Application of Halogen Bonding to Organocatalysis: A Theoretical Perspective



Abstract
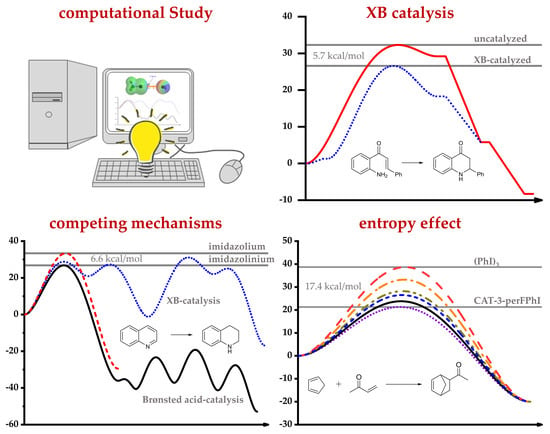
:
1. Introduction
2. Computational Methods for XB Organocatalysis
3. Computational XB Organocatalysis Studies
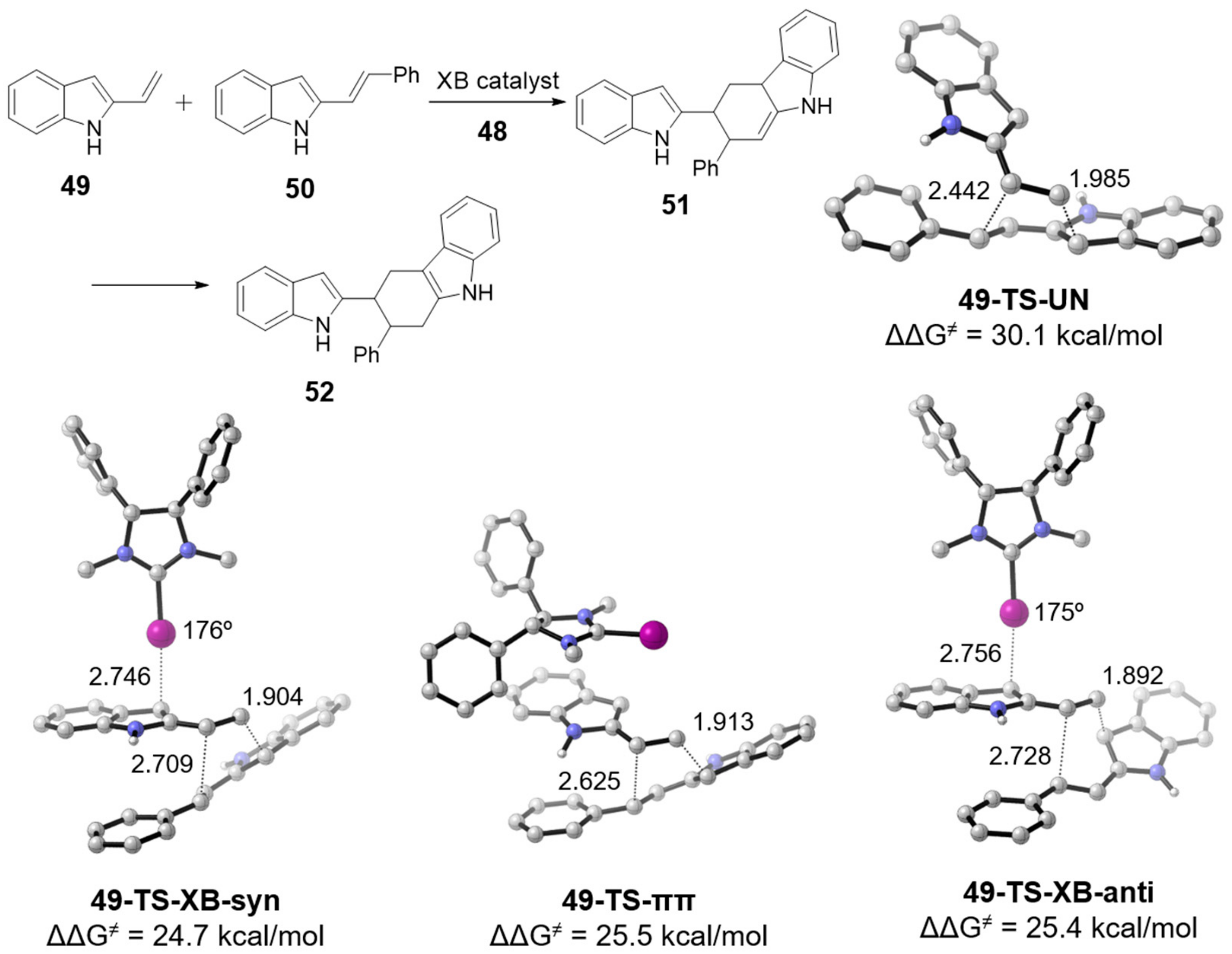
3.1. Transition State and Binding XB Complex Studies
3.2. Neutral Halogen Bond Catalysts
3.2.1. Computational Study of XB-Catalyzed Hydrocyanation of Imines
3.2.2. Computational Design of Neutral XB Catalysts
3.2.3. I2 as an XB Catalyst in Organic Reactions
3.2.4. I2-Catalyzed iso-Nazarov Cyclization of Conjugated Dienals
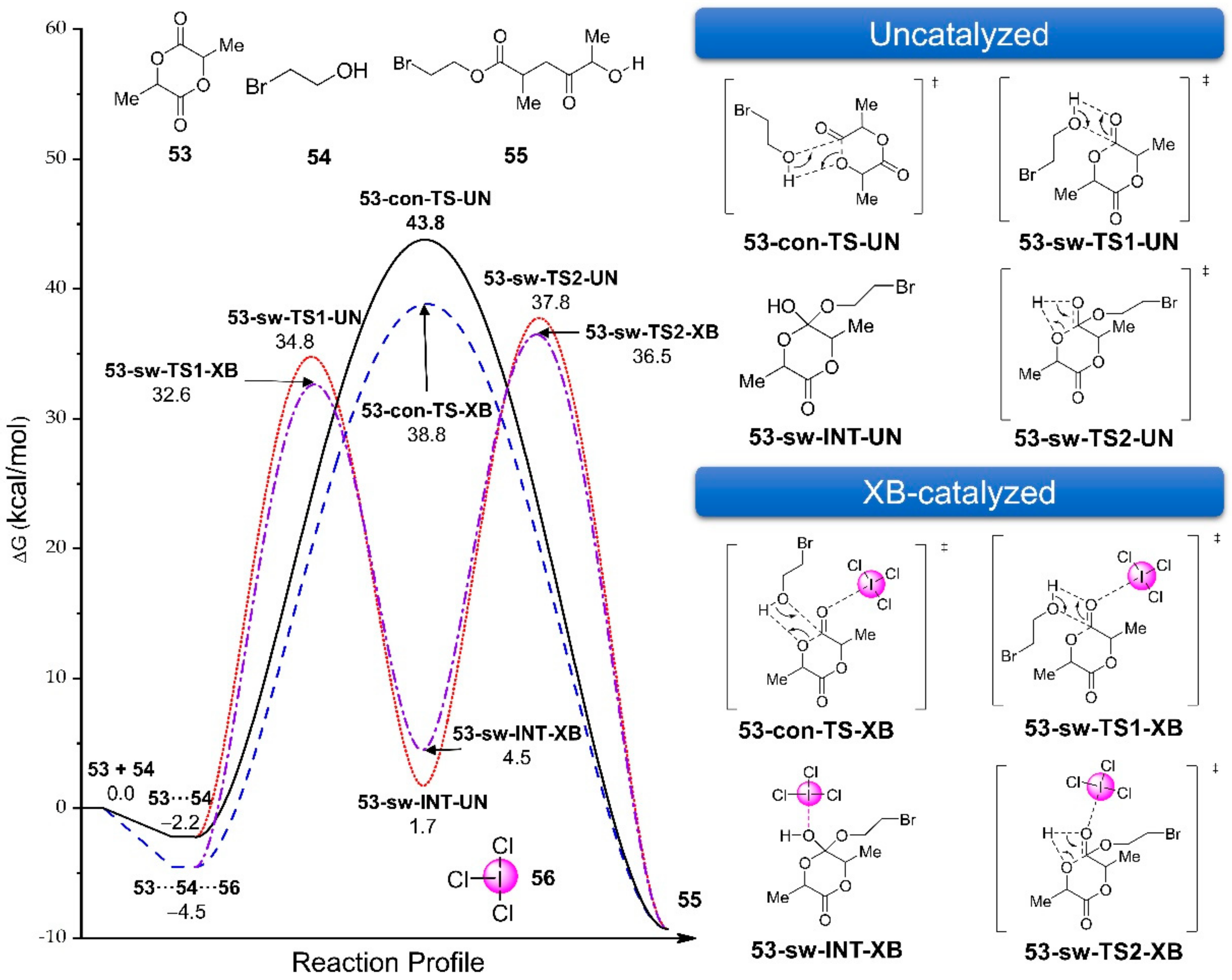
3.2.5. Dihalogen-Catalyzed Michael Addition Reactions
3.2.6. XB in Metal Acetate-Catalyzed Halolactonization
3.3. Cationic XB Catalysts
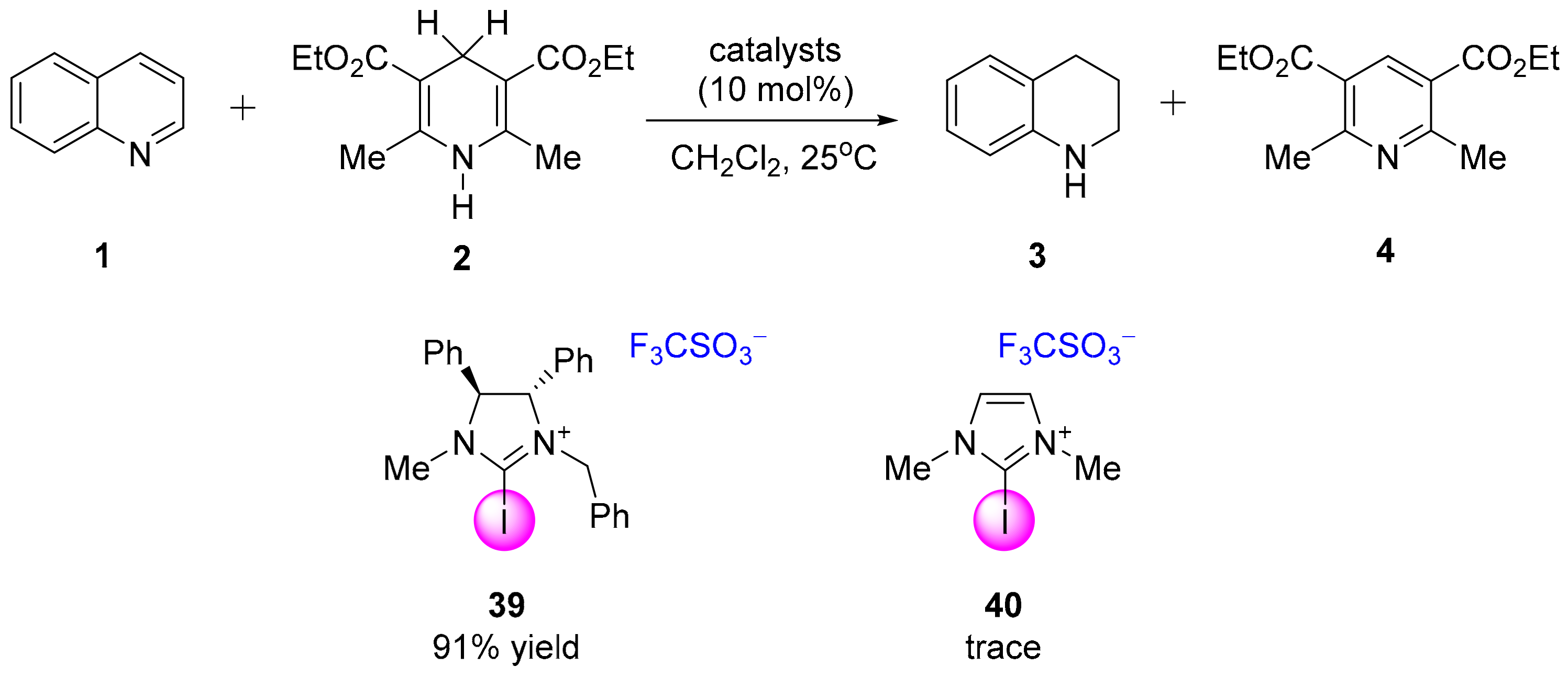
3.3.1. XB-Catalyzed Reduction of Quinolines by Hantzsch Ester
3.3.2. XB-Catalyzed Conjugate Addition of Thiophenes to Enones and Enals
3.3.3. Catalysis by C–I···π Type of XB Interaction
3.4. Hypervalent Iodine(III)-Catalyzed Reactions
ICl3-Catalyzed Ring-Opening Polymerization of l-Lactide
4. Summary and Outlook
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. Proceedings of “Modeling interactions in biomolecules II”, Prague, September 5th-9th, 2005. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Ang, S.J.; Mak, A.M.; Sullivan, M.B.; Wong, M.W. Site specificity of halogen bonding involving aromatic acceptors. Phys. Chem. Chem. Phys. 2018, 20, 8685–8694. [Google Scholar] [CrossRef] [PubMed]
- Angarov, V.; Kozuch, S. On the σ, π and δ hole interactions: A molecular orbital overview. New J. Chem. 2018, 42, 1413–1422. [Google Scholar] [CrossRef]
- Wolters, L.P.; Bickelhaupt, F.M. Halogen bonding versus hydrogen bonding: A molecular orbital perspective. ChemistryOpen 2012, 1, 96–105. [Google Scholar] [CrossRef]
- Ang, S.J.; Mak, A.M.; Wong, M.W. Nature of halogen bonding involving π-systems, nitroxide radicals and carbenes: A highlight of the importance of charge transfer. Phys. Chem. Chem. Phys. 2018, 20, 26463–26478. [Google Scholar] [CrossRef]
- Thirman, J.; Engelage, E.; Huber, S.M.; Head-Gordon, M. Characterizing the interplay of Pauli repulsion, electrostatics, dispersion and charge transfer in halogen bonding with energy decomposition analysis. Phys. Chem. Chem. Phys. 2018, 20, 905–915. [Google Scholar] [CrossRef]
- Wang, C.; Danovich, D.; Mo, Y.; Shaik, S. On the nature of the halogen bond. J. Chem. Theory Comput. 2014, 10, 3726–3737. [Google Scholar] [CrossRef]
