
3-Aryl-4-nitrobenzothiochromans S,S-dioxide: From Calcium-Channel Modulators Properties to Multidrug-Resistance Reverting Activity

 ,
,  , , ,
, , ,  ,
,
Abstract
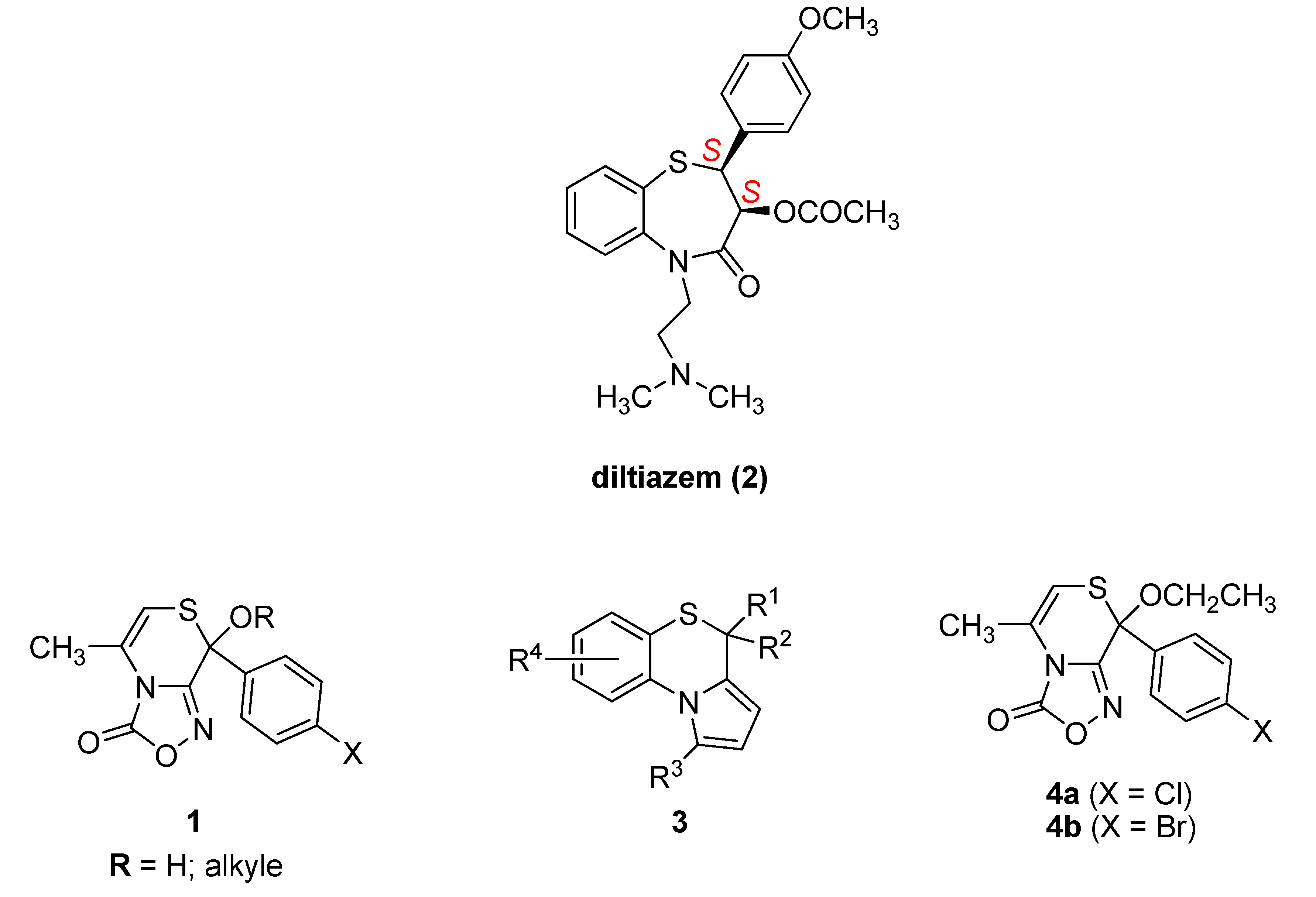
:1. Introduction
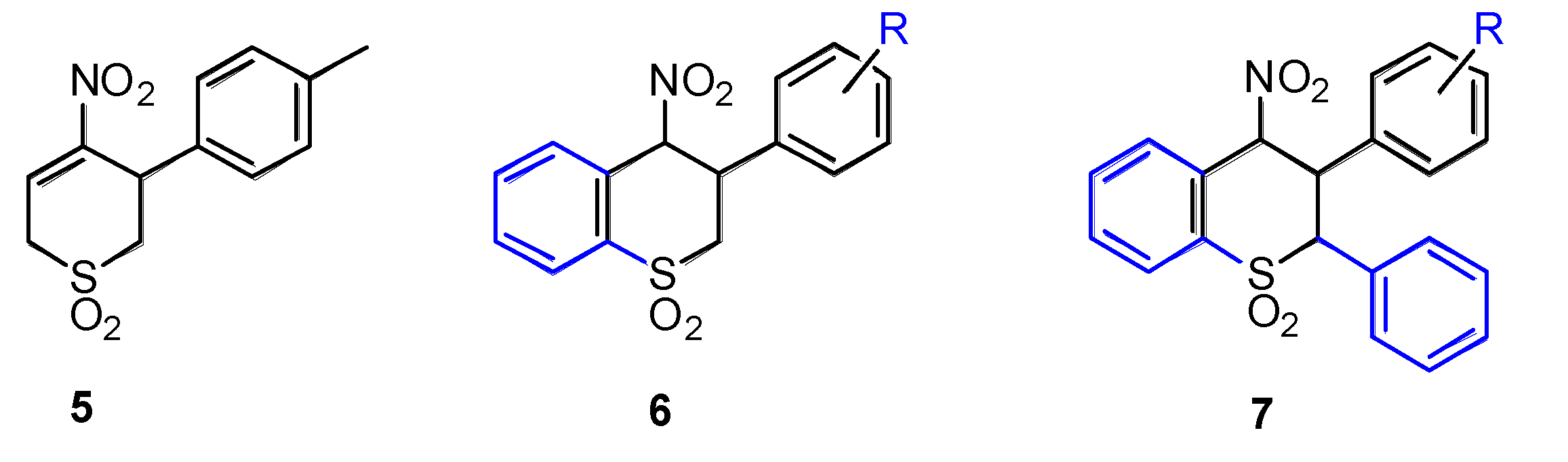
2. Results
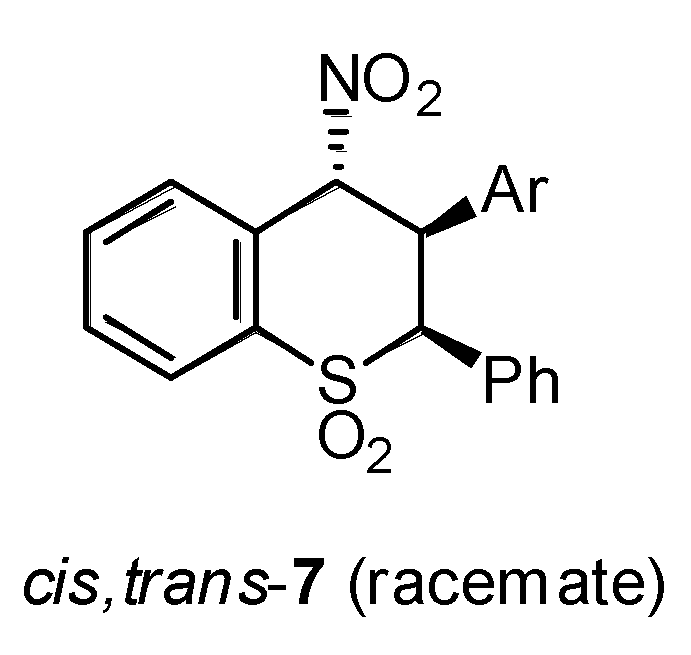
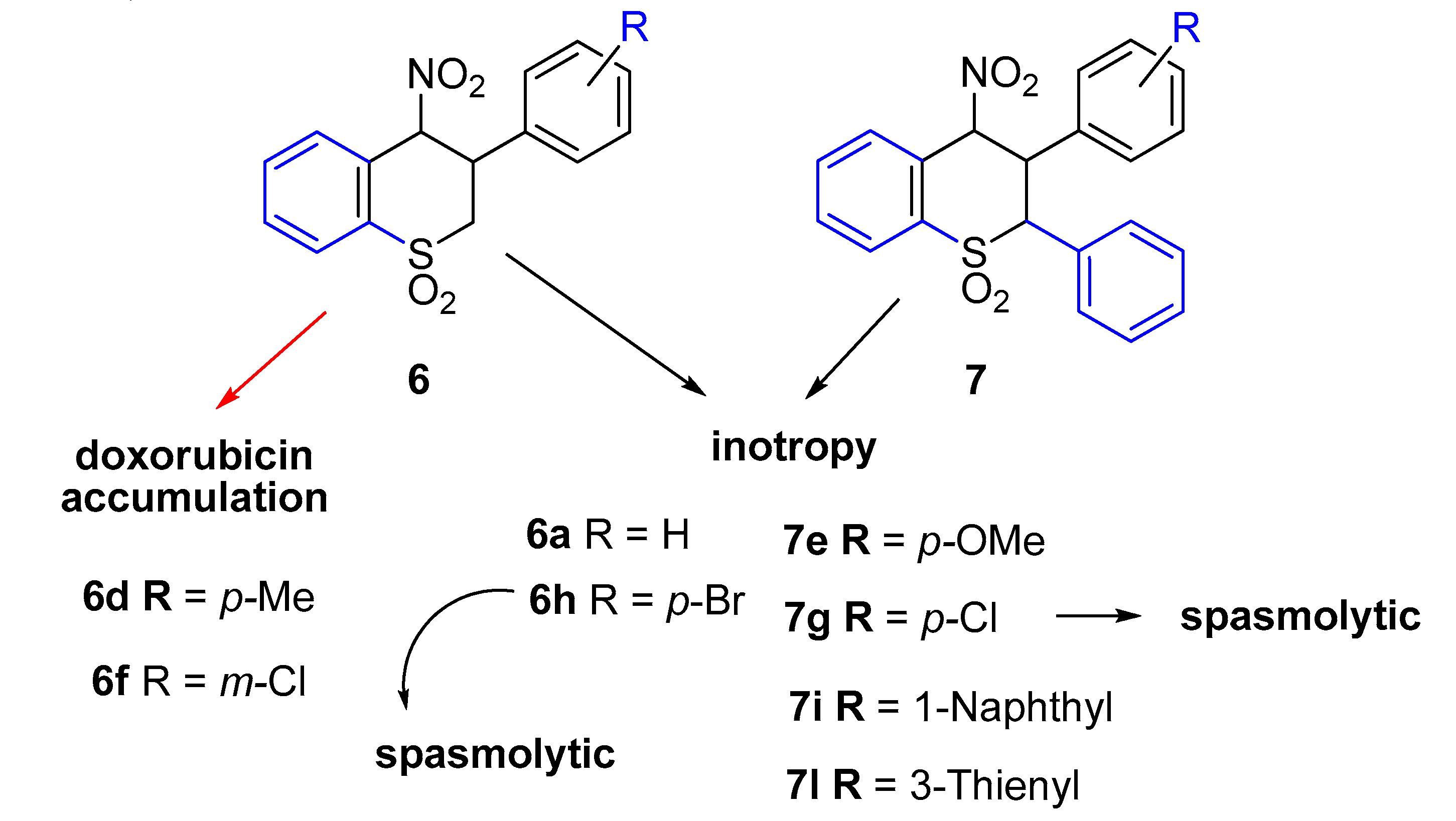
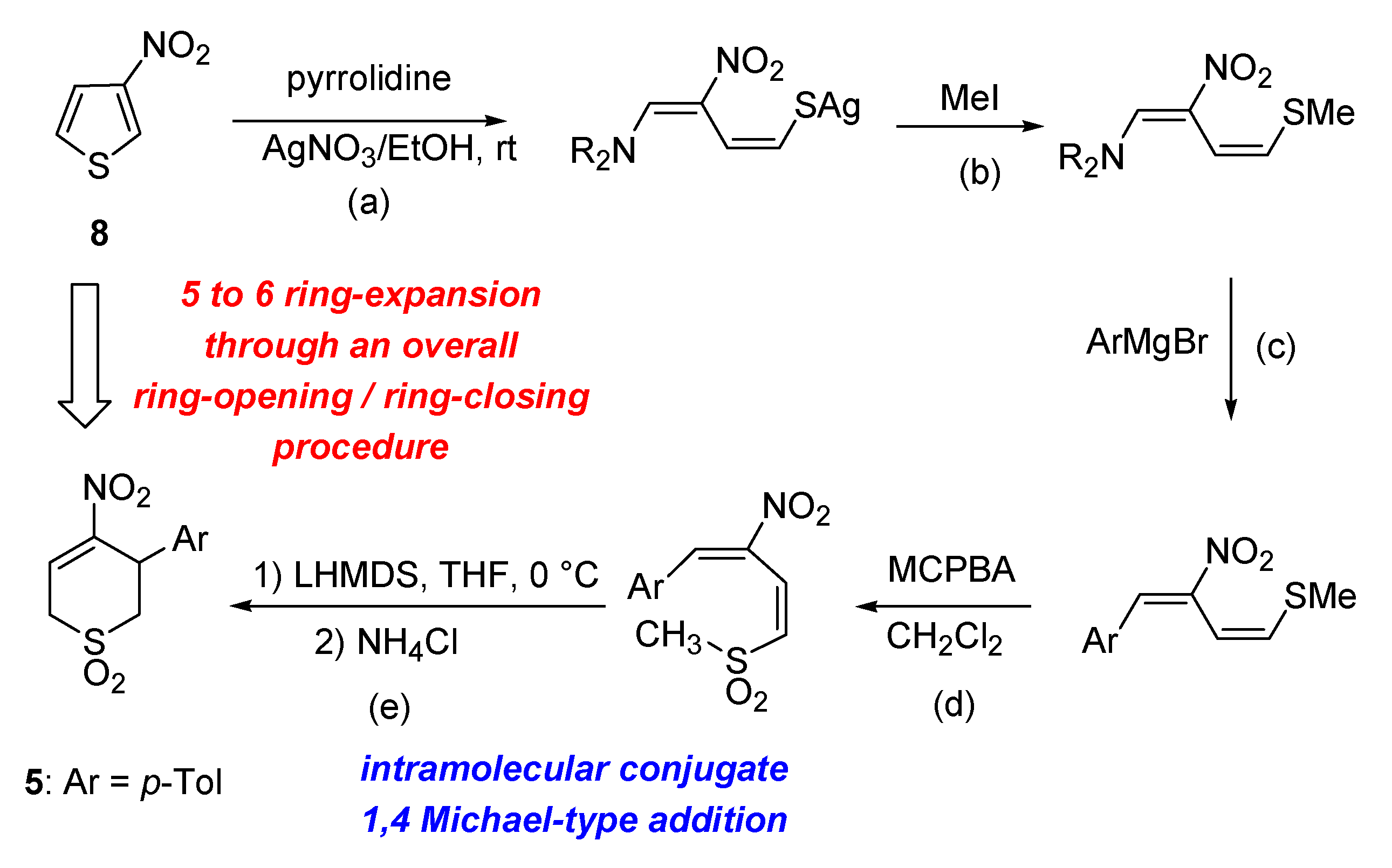
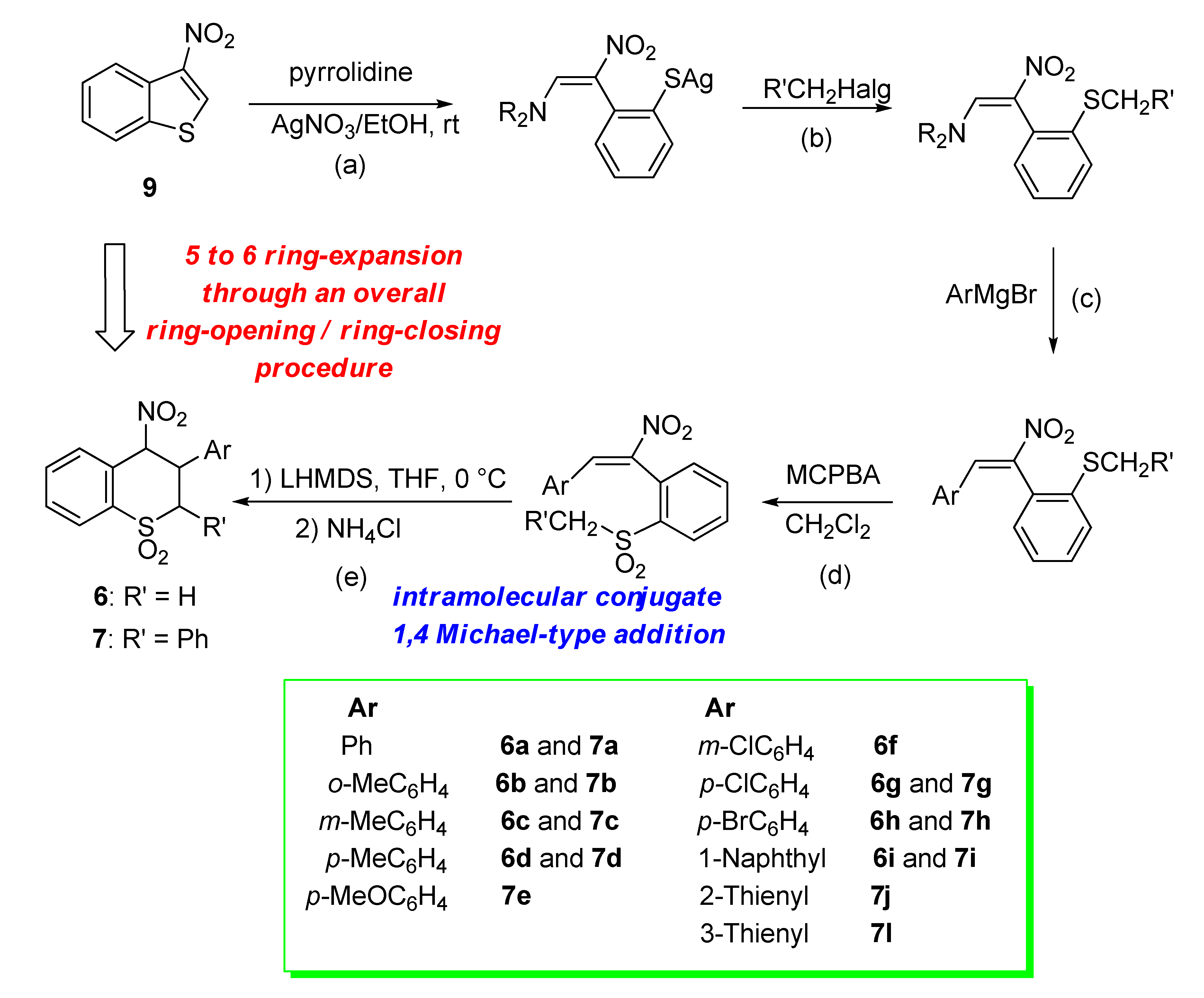
2.1. Chemistry
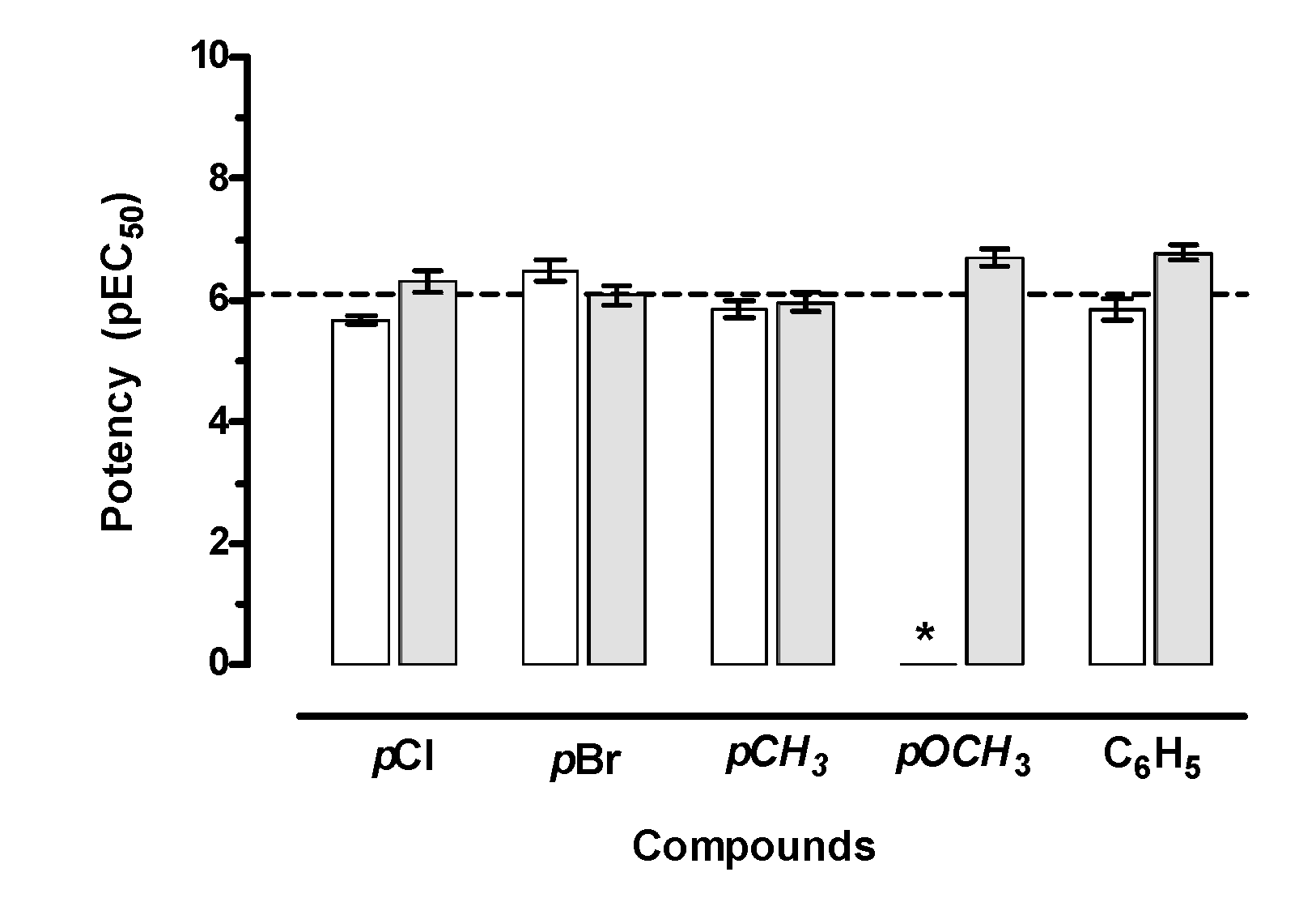
2.2. Ex Vivo Functional Studies
2.2.1. Cardiac Activity
2.2.2. Smooth Muscle Spasmolytic Activity
2.3. Antiproliferative Activity of Compounds 6 and 7
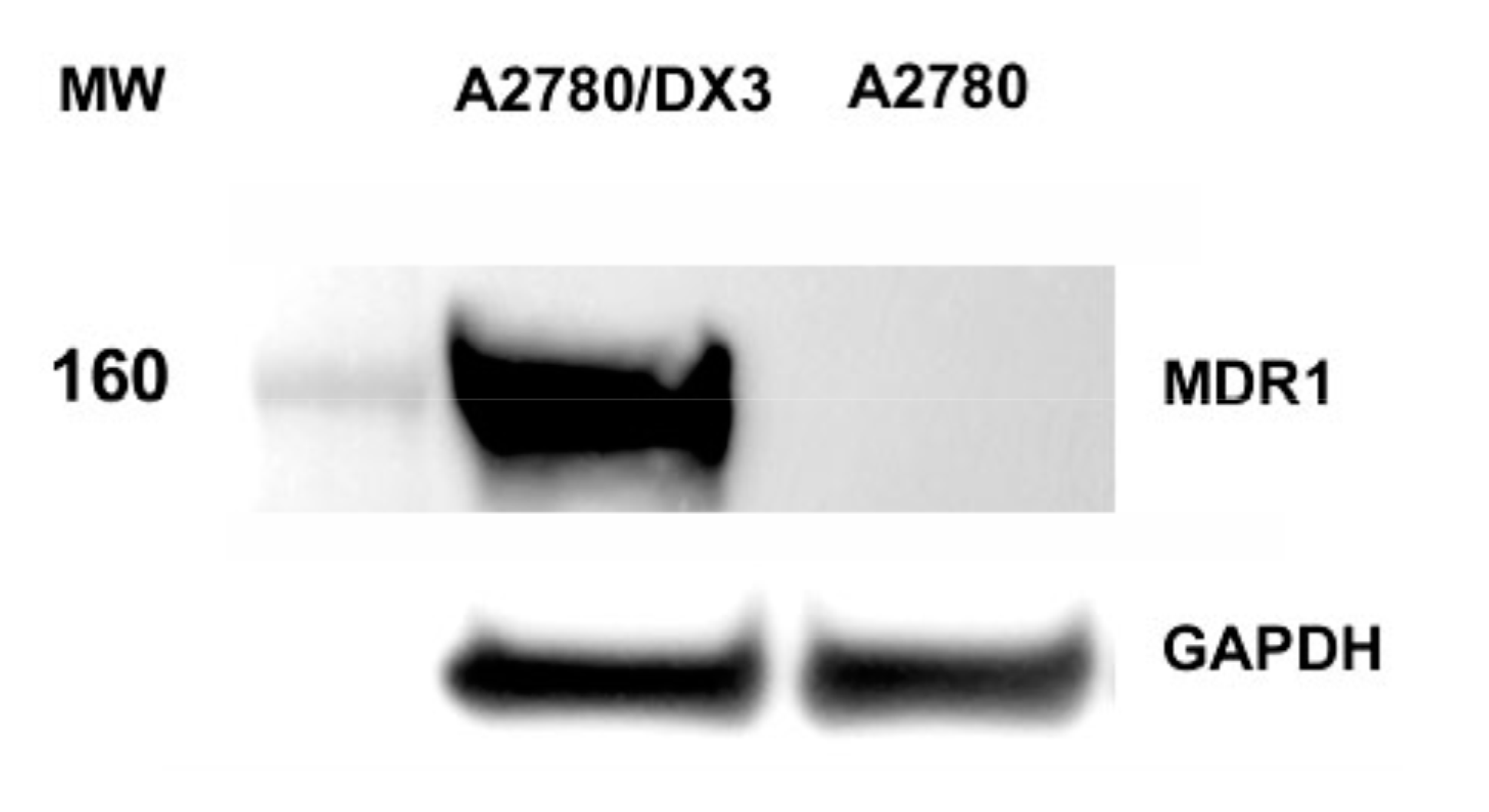
2.4. Determination of Doxorubicin Accumulation by Flow Cytometry
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.2. Ex Vivo Studies
4.3. In Vitro Studies
4.3.1. Chemicals
4.3.2. Cell Lines
4.3.3. MTT Assay
4.3.4. Study of Doxorubicin Intracellular Accumulation
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gillet, J.P.; Gottesman, M.M. Mechanisms of multidrug resistance in cancer. Methods Mol. Biol. 2010, 596, 47–76. [Google Scholar] [PubMed]
- Trédan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Beck, W.T. Strategies to circumvent multidrug resistance due to P-glycoprotein or to altered DNA topoisomerase II. Bull. Cancer 1990, 77, 1131–1141. [Google Scholar] [PubMed]
- Breier, A.; Barancík, M.; Sulová, Z.; Uhrík, B. P-glycoprotein–implications of metabolism of neoplastic cells and cancer therapy. Curr. Cancer Drug Targets 2005, 6, 457–468. [Google Scholar] [CrossRef]
- Kessel, D.; Botterill, V.; Wodinsky, I. Uptake and retention of daunomycin by mouse leukemic cells as factors in drug response. Cancer Res. 1968, 28, 938–941. [Google Scholar]
