Mono-, Di- and Tetra-iron Complexes with Selenium or Sulphur Functionalized Vinyliminium Ligands: Synthesis, Structural Characterization and Antiproliferative Activity

 , ,
, ,  , , ,
, , , 
Abstract
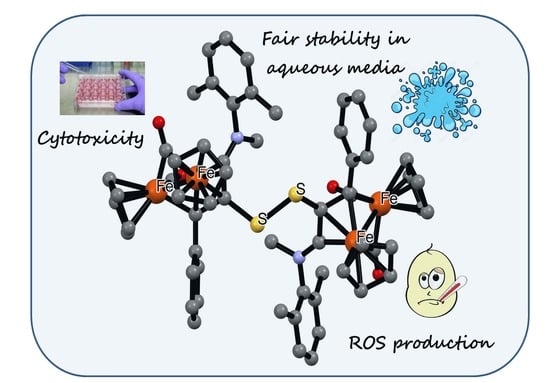
:
1. Introduction
2. Results and Discussion
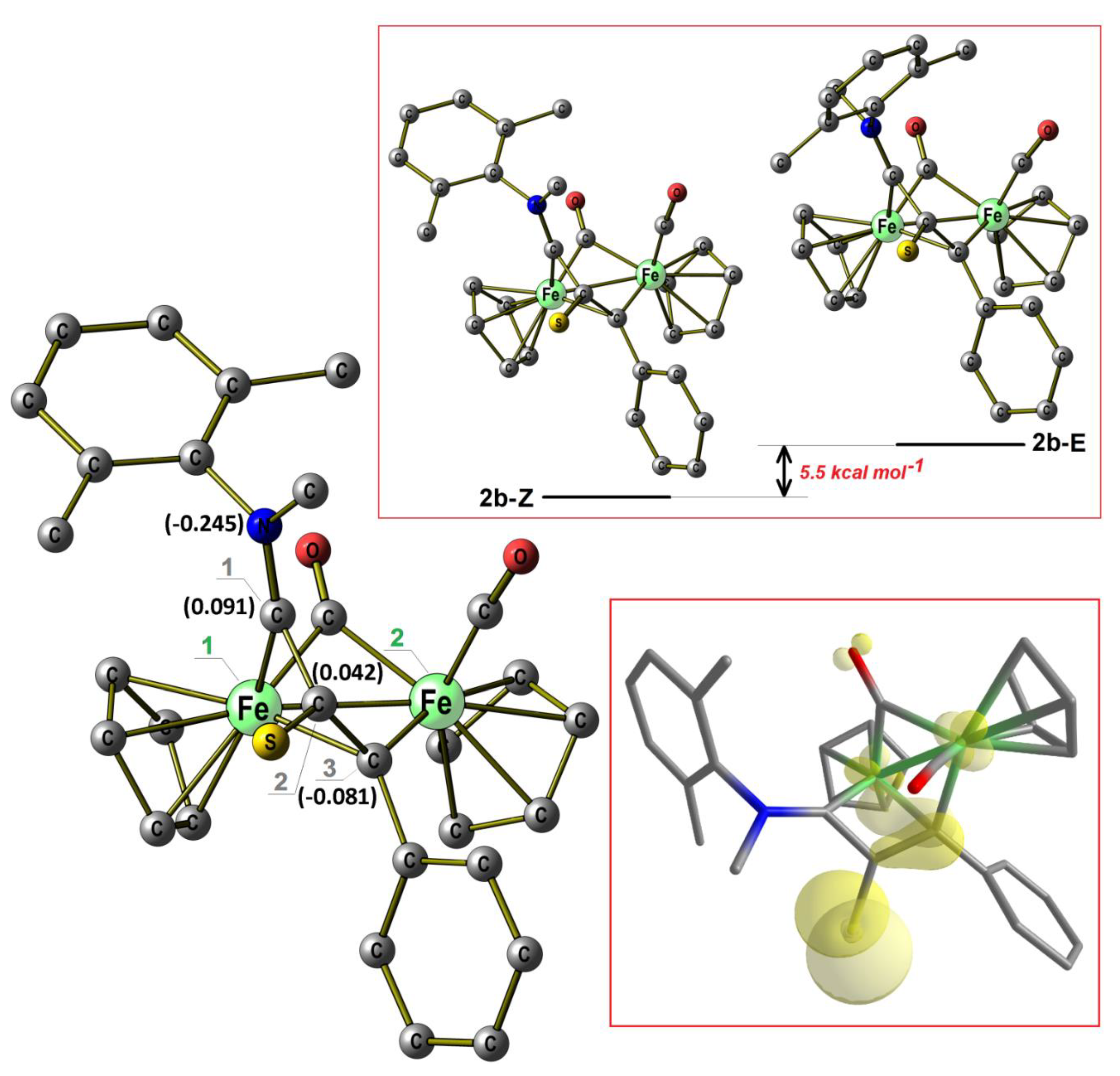
