily available terminal alkyne
58 with an unsymmetrical (pinB–Bdan) diboron reagent for the synthesis of stereoselective trisubstituted 1,1-bis(boryl)alkenes (
59a–d), with good yields (
Scheme 23) [
39].
The mechanism proposed by Chirik’s group involved the initial formation of cobalt acetylide (X), which, upon reacting with pinacolborane, yielded compound XI, which had more Lewis acidic boron substituent (Bpin). This could transfer to the alkyne, and the resulting alkynyl−BPin cobalt complex (XII) underwent syn-borylcobaltation, selectively affording XIII, which finally produced the stereoselective alkene 59.
Taking advantage of the different chemical reactivities of two boron moieties (Bpin, Bdan) in 1,1-unsymmetrical bis(boryl)alkenes (
59), an SMCC reaction was carried out with aryl iodides to afford corresponding (
Z)-alkenes (
60), which had an extended conjugation and good yields. Interestingly, they observed that the cross-coupling took place selectively at the Bpin moiety over Bdan (
Scheme 24) [
39]. The whole methodology, which includes the 1,1-diboration of alkynes (
Scheme 23) and the cross-coupling reaction of 1,1-unsymmetrical bis(boryl)alkenes (
Scheme 24), represents a formal 1,1-carboboration of hept-1-yne with Ar–Bdan [
40,
41,
42].
In 2018, the Molander group reported the borylation of 3-bromo-2,1-borazaronaphthalenes (
61) with boronic acid pinacol esters, affording 3-boryl-2,1-borazaronaphthalene
62a–f (1,1-unsymmetrical bis(boryl)alkenes) [
43]. These borazaronaphthalenes (
62) also exhibited an umpolung character in cross-coupling reactions. This method allows for the synthesis of a wide range of heterocycles with different substituents at the boron center, with electron-rich and electron-poor aryl and heteroaryl groups and up to an 83% yield (
Scheme 25A). Next, compound
62 was converted into organotrifluroborate salt (
63) by treating it with commercially available KHF
2 as a fluoride ion source (
Scheme 25B).
Later, they also utilized the bis-boryl compounds
62a and
63d for a palladium-catalyzed cross-coupling strategy with a variety of aryl halides containing an electron-withdrawing group or an electron-donating group, which yielded the corresponding coupling products
64a–h (
Scheme 26) [
43].
In 2014, the Nishihara research group reported the platinum-catalyzed diborylation of 1-phenylethynyl MIDA boronate
65a with bis(pinacolato)diboron, affording stereoselective 1,1,2-triboryl-2-phenylethene
66a with an 86% yield [
44]. Under similar reaction conditions, they also extended diboration with the aliphatic 1-alkynyl MIDA boronate
65b, yielding the 1,1,2-triboryl-2-hexylethene
66b, as shown in
Scheme 27A. Furthermore, 1,1,2-triboryl-2-phenylethene,
66, was successfully applied to chemoselective palladium-catalyzed Suzuki−Miyaura coupling with aryl halides bearing electron-donating and electron-withdrawing groups. Under optimized reaction conditions, they synthesized a library of synthetically useful 1,1-bis(boryl)olefins,
67a–f, with up to 91% yields (
Scheme 27B).
To determine the
(Z)-configuration of the chemoselective arylated product
67, they carried out Suzuki−Miyaura coupling of 1,1,2-triboryl-2-phenylethene
66a and iodobenzene to afford the arylated unsymmetrical 1,1-bis(boryl)-2,2-diphenylethene
67g, with 82% yield. Then, transformation of the BMIDA group into Bpin afforded the symmetrical 1,1-bis(boryl)-2,2-diphenylethene
68a at a 98% yield, which matched with earlier reported spectroscopic data. This experimental result clearly suggests that selective cross-coupling takes place at the Bpin group, which is geminal to the aryl moiety (
Scheme 28).
Furthermore, they changed the electrophile ArI into BnCl (1.5 equiv) for an SMCC reaction with compound
66a and observed only one isomer of unsymmetrical 1,1-bis(boryl) alkene
67h, with a moderate yield (
Scheme 29) [
44].
In 2019, the Tsuchimoto group reported that the Pt-catalyzed diboration of alkyne had terminal Bdan with pinB–Bpin, resulting in the 1,1,2-triboryalkene
66c, which had perfect stereoselectivity without suffering direct activation by the platinum complex [
45]. Compared to other reports on the diboration of alkynes [
46,
47] with pinB–Bpin, this method afforded an excellent yield (
Scheme 30A).
In 2019, utilizing the
tetrasubstituted triborylalkene
66c, for the first time the Tsuchimoto group successfully synthesized
tetrasubstituted aryl alkenes (
72) through sequential SMCC reactions. In the triborylalkene
66c, the C
sp2-B bond is inactive; thus, it is very unreactive under SMCC conditions. Of the remaining two C
sp2-B bonds, one is at the transposition of the aryl group, which can possibly increase the nucleophilicity of the boron atom through electron flow: it reacts more facilely than does the other under Pd(II) SMCC conditions. In the first SMCC reaction, 4-iodobenzotrifluoride was treated under ligand-free conditions, affording
69 in very good yield (with regioselectivity). In the second SMCC arylation, 4-iodoanisole reacted preferentially with C
sp2-Bpin over C
sp2-Bdan, affording
70. Since Bdan is unreactive toward SMCC, it was converted into Bpin under acidic conditions with pinacol. In the final sequence of SMCC arylation, 4-iodobenzonitrile was treated with C
sp2-Bpin to produce the tetra-arylalkene
72. Importantly, during all of the SMCC reactions observed, there was only one stereoisomer with a high yield (
Scheme 30B) [
47].