- Robinson, S.W.; Mustoe, C.L.; White, N.G.; Brown, A.; Thompson, A.L.; Kennepohl, P.; Beer, P.D. Evidence for halogen bond covalency in acyclic and interlocked halogen-bonding receptor anion recognition. J. Am. Chem. Soc. 2015, 137, 499–507. [Google Scholar] [CrossRef]
- Mustoe, C.L.; Gunabalasingam, M.; Yu, D.; Patrick, B.O.; Kennepohl, P. Probing covalency in halogen bonds through donor K-edge X-ray absorption spectroscopy: Polyhalides as coordination complexes. Faraday Discuss. 2017, 203, 79–91. [Google Scholar] [CrossRef]
- Erakovic, M.; Cincic, D.; Molcanov, K.; Stilinovic, V. A crystallographic charge density study of the partial covalent nature of strong NBr halogen bonds. Angew. Chem. Int. Ed. 2019, 58, 15702–15706. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen and halogen bonds are ruled by the same mechanisms. Phys. Chem. Chem. Phys. 2013, 15, 7249–7259. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Lane, P. σ-Hole bonding and hydrogen bonding: Competitive interactions. Int. J. Quantum Chem. 2007, 107, 3046–3052. [Google Scholar] [CrossRef]
- Bolm, C.; Bruckmann, A.; Pena, M. Organocatalysis through halogen-bond activation. Synlett 2008, 2008, 900–902. [Google Scholar] [CrossRef]
- Bulfield, D.; Huber, S.M. Halogen bonding in organic synthesis and organocatalysis. Chem. Eur. J. 2016, 22, 14434–14450. [Google Scholar] [CrossRef]
- Sutar, R.L.; Huber, S.M. Catalysis of organic reactions through halogen bonding. ACS Catal. 2019, 9, 9622–9639. [Google Scholar] [CrossRef]
- Walter, S.M.; Kniep, F.; Herdtweck, E.; Huber, S.M. Halogen-bond-induced activation of a carbon–heteroatom bond. Angew. Chem. Int. Ed. 2011, 50, 7187–7191. [Google Scholar] [CrossRef]
- Kniep, F.; Jungbauer, S.H.; Zhang, Q.; Walter, S.M.; Schindler, S.; Schnapperelle, I.; Herdtweck, E.; Huber, S.M. Organocatalysis by neutral multidentate halogen-bond donors. Angew. Chem. Int. Ed. 2013, 52, 7028–7032. [Google Scholar] [CrossRef]
- He, W.; Ge, Y.C.; Tan, C.H. Halogen-bonding-induced hydrogen transfer to C=N bond with Hantzsch ester. Org. Lett. 2014, 16, 3244–3247. [Google Scholar] [CrossRef]
- Jungbauer, S.H.; Bulfield, D.; Kniep, F.; Lehmann, C.W.; Herdtweck, E.; Huber, S.M. Toward molecular recognition: Three-point halogen bonding in the solid state and in solution. J. Am. Chem. Soc. 2014, 136, 16740–16743. [Google Scholar] [CrossRef]
- Jungbauer, S.H.; Huber, S.M. Cationic multidentate halogen-bond donors in halide abstraction organocatalysis: Catalyst optimization by preorganization. J. Am. Chem. Soc. 2015, 137, 12110–12120. [Google Scholar] [CrossRef]