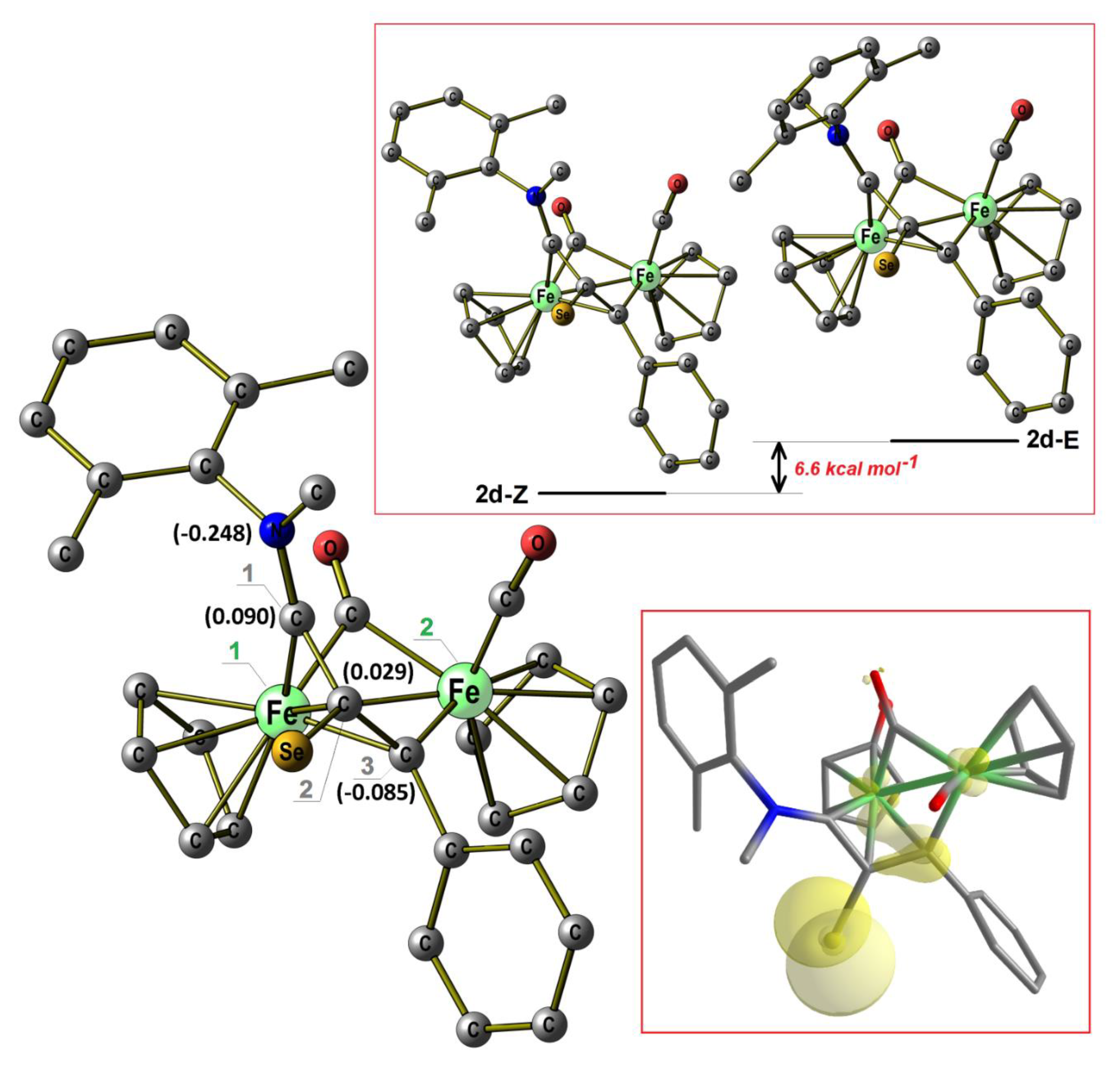
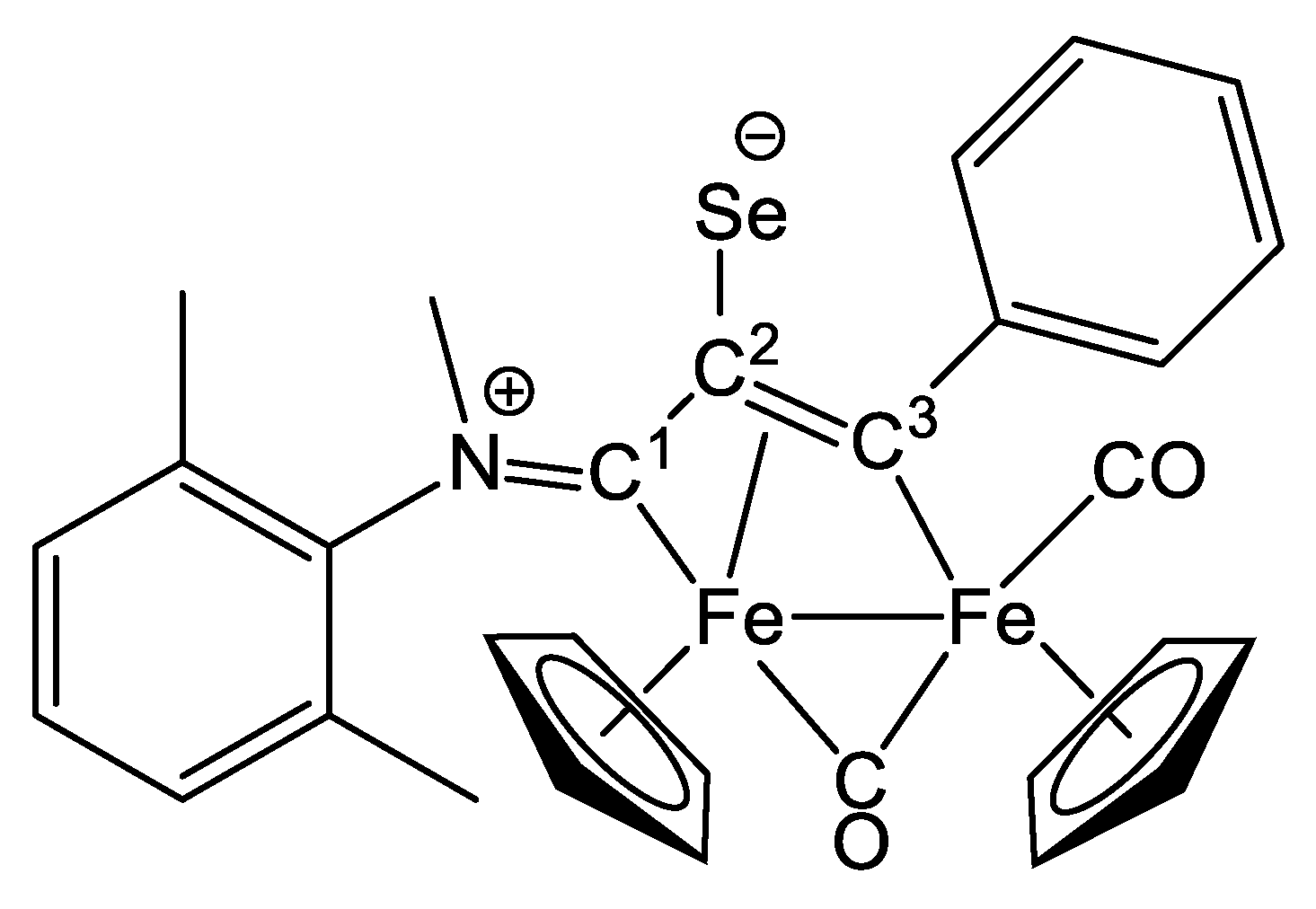
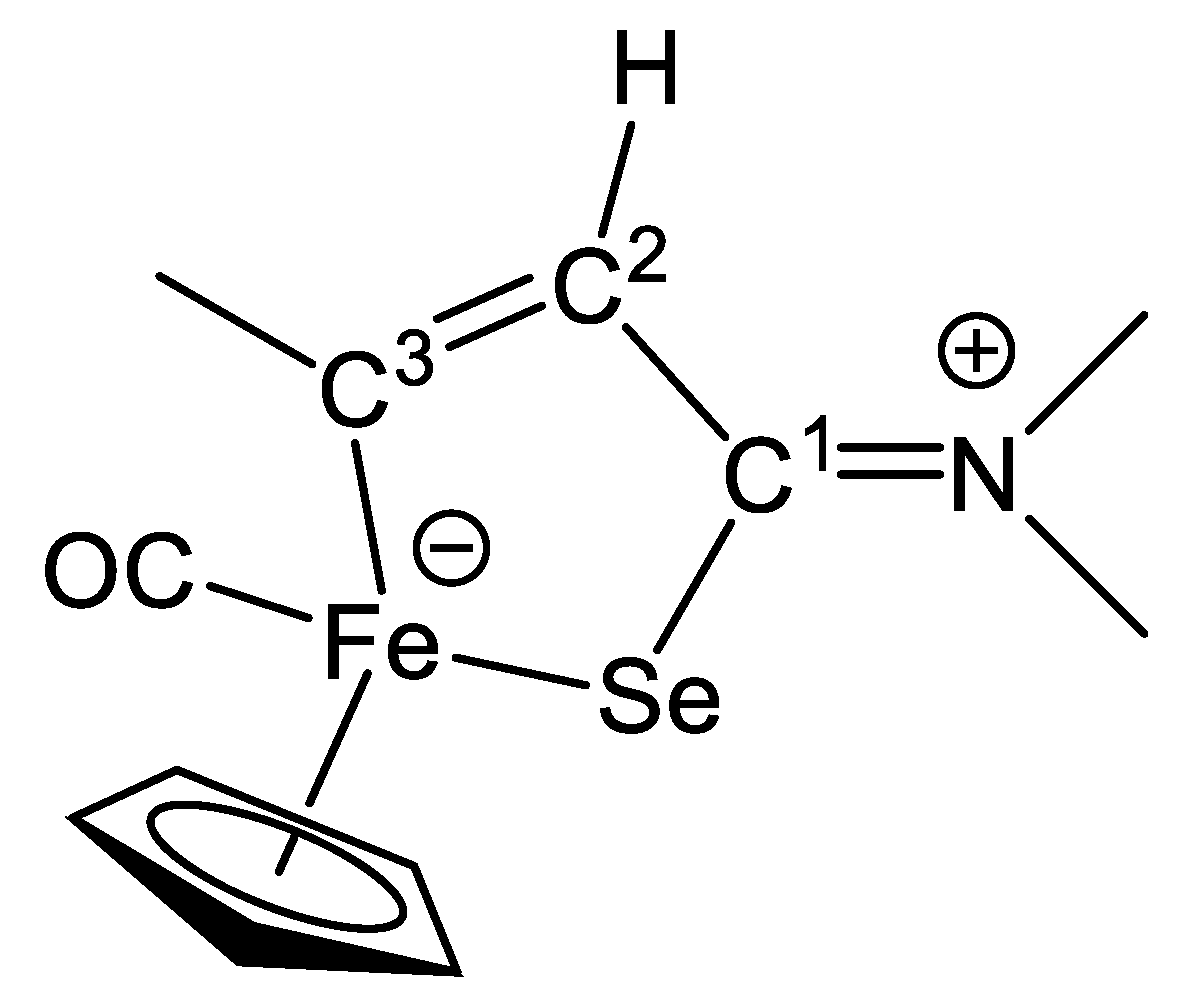