- Carosati, E.; Cosimelli, B.; Ioan, P.; Severi, E.; Katneni, K.; Chiu, F.C.; Saponara, S.; Fusi, F.; Frosini, M.; Matucci, R.; et al. Understanding Oxadiazolothiazinone Biological Properties: Negative Inotropic Activity versus Cytochrome P450-Mediated Metabolism. J. Med. Chem. 2016, 59, 3340–3352. [Google Scholar] [CrossRef] [Green Version]
- Leoni, A.; Frosini, M.; Locatelli, A.; Micucci, M.; Carotenuto, C.; Durante, M.; Budriesi, R. 4-Imidazo[2,1-b]Thiazole-1,4-DHPs and Neuroprotection: Preliminary Study in Hits Searching. Eur. J. Med. Chem. 2019, 169, 89–102. [Google Scholar] [CrossRef]
- Budriesi, R.; Cosimelli, B.; Ioan, P.; Lanza, C.Z.; Spinelli, D.; Chiarini, A. Cardiovascular characterization of [1,4]thiazino[3,4-c][1,2,4]oxadiazol-1-one derivatives: Selective myocardial calcium channel modulators. J. Med. Chem. 2002, 45, 3475–3481. [Google Scholar] [CrossRef]
- Dustan, H.P. Calcium channel blockers. Potential medical benefits and side effects. Hypertension 1989, 13, I137–I140. [Google Scholar] [CrossRef] [Green Version]
- Corelli, F.; Manetti, F.; Tafi, A.; Campiani, G.; Nacci, V.; Botta, M. Diltiazem-like calcium entry blockers: A hypothesis of the receptor-binding site based on a comparative molecular field analysis model. J. Med. Chem. 1997, 40, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Tsuruo, T.; Iida, H.; Tsukagoshi, S.; Sakurai, Y. Potentiation of vincristine and Adriamycin effects in human hemopoietic tumor cell lines by calcium antagonists and calmodulin inhibitors. Cancer Res. 1983, 43, 2267–2272. [Google Scholar] [PubMed]
- Tsuruo, T.; Iida, H.; Tsukagoshi, S.; Sakurai, Y. Vincristine-resistant P388 leukemia cells contain a large amount of calcium. GANN Jpn. J. Cancer Res. 1983, 74, 619–621. [Google Scholar] [PubMed]