- Bergamaschi, G.; Lascialfari, L.; Pizzi, A.; Martinez Espinoza, M.I.; Demitri, N.; Milani, A.; Gori, A.; Metrangolo, P. A halogen bond-donor amino acid for organocatalysis in water. Chem. Commun. 2018, 54, 10718–10721. [Google Scholar] [CrossRef]
- Dreger, A.; Engelage, E.; Mallick, B.; Beer, P.D.; Huber, S.M. The role of charge in 1,2,3-triazol(ium)-based halogen bonding activators. Chem. Commun. 2018, 54, 4013–4016. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, K.; Uno, H.; Tokunaga, E.; Shibata, N. Fluorobissulfonylmethyl iodides: An efficient scaffold for halogen bonding catalysts with an sp3-hybridized carbon–iodine moiety. ACS Catal. 2018, 8, 6601–6605. [Google Scholar] [CrossRef]
- Schulz, N.; Sokkar, P.; Engelage, E.; Schindler, S.; Erdelyi, M.; Sanchez-Garcia, E.; Huber, S.M. The interaction modes of haloimidazolium salts in solution. Chem. Eur. J. 2018, 24, 3464–3473. [Google Scholar] [CrossRef]
- Squitieri, R.A.; Fitzpatrick, K.P.; Jaworski, A.A.; Scheidt, K.A. Synthesis and evaluation of azolium-based halogen-bond donors. Chem. Eur. J. 2019, 25, 10069–10073. [Google Scholar] [CrossRef]
- Szell, P.M.J.; Zablotny, S.; Bryce, D.L. Halogen bonding as a supramolecular dynamics catalyst. Nat. Commun. 2019, 10, 916. [Google Scholar] [CrossRef] [Green Version]
- Erdélyi, M. Halogen bonding in solution. Chem. Soc. Rev. 2012, 41, 3547–3557. [Google Scholar]
- Beale, T.M.; Chudzinski, M.G.; Sarwar, M.G.; Taylor, M.S. Halogen bonding in solution: Thermodynamics and applications. Chem. Soc. Rev. 2013, 42, 1667–1680. [Google Scholar] [CrossRef]
- Walter, S.M.; Kniep, F.; Rout, L.; Schmidtchen, F.P.; Herdtweck, E.; Huber, S.M. Isothermal calorimetric titrations on charge-assisted halogen bonds: Role of entropy, counterions, solvent, and temperature. J. Am. Chem. Soc. 2012, 134, 8507–8512. [Google Scholar] [CrossRef]
- Von der Heiden, D.; Bozkus, S.; Klussmann, M.; Breugst, M. Reaction mechanism of iodine-catalyzed Michael additions. J. Org. Chem. 2017, 82, 4037–4043. [Google Scholar] [CrossRef]
- Chan, Y.C.; Yeung, Y.Y. Halogen-bond-catalyzed addition of carbon-based nucleophiles to N-acylimminium ions. Org. Lett. 2019, 21, 5665–5669. [Google Scholar] [CrossRef]
- Purvis, G.D.; Bartlett, R.J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Kozuch, S.; Martin, J.M.L. Halogen bonds: Benchmarks and theoretical analysis. J. Chem. Theory Comput. 2013, 9, 1918–1931. [Google Scholar] [CrossRef]