In 2008, the Walsh group described the reaction of stable pinB-substituted alkynes (
65) with dicyclohexyl borane to afford unsymmetrical 1,1-bis(boryl)alkene species (
73) [
48]. Compound
73 was not isolated and exhibited two peaks at 30 ppm and 80 ppm in a crude
11B-NMR spectrum. In addition, crude
1H-NMR of
73 exhibited only one isomer in the hydroboration reaction. Later on, they successfully utilized a 1,1-bis(boryl)alkane species for chemoselective transmetallation with an organozinc reagent (in place of Cy
2B) to afford the boron/zinc heterobimetallic reagent
74; they then added it to an aldehyde to obtain pinB-substituted
(E)-allylic alcohols (
75) with good yields. By utilizing this method, they synthesized a library of secondary alcohols via hydroboration and transmetallation, followed by aldehyde treatment with pinB-substituted alkynes (
65) (
Scheme 31) [
48].
In 2013, the Braunschweig group demonstrated a photolytic approach to creating a new class of three-membered borirenes using an aminoboryl complex [(OC)
5Cr=B=N(SiMe
3)
2] in the presence of a series of mono- or bis(boryl) alkynes: 1-phenyl-2-bis-(dimethylamino)borylethyne, bis{bis-(dimethylamino)boryl}ethyne, and 1-trimethylsilyl-2-bis-(dimethylamino)borylethyne [
49]. As depicted in
Scheme 32A, aminoboryl complex
76 was irradiated with alkyne
77 at room temperature to produce the desired unsymmetrical 1,1-bis(boryl) alkenes (i.e., iminoboranes) (
78a–c) (
Scheme 32A).
Generally, aminoboranes (i.e., compound
80) do not follow quaternization, since the
p-orbital of boron is filled by the π-basic amino group. The utility of iminoboranes (
78) was established by the synthesis of quaternary aminoboranes (
79): these kinds of aminoboranes are very rare, according to the literature (
Scheme 32B). The iminoborane
78 did not react with DMAP (4-(dimethylamino)pyridine), pyridine, PCy
3, or PMe
3 even at 80 °C, whereas it reacted with IMe (IMe = 1,3-dimethyl-2,3-dihydro-1
H-imidazole) at ambient temperature to produce quaternization in the endocyclic boryl group. Interestingly, the Braunschweig group observed that there was no quaternization of the exocyclic boryl groups even upon the addition of an excess of IMe (
Scheme 32B).
The dequarternization of borirenes also afforded the parental unsymmetrical 1,1-bis(boryl)alkanes and alkenes (
80) in the presence of B(C
6F
5)
3 (
Scheme 32C) [
49].
In 2013, Weber et al. reported the hydroboration of 2-alkyl- and 2-aryl-ethynyl-1,3,2-benzodiazaboroles with dicyclohexylborane (DCB) under metal-free conditions at room temperature, affording the
cis-1,1-bis(boryl)alkene
82 as a major regioisomer, with quantitative yields [
50]. Interestingly, they also observed the hydroboration of 2-silylethynyl-1,3,2-benzodiazaborole with DCB, which afforded 1,1-bis(boryl)alkene (
82) as a minor regioisomer and
trans-1,2-bis(boryl)alkene as a major regioisomer (
83), with quantitative yields (
Scheme 33) [
50].
In 2015, the Erker group reported the preparation of 1,2,5-trisubstituted boroles containing 1,1-bis(boryl) groups from the bis(ethynyl)borane
84 and B(C
6F
5)
3 via a 1,1-carboboration sequence followed by di-π-borane rearrangement [
51]. Initially, the reaction of bis(ethynyl)borane with B(C
6F
5)
3 resulted in 44:56 mixtures of the 1,1-carboboration product
Z-85 and the trisubstituted borole
87 via
86, which was isolated after treatment with pyridine (
Scheme 34A).
Under thermal conditions, a mixture of
Z-85 and
87 afforded the [4 + 2] cycloaddition product
88, which was formed after the dimerization of reactive borole
87. The Erker group observed that the unreacted
Z-85 remained under thermal conditions (
Scheme 34B). Since compound
Z-85 was not isomerized thermally, they tried photolysis. Under photolysis,
Z-85 isomerized to
E-85, which can easily be converted into
87, as shown in
Scheme 34A, and compound
87 also cleanly isomerized to the new trisubstituted borole
90 via di-π-borane rearrangements (two formal 1,3-boron migrations) followed by ring opening. Consequently, the photolysis of a solution of the
Z-85 +
87 mixture resulted in 1,1-bis(boryl)alkanes and alkenes (
90) (1,2,5-trisubstituted boroles) in quantitative conversion, which was isolated after treatment with pyridine (
Scheme 34B) [
51].
In 2018, the same group prepared a 1,1-bis(boryl)alkene by reacting tetraaryldiborane(4)
92 with 1-pentyne (
Scheme 35) [
52].
Under thermal conditions, diborane(4) underwent 1,2-migration in its acetylenic hydrogen along the alkyne π-bond to afford 1,1-bis(boryl)alkene
93, along with
94, in a 42:37 ratio. During the reaction, one B–B bond was cleaved, and new C–B bonds were formed (
Scheme 35) [
52].


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


