- Viale, M.; Cordazzo, C.; Cosimelli, B.; de Totero, D.; Castagnola, P.; Aiello, C.; Severi, E.; Petrillo, G.; Cianfriglia, M.; Spinelli, D. Inhibition of MDR1 activity in vitro by a novel class of diltiazem analogues: Toward new candidates. J. Med. Chem. 2009, 52, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Budriesi, R.; Carosati, E.; Chiarini, A.; Cosimelli, B.; Cruciani, G.; Ioan, P.; Spinelli, D.; Spisani, R. A new class of selective myocardial calcium channel modulators. 2. Role of the acetal chain in oxadiazol-3-one derivatives. J. Med. Chem. 2005, 48, 2445–2456. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, L.; Maccagno, M.; Petrillo, G.; Rizzato, E.; Sancassan, F.; Severi, E.; Spinelli, D.; Stenta, M.; Galatini, A.; Tavani, C. A new route to thiopyran S,S-dioxide derivatives via an overall ring-enlargement protocol from 3-nitrothiophene. Tetrahedron 2009, 65, 336–343. [Google Scholar] [CrossRef]
- Bianchi, L.; Dell’Erba, C.; Maccagno, M.; Morganti, S.; Novi, M.; Petrillo, G.; Rizzato, E.; Sancassan, F.; Severi, E.; Spinelli, D.; et al. Easy access to 4-nitrothiochroman S,S-dioxides via ring-enlargement from 3-nitrobenzo[b]thiophene. Tetrahedron 2004, 60, 4967–4973. [Google Scholar] [CrossRef]
- Bianchi, L.; Maccagno, M.; Petrillo, G.; Rizzato, E.; Sancassan, F.; Spinelli, D.; Tavani, C. Access to 2,3-diaryl-4-nitrothiochroman S,S-dioxides from 3-nitrobenzo[b]thiophene. Tetrahedron 2011, 67, 8160–8169. [Google Scholar] [CrossRef]
- Dell’Erba, C.; Spinelli, D.; Leandri, G. Ring-opening Reaction in the Thiophen Series: Reaction between 3,4-Dinitrothiophen and Secondary Amines. Chem. Commun. 1969, 10, 549–550. [Google Scholar]
- Bianchi, L.; Maccagno, M.; Petrillo, G.; Sancassan, F.; Spinelli, D.; Tavani, C. 2,3-Dinitro-1,3-butadienes: Versatile building-blocks from the ring opening of 3,4-dinitrothiophene. In Targets in Heterocyclic Systems: Chemistry and Properties; Attanasi, O.A., Spinelli, D., Eds.; Società Chimica Italiana: Rome, Italy, 2006; Volume 10, pp. 1–23. [Google Scholar]