- Kesharwani, M.K.; Manna, D.; Sylvetsky, N.; Martin, J.M.L. The X40 × 10 halogen bonding benchmark revisited: Surprising importance of (n-1)d subvalence correlation. J. Phys. Chem. A 2018, 122, 2184–2197. [Google Scholar] [CrossRef]
- Anderson, L.N.; Aquino, F.W.; Raeber, A.E.; Chen, X.; Wong, B.M. Halogen bonding interactions: Revised benchmarks and a new assessment of exchange vs. dispersion. J. Chem. Theory Comput. 2018, 14, 180–190. [Google Scholar] [CrossRef] [Green Version]
- Ang, S.J.; Ser, C.T.; Wong, M.W. Modeling halogen bonding with planewave density functional theory: Accuracy and challenges. J. Comput. Chem. 2019, 40, 1829–1835. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.S.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn-Sham global-hybrid exchange-correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. [Google Scholar] [CrossRef] [Green Version]
- Ser, C.T.; Yang, H.; Wong, M.W. Iodoimidazolinium-catalyzed reduction of quinoline by Hantzsch ester: Halogen bond or Brønsted acid catalysis. J. Org. Chem. 2019, 84, 10338–10348. [Google Scholar] [CrossRef] [Green Version]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Engelage, E.; Schulz, N.; Heinen, F.; Huber, S.M.; Truhlar, D.G.; Cramer, C.J. Refined SMD parameters for bromine and iodine accurately model halogen-bonding interactions in solution. Chem. Eur. J. 2018, 24, 15983–15987. [Google Scholar] [CrossRef]
- Riplinger, C.; Neese, F. An efficient and near linear scaling pair natural orbital based local coupled cluster method. J. Chem. Phys. 2013, 138, 034106. [Google Scholar] [CrossRef]
- Yepes, D.; Neese, F.; List, B.; Bistoni, G. Unveiling the delicate balance of steric and dispersion interactions in organocatalysis using high-level computational methods. J. Am. Chem. Soc. 2020, 142, 3613–3625. [Google Scholar] [CrossRef] [Green Version]
- Liakos, D.G.; Neese, F. Is it possible to obtain coupled cluster quality energies at near density functional theory cost? Domain-based local pair natural orbital coupled cluster vs. modern density functional theory. J. Chem. Theory Comput. 2015, 11, 4054–4063. [Google Scholar] [CrossRef]
- Hamlin, T.A.; Fernandez, I.; Bickelhaupt, F.M. How dihalogens catalyze Michael addition reactions. Angew. Chem. Int. Ed. 2019, 58, 8922–8926. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Sarwar, M.G.; Dragisic, B.; Salsberg, L.J.; Gouliaras, C.; Taylor, M.S. Thermodynamics of halogen bonding in solution: Substituent, structural, and solvent effects. J. Am. Chem. Soc. 2010, 132, 1646–1653. [Google Scholar] [CrossRef] [PubMed]