- Bianchi, L.; Maccagno, M.; Petrillo, G.; Rizzato, E.; Sancassan, F.; Severi, E.; Spinelli, D.; Tavani, C.; Viale, M. Versatile nitrobutadienic building-blocks from the ring-opening of 2- and 3-nitrothiophenes. In Targets in Heterocyclic Systems: Chemistry and Properties; Attanasi, O.A., Spinelli, D., Eds.; Società Chimica Italiana: Rome, Italy, 2007; Volume 11, pp. 1–20. [Google Scholar]
- Tallarida, R.J.; Murray, R.B. Manual of Pharmacologic Calculations with Computer Programs, 2nd ed.; Springer: New York, NY, USA, 1987. [Google Scholar]
- Hirose, M.; Hosoi, E.; Hamano, S.; Jalili, A. Multidrug Resistance in Hematological Malignancy. J. Med. Investig. 2003, 50, 126–135. [Google Scholar]
- Sonneveld, P. Multidrug Resistance in Haematological Malignancies. J. Intern. Med. 2000, 247, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.B. Multidrug Resistance: Clinical Relevance in Solid Tumours and Strategies for Circumvention. Curr. Opin. Oncol. 1998, 10, S15–S19. [Google Scholar] [PubMed]
- Ren, F.; Shen, J.; Shi, H.; Hornicek, F.J.; Kan, Q.; Duan, Z. Novel mechanisms and approaches to overcome multidrug resistance in the treatment of ovarian cancer. Biochim. Biophys. Acta 2016, 1866, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Li., Z.; Zhang, Q.; De Amorim Bernstein, K.; Lozano-Calderon, S.; Choy, E.; Hornicek, F.J.; Duan, Z. Targeting ABCB1 (MDR1) in multi-drug resistant osteosarcoma cells using the CRISPR-Cas9 system to reverse drug resistance. Oncotarget 2016, 50, 83502–83513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breier, A.; Drobná, Z.; Docolomansky, P.; Barancik, M. Cytotoxic activity of several unrelated drugs on L1210 mouse leukemic cell sublines with P-glycoprotein (PGP) mediated multidrug resistance (MDR) phenotype. A QSAR study. Neoplasma 2000, 2, 100–106. [Google Scholar]
- Triggle, D.J. Calcium channel antagonists: Clinical uses: Past, present and future. Biochem. Pharm. 2007, 74, 1–9. [Google Scholar] [CrossRef]
- Palmeira, A.; Sousa, E.; Vasconcelos, M.T.; Pinto, M.M. Tree decades of P-gp inhibitors: Skimming through several generations and scaffolds. Curr. Med. Chem. 2012, 19, 1946–2025. [Google Scholar] [CrossRef]
- Spinelli, D.; Budriesi, R.; Cosimelli, B.; Severi, E.; Micucci, M.; Baroni, M.; Fusi, F.; Joan, P.; Cross, S.; Frosini, M.; et al. Playing with Opening and Closing of Heterocycles: Using the Cusmano-Ruccia Reaction to Develop a Novel Class of Oxadiazolothiazinones, Active as Calcium Channel Modulators and P-Glycoprotein Inhibitors. Molecules 2014, 19, 16543–16572. [Google Scholar] [CrossRef] [Green Version]
- Viale, M.; Cordazzo, C.; de Torero, D.; Budriesi, R.; Rosano, C.; Leoni, A.; Ioan, P.; Aiello, C.; Croce, M.; Andreani, A.; et al. Inhibition of MDR1 activity and induction of apoptosis by analogues of nifedipine and diltiazem: An in vitro analysis. Investig. New Drugs 2011, 29, 98–109. [Google Scholar] [CrossRef]
- Teodori, E.; Dei, S.; Quidu, P.; Budriesi, R.; Chiarini, A.; Garnier-Suillerot, A.; Gualtieri, F.; Manetti, D.; Romanelli, M.N.; Scapecchi, S. Design, synthesis, and in vitro activity of catamphiphilic reverters of multidrug resistance: Discovery of a selective, 37highly efficacious chemosensitizer with potency in the nanomolar range. J. Med. Chem. 1999, 42, 1687–1697. [Google Scholar] [CrossRef]
- Sulová, Z.; Seres, M.; Barancík, M.; Gibalová, L.; Uhrík, B.; Poleková, L.; Breier, A. Does any relationship exist between P-glycoprotein-mediated multidrug resistance and intracellular calcium homeostasis. Gen. Physiol. Biophys. 2009, 28, F89–F95. [Google Scholar]
- Carosati, E.; Cruciani, G.; Chiarini, A.; Budriesi, R.; Ioan, P.; Spisani, R.; Spinelli, D.; Cosimelli, B.; Fusi, F.; Frosini, M.; et al. Calcium channel antagonists discovered by a multidisciplinary approach. J. Med. Chem. 2006, 49, 5206–5216. [Google Scholar] [CrossRef] [PubMed]