- Jungbauer, S.H.; Walter, S.M.; Schindler, S.; Rout, L.; Kniep, F.; Huber, S.M. Activation of a carbonyl compound by halogen bonding. Chem. Commun. 2014, 50, 6281–6284. [Google Scholar] [CrossRef] [PubMed]
- Gliese, J.P.; Jungbauer, S.H.; Huber, S.M. A halogen-bonding-catalyzed Michael addition reaction. Chem. Commun. 2017, 53, 12052–12055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dreger, A.; Wonner, P.; Engelage, E.; Walter, S.M.; Stoll, R.; Huber, S.M. A halogen-bonding-catalysed Nazarov cyclisation reaction. Chem. Commun. 2019, 55, 8262–8265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoesser, J.; Rojas, G.; Bulfield, D.; Hidalgo, P.I.; Pasán, J.; Ruiz-Pérez, C.; Jiménez, C.A.; Huber, S.M. Halogen bonding two-point recognition with terphenyl derivatives. New J. Chem. 2018, 42, 10476–10480. [Google Scholar] [CrossRef]
- Linke, A.; Jungbauer, S.H.; Huber, S.M.; Waldvogel, S.R. Potent affinity material for tracing acetone and related analytes based on molecular recognition by halogen bonds. Chem. Commun. 2015, 51, 2040–2043. [Google Scholar] [CrossRef] [Green Version]
- Coulembier, O.; Meyer, F.; Dubois, P. Controlled room temperature ROP of L-lactide by ICl3: A simple halogen-bonding catalyst. Polym. Chem. 2010, 1, 434–437. [Google Scholar] [CrossRef]
- Chan, Y.C.; Yeung, Y.Y. Halogen bond catalyzed bromocarbocyclization. Angew. Chem. Int. Ed. 2018, 57, 3483–3487. [Google Scholar] [CrossRef]
- Togo, H.; Iida, S. Synthetic use of molecular iodine for organic synthesis. Synlett 2006, 2006, 2159–2175. [Google Scholar] [CrossRef]
- Thakur, A.; Das, S.; Borah, R.; Devi, R. Molecular iodine in protection and deprotection chemistry. Synlett 2008, 2008, 2741–2762. [Google Scholar] [CrossRef]
- Jereb, M.; Vražič, D.; Zupan, M. Iodine-catalyzed transformation of molecules containing oxygen functional groups. Tetrahedron 2011, 67, 1355–1387. [Google Scholar] [CrossRef]
- Parvatkar, P.T.; Parameswaran, P.S.; Tilve, S.G. Recent developments in the synthesis of five- and six-membered heterocycles using molecular iodine. Chem. Eur. J. 2012, 18, 5460–5489. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.-M.; Cai, C.; Yang, R.-C. Molecular iodine-catalyzed multicomponent reactions: An efficient catalyst for organic synthesis. RSC Adv. 2013, 3, 7182–7204. [Google Scholar] [CrossRef]
- Breugst, M.; von der Heiden, D. Mechanisms in iodine catalysis. Chem. Eur. J. 2018, 24, 9187–9199. [Google Scholar] [CrossRef]