- Rosano, C.; Viale, M.; Cosimelli, B.; Strict, E.; Gangemi, R.; Ciogli, A.; De Totero, D.; Spinelli, D. ABCB1 structural models, molecular docking, and synthesis of new oxadiazolothiazin-3-one inhibitors. ACS Med. Chem. Lett. 2013, 4, 694–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisi, A.; Micucci, M.; Gobbi, S.; Belluti, F.; Budriesi, R.; Rampa, A. Cardiovascular Profile of Xanthone-Based 1,4 Dihydropyridines Bearing a Lidoflazine Pharmacophore Fragment. Molecules 2018, 23, 3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motulsky, H.; Christopoulos, A. Fitting Models to Biological Data Using Linear and Non Linear Regression: A Practical Guide to Curve Fitting; Oxford University Press: New York, NY, USA, 2003. [Google Scholar]
- Motulsky, H.J. Prism 5 Statistics Guide; GraphPad Software Inc.: San Diego, CA, USA, 2007. [Google Scholar]
Sample Availability: Samples of the compounds are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | Left Atrium | Right Atrium | |||||
|---|---|---|---|---|---|---|---|
| Negative Inotropy | Negative Inotropy | Negative Chronotropy | |||||
| Activity a M ± S.E.M. | EC50 b (µM) | 95% c.l. | Activity c M ± S.E.M. | EC50 b (µM) | 95% c.l. | Activity d M ± S.E.M. | |
| Diltiazem | 78 ± 3.5 e | 0.79 | 0.70–0.85 | 94 ± 5.6 h,i | |||
| 5 | 40 ± 2.3 | 31 ± 2.1 | 3 ± 0.2 | ||||
| 6a | 77 ± 1.5 f | 0.42 | 0.28–0.63 | 79 ± 3.3 | 1.88 | 1.13–2.12 | 15 ± 1.3 |
| 7a | 76 ± 0.9 | 0.95 | 0.66–1.37 | 80 ± 1.6 | 1.28 | 0.95–1.92 | 6 ± 0.4 |
| 6b | 90 ± 2.7 | 1.17 | 0.80–1.31 | 77 ± 2.8 | 4.17 | 3.79–5.23 | 3 ± 0.2 |
| 7b | 74 ± 2.2 f | 0.98 | 0.63–1.34 | 78 ± 2.1 | 1.45 | 1.03–2.03 | 6 ± 0.4 |
| 6c | 83 ± 2.4 | 0.90 | 0.57–1.21 | 78 ± 3.5 | 3.34 | 2.66–4.01 | 7 ± 0.5 |
| 7c | 85 ± 5.9 | 0.66 | 0.48–0.88 | 77 ± 1.3 | 1.13 | 0.78–1.65 | 6 ± 0.5 |
| 6d | 95 ± 2.6 | 1.38 | 0.97–1.96 | 80 ± 2.2 | 9.52 | 8.73–10.38 | 8 ± 0.4 |
| 7d | 86 ± 3.3 | 1.08 | 0.71–1.50 | 79 ± 4.5 | 4.19 | 3.90–5.04 | 1 ± 0.1 |
| 7e | 88 ± 3.4 g | 0.19 | 0.14–0.26 | 56 ± 2.4 | 1.62 | 0.98–2.06 | 2 ± 0.2 |
| 6f | 89 ± 0.9 | 3.56 | 2.37–5.34 | 60 ± 0.7 e | 0.28 | 0.17–0.38 | 14 ± 0.8 |
| 6g | 94 ± 2.5 | 2.05 | 1.76–2.38 | 84 ± 3.6 | 4.21 | 3.87–5.03 | 25 ± 1.0 |
| 7g | 82 ± 3.6 | 0.48 | 0.32–0.74 | 72 ± 2.8 | 2.61 | 2.03–3.04 | 6 ± 0.3 |
| 6h | 74 ± 3.4 | 0.32 | 0.21–0.48 | 72 ± 1.9 | 2.24 | 1.27–3.95 | 5 ± 0.3 |
| 7h | 69 ± 1.6 | 0.79 | 0.55–1.15 | 71 ± 1.6 | 7.29 | 4.13–10.89 | 25 ± 0.7 |
| 6i | 78 ± 2.6 | 1.41 | 0.90–2.00 | 92 ± 0.8 | 8.12 | 5.76–11.44 | 40 ± 2.4 |
| 7i | 82 ± 0.3 e | 0.17 | 0.12–0.21 | 73 ± 1.9 e | 0.66 | 0.49–0.90 | 2 ± 0.1 f |
| 7j | 80 ± 1.9 f | 0.57 | 0.42–0.78 | 56 ± 2.2 g | 0.19 | 0.13–0.28 | 5 ± 0.1 |
| 7l | 90 ± 3.4 | 0.31 | 0.21–0.45 | 86 ± 2.4 | 2.05 | 1.57–2.98 | 29 ± 2.4 |
| Compd | Aorta | Ileum | ||||
|---|---|---|---|---|---|---|
| Activity a M ± S.E.M. | EC50 b (µM) | 95% c.l. | Activity a M ± S.E.M. | EC50 b (µM) | 95% c.l. | |
| Diltiazem | 88 ± 2.3 | 2.6 | 2.2–3.1 | 98 ± 1.5 c | 0.11 | 0.085–0.13 |
| 5 | 15 ± 0.9 | 26 ± 2.1 | ||||
| 6a | 3 ± 0.2 | |||||
| 7a | 18 ± 0.9 | |||||
| 6b | 5 ± 0.3 | 88 ± 2.7 | 12.89 | 10.06–15.66 | ||
| 7b | 2 ± 0.1 | |||||
| 6c | 4 ± 0.2 | |||||
| 7c | 10 ± 0.1 | 96 ± 3.5 | 7.81 | 6.73–9.06 | ||
| 6d | 14 ± 0.3 | |||||
| 7d | 28 ± 1.4 | |||||
| 7e | 17 ± 0.9 | 59 ± 3.9 | 13.38 | 10.12–17.68 | ||
| 6f | 3 ± 0.2 | |||||
| 6g | 2 ± 0.1 | 79 ± 1.6 d | 0.17 | 0.10–0.21 | ||
| 7g | 14 ± 0.6 | 38 ± 1.7 | ||||
| 6h | 2 ± 0.1 | 84 ± 3.1 e | 0.00018 | 0.00014–0.00024 | ||
| 7h | 2 ± 0.1 | 58 ± 2.3 d | 0.82 | 0.53–1.27 | ||
| 6i | 2 ± 0.1 | |||||
| 7i | 2 ± 0.1 | |||||
| 7j | 1 ± 0.09 | |||||
| 7l | 4 ± 0.2 | 83 ± 3.3 | 2.79 | 2.03–3.82 | ||
| Compd | A2780 | A2780/DX3 | MDA-MB-453 | SHSY5Y | |
|---|---|---|---|---|---|
| IC50 a | IC50 | IC5 a | IC50 | IC50 | |
| 6a | >30 | >30 | 4.7 | >30 | >30 |