- Hibbert, H. Use of iodine as a dehydrating and condensing agent. J. Am. Chem. Soc. 1915, 37, 1748–1763. [Google Scholar] [CrossRef] [Green Version]
- Heinen, F.; Engelage, E.; Dreger, A.; Weiss, R.; Huber, S.M. Iodine(III) derivatives as halogen bonding organocatalysts. Angew. Chem. Int. Ed. 2018, 57, 3830–3833. [Google Scholar] [CrossRef]
- Corey, E.J.; Grogan, M.J. Enantioselective synthesis of α-amino nitriles from N-benzhydryl imines and HCN with a chiral bicyclic guanidine as catalyst. Org. Lett. 1999, 1, 157–160. [Google Scholar] [CrossRef]
- Heinz, N.; Dolg, M.; Berkessel, A. A theoretical study of imine hydrocyanation catalyzed by halogen-bonding. J. Comput. Chem. 2015, 36, 1812–1817. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef]
- Matsuzawa, A.; Takeuchi, S.; Sugita, K. Iodoalkyne-based catalyst-mediated activation of thioamides through halogen bonding. Chem. Asian J. 2016, 11, 2863–2866. [Google Scholar] [CrossRef]
- Kee, C.W.; Wong, M.W. In silico design of halogen-bonding-based organocatalyst for Diels–Alder reaction, Claisen rearrangement, and Cope-type hydroamination. J. Org. Chem. 2016, 81, 7459–7470. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S.; Shaik, S. A combined kinetic−quantum mechanical model for assessment of catalytic cycles: Application to cross-coupling and Heck reactions. J. Am. Chem. Soc. 2006, 128, 3355–3365. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S.; Shaik, S. How to conceptualize catalytic cycles? The energetic span model. Acc. Chem. Res. 2011, 44, 101–110. [Google Scholar] [CrossRef]
- Uhe, A.; Kozuch, S.; Shaik, S. Automatic analysis of computed catalytic cycles. J. Comput. Chem. 2011, 32, 978–985. [Google Scholar] [CrossRef]
- Mitsumori, S.; Zhang, H.; Ha-Yeon Cheong, P.; Houk, K.N.; Tanaka, F.; Barbas, C.F. Direct asymmetric anti-Mannich-type reactions catalyzed by a designed amino acid. J. Am. Chem. Soc. 2006, 128, 1040–1041. [Google Scholar] [CrossRef] [PubMed]
- Shinisha, C.B.; Sunoj, R.B. Bicyclic proline analogues as organocatalysts for stereoselective aldol reactions: An in silico DFT study. Org. Biomol. Chem. 2007, 5, 1287–1294. [Google Scholar] [CrossRef] [PubMed]
- Malerich, J.P.; Hagihara, K.; Rawal, V.H. Chiral squaramide derivatives are excellent hydrogen bond donor catalysts. J. Am. Chem. Soc. 2008, 130, 14416–14417. [Google Scholar] [CrossRef] [Green Version]