| 6d | >30 | >30 | 12.7 | >30 | >30 |
| 6f | >30 | >30 | 1.0 | >30 | >30 |
| 7a | >30 | >30 | 3.6 | >30 | >30 |
| 7d | >30 | >30 | 6.8 | >30 | >30 |
| 7h | >30 | >30 | 6.4 | >30 | >30 |
| 7i | >30 | >30 | 34.7 | >30 | >30 |
| 7l | >30 | >30 | 1.0 | >30 | >30 |
| Compd | ||||||||
|---|---|---|---|---|---|---|---|---|
| 6a | 6d | 6f | 7a | 7d | 7h | 7i | 7l | |
| % doxorubicin accumulation a | 102 ± 12 b | 128 ± 9 | 124 ± 4 | 104 ± 7 | 111 ± 5 | 98 ± 8 | 102 ± 10 | 103 ± 8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Micucci, M.; Viale, M.; Chiarini, A.; Spinelli, D.; Frosini, M.; Tavani, C.; Maccagno, M.; Bianchi, L.; Gangemi, R.; Budriesi, R. 3-Aryl-4-nitrobenzothiochromans S,S-dioxide: From Calcium-Channel Modulators Properties to Multidrug-Resistance Reverting Activity. Molecules 2020, 25, 1056. https://doi.org/10.3390/molecules25051056
Micucci M, Viale M, Chiarini A, Spinelli D, Frosini M, Tavani C, Maccagno M, Bianchi L, Gangemi R, Budriesi R. 3-Aryl-4-nitrobenzothiochromans S,S-dioxide: From Calcium-Channel Modulators Properties to Multidrug-Resistance Reverting Activity. Molecules. 2020; 25(5):1056. https://doi.org/10.3390/molecules25051056
Chicago/Turabian StyleMicucci, Matteo, Maurizio Viale, Alberto Chiarini, Domenico Spinelli, Maria Frosini, Cinzia Tavani, Massimo Maccagno, Lara Bianchi, Rosaria Gangemi, and Roberta Budriesi. 2020. "3-Aryl-4-nitrobenzothiochromans S,S-dioxide: From Calcium-Channel Modulators Properties to Multidrug-Resistance Reverting Activity" Molecules 25, no. 5: 1056. https://doi.org/10.3390/molecules25051056
APA StyleMicucci, M., Viale, M., Chiarini, A., Spinelli, D., Frosini, M., Tavani, C., Maccagno, M., Bianchi, L., Gangemi, R., & Budriesi, R. (2020). 3-Aryl-4-nitrobenzothiochromans S,S-dioxide: From Calcium-Channel Modulators Properties to Multidrug-Resistance Reverting Activity. Molecules, 25(5), 1056. https://doi.org/10.3390/molecules25051056