- Neel, A.J.; Hehn, J.P.; Tripet, P.F.; Toste, F.D. Asymmetric cross-dehydrogenative coupling enabled by the design and application of chiral triazole-containing phosphoric acids. J. Am. Chem. Soc. 2013, 135, 14044–14047. [Google Scholar] [CrossRef] [Green Version]
- Markad, D.; Mandal, S.K. Design of a primary-amide-functionalized highly efficient and recyclable hydrogen-bond-donating heterogeneous catalyst for the Friedel–Crafts alkylation of indoles with β-nitrostyrenes. ACS Catal. 2019, 9, 3165–3173. [Google Scholar] [CrossRef]
- Yang, H.; Wong, M.W. b-amino acid catalyzed asymmetric Michael additions: Design of organocatalysts with catalytic acid/base dyad inspired by serine proteases. J. Org. Chem. 2011, 76, 7399–7405. [Google Scholar] [CrossRef]
- Breugst, M.; Detmar, E.; von der Heiden, D. Origin of the catalytic effects of molecular iodine: A computational analysis. ACS Catal. 2016, 6, 3203–3212. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Marsili, L.A.; Pergomet, J.L.; Gandon, V.; Riveira, M.J. Iodine-catalyzed iso-Nazarov cyclization of conjugated dienals for the synthesis of 2-cyclopentenones. Org. Lett. 2018, 20, 7298–7303. [Google Scholar] [CrossRef]
- Koenig, J.J.; Arndt, T.; Gildemeister, N.; Neudorfl, J.M.; Breugst, M. Iodine-catalyzed Nazarov cyclizations. J. Org. Chem. 2019, 84, 7587–7605. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- Van Zeist, W.J.; Bickelhaupt, F.M. The activation strain model of chemical reactivity. Org. Biomol. Chem. 2010, 8, 3118–3127. [Google Scholar] [CrossRef]
- Denmark, S.E.; Kuester, W.E.; Burk, M.T. Catalytic, asymmetric halofunctionalization of alkenes—A critical perspective. Angew. Chem. Int. Ed. 2012, 51, 10938–10953. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.A.; Yu, W.Z.; Yeung, Y.-Y. Recent advances in asymmetric intra- and intermolecular halofunctionalizations of alkenes. Org. Biomol. Chem. 2014, 12, 2333–2343. [Google Scholar] [CrossRef]
- Guha, S.; Kazi, I.; Nandy, A.; Sekar, G. Role of Lewis-base-coordinated halogen(I) intermediates in organic synthesis: The journey from unstable intermediates to versatile reagents. Eur. J. Org. Chem. 2017, 2017, 5497–5518. [Google Scholar] [CrossRef]
- Arai, T.; Horigane, K.; Watanabe, O.; Kakino, J.; Sugiyama, N.; Makino, H.; Kamei, Y.; Yabe, S.; Yamanaka, M. Association of halogen bonding and hydrogen bonding in metal acetate-catalyzed asymmetric halolactonization. iScience 2019, 12, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.-C.; Yang, H.; Heusler, A.; Chua, Z.; Wong, M.W.; Tan, C.-H. Halogen-bonding-induced conjugate addition of thiophenes to enones and enals. Chem. Asian J. 2019, 14, 2656–2661. [Google Scholar] [CrossRef] [PubMed]
- Kuwano, S.; Suzuki, T.; Yamanaka, M.; Tsutsumi, R.; Arai, T. Catalysis based on C−I⋅⋅⋅π halogen bonds: Electrophilic activation of 2-alkenylindoles by cationic halogen-bond donors for [4+2] cycloadditions. Angew. Chem. Int. Ed. 2019, 58, 10220–10224. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Sun, C.; Meng, L.; Zeng, Y. The mechanism of ring-opening polymerization of L-lactide by ICl3 catalysts: Halogen bond catalysis or participating in reactions? J. Comput. Chem. 2019, 40, 2827–2833. [Google Scholar] [CrossRef]
- Sutar, R.L.; Engelage, E.; Stoll, R.; Huber, S.M. Bidentate chiral bis(imidazolium)-based halogen bond donors: Synthesis and first applications in enantioselective recognition and catalysis. Angew. Chem. Int. Ed. 2020. [Google Scholar] [CrossRef] [Green Version]






















{kind=link}
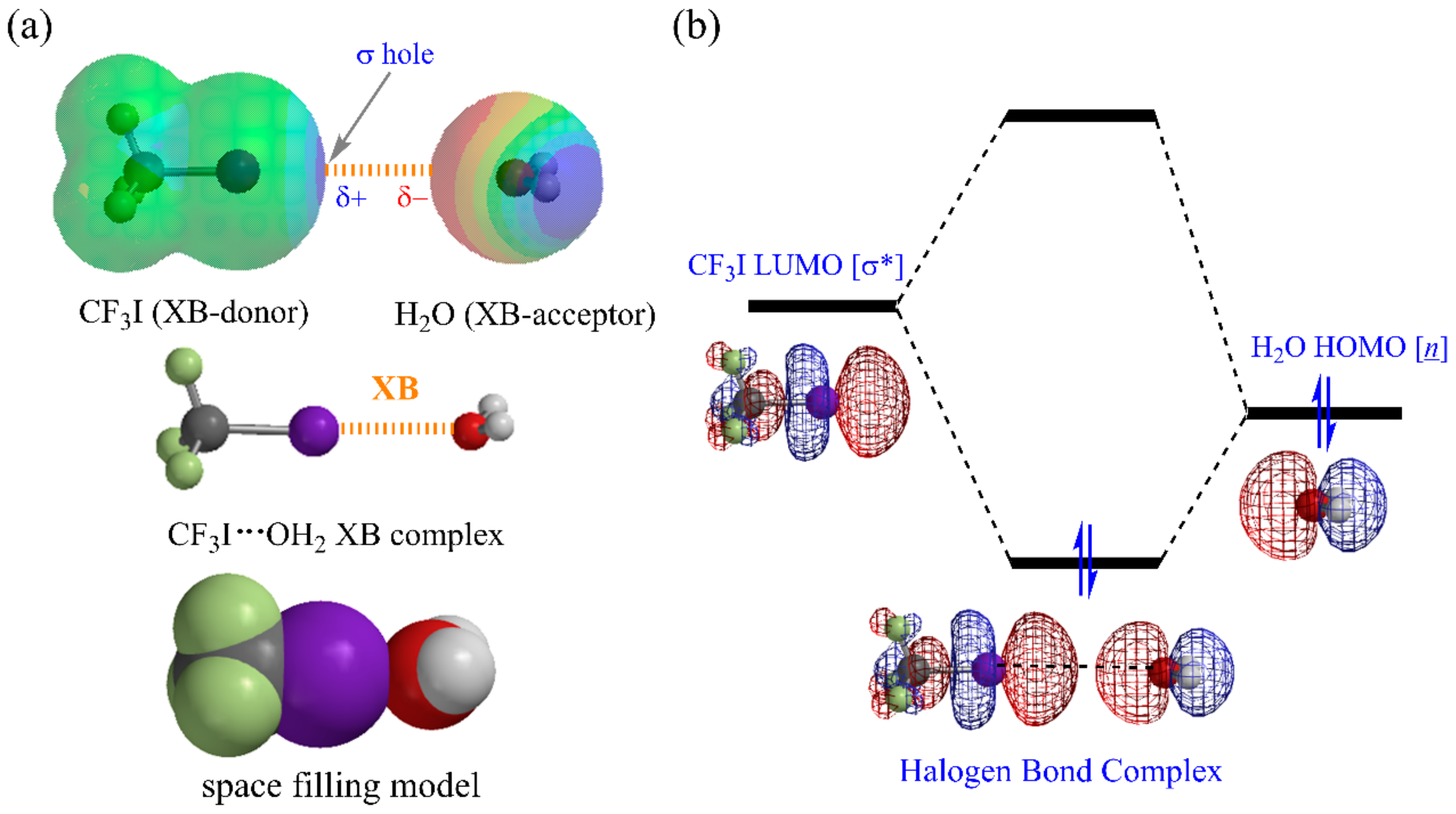{kind=link}
{kind=link}
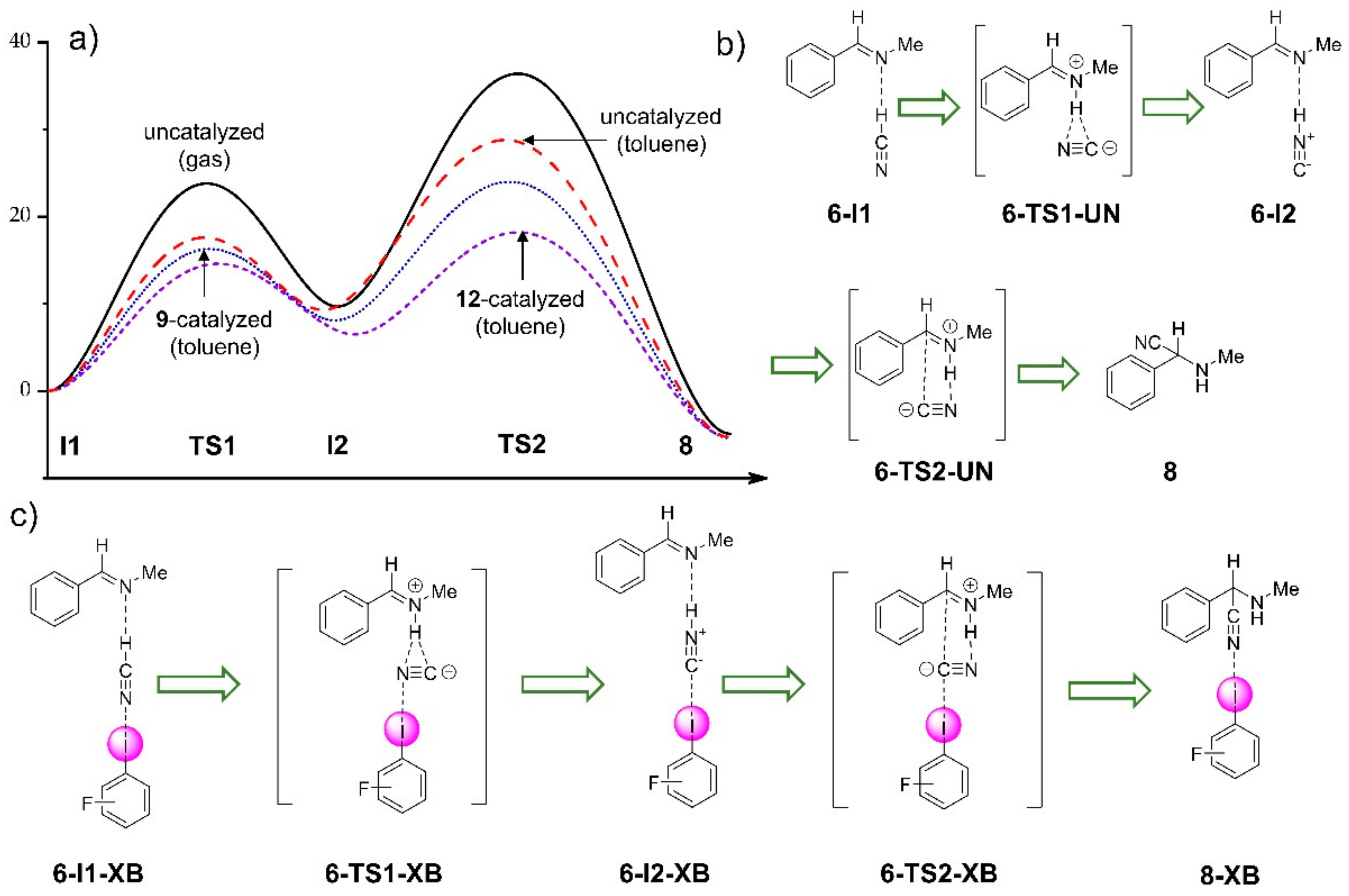{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
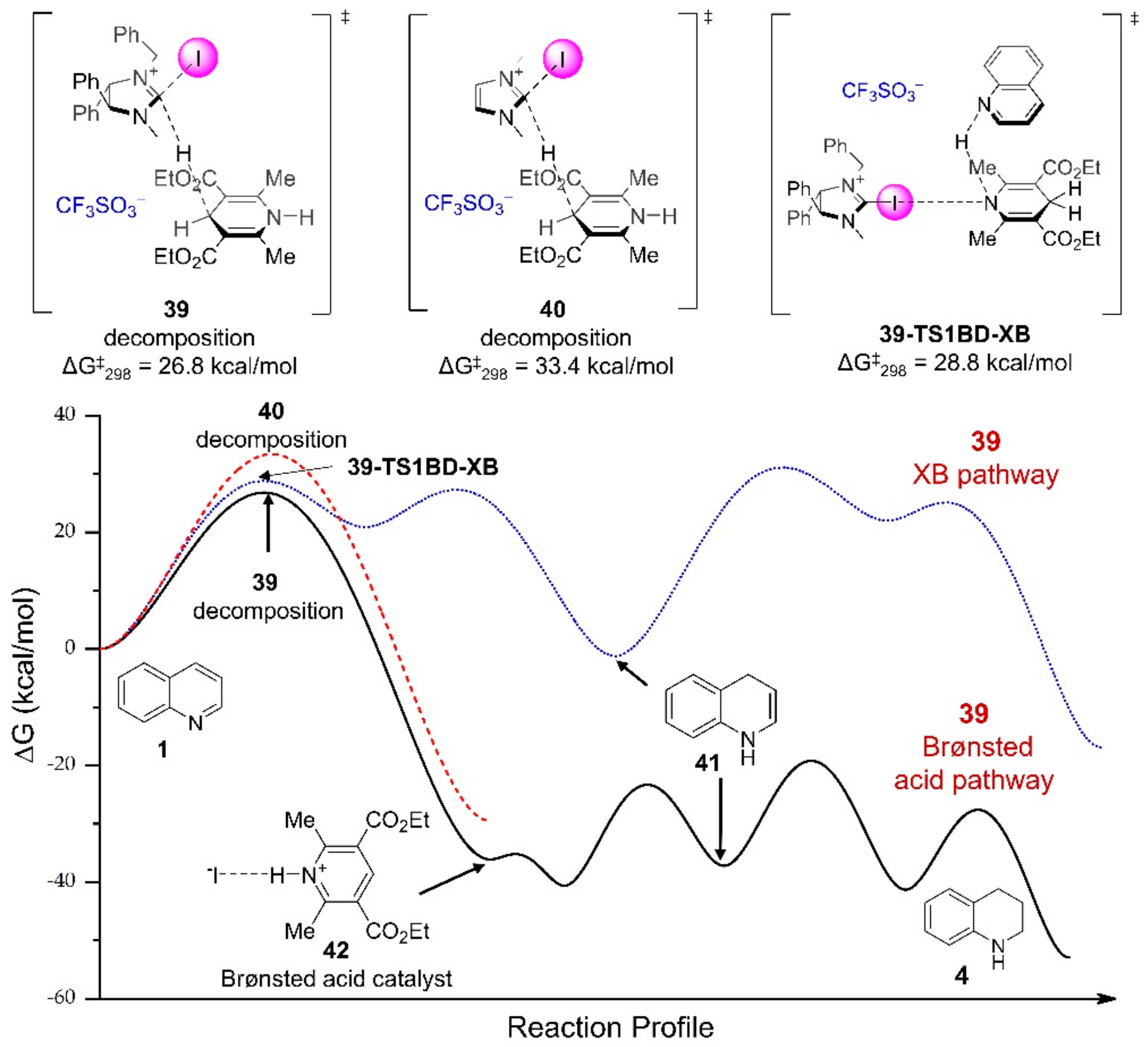{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DFT a | ΔH298 (kcal/mol) | ΔG298 b (kcal/mol) | DXB (Å) |
|---|---|---|---|
| MN15 | −10.1 | −1.0 | 2.75 |
| B3LYP | −1.6 | 7.2 | 2.87 |
| B3LYP-D3 | −8.1 | 1.4 | 2.79 |
| M06-2X | −8.2 | 1.2 | 2.80 |
| ω-B97XD | −6.9 | 2.4 | 2.85 |
| PBE0 | −4.1 | 5.0 | 2.78 |
| PBE0-D3 | −8.2 | 1.1 | 2.74 |

| Uncatalyzed | 18-Catalyzed | |||
|---|---|---|---|---|
| T(k) | ΔG≠ (kcal/mol) | kKeq (M h−1) | ΔG≠n-hexane (kcal/mol) | TOF (h−1) |
| 318.15 | 26.5 | 5.3×10−4 | 28.3 | 8.6×10−4 |
| 298.15 | 26.5 | 3.3×10−5 | 27.3 | 2.3×10−4 |
| 273.15 | 26.4 | 5.3×10−7 | 26.0 | 2.5×10−5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, H.; Wong, M.W. Application of Halogen Bonding to Organocatalysis: A Theoretical Perspective. Molecules 2020, 25, 1045. https://doi.org/10.3390/molecules25051045
Yang H, Wong MW. Application of Halogen Bonding to Organocatalysis: A Theoretical Perspective. Molecules. 2020; 25(5):1045. https://doi.org/10.3390/molecules25051045
Chicago/Turabian StyleYang, Hui, and Ming Wah Wong. 2020. "Application of Halogen Bonding to Organocatalysis: A Theoretical Perspective" Molecules 25, no. 5: 1045. https://doi.org/10.3390/molecules25051045
APA StyleYang, H., & Wong, M. W. (2020). Application of Halogen Bonding to Organocatalysis: A Theoretical Perspective. Molecules, 25(5), 1045. https://doi.org/10.3390/molecules25051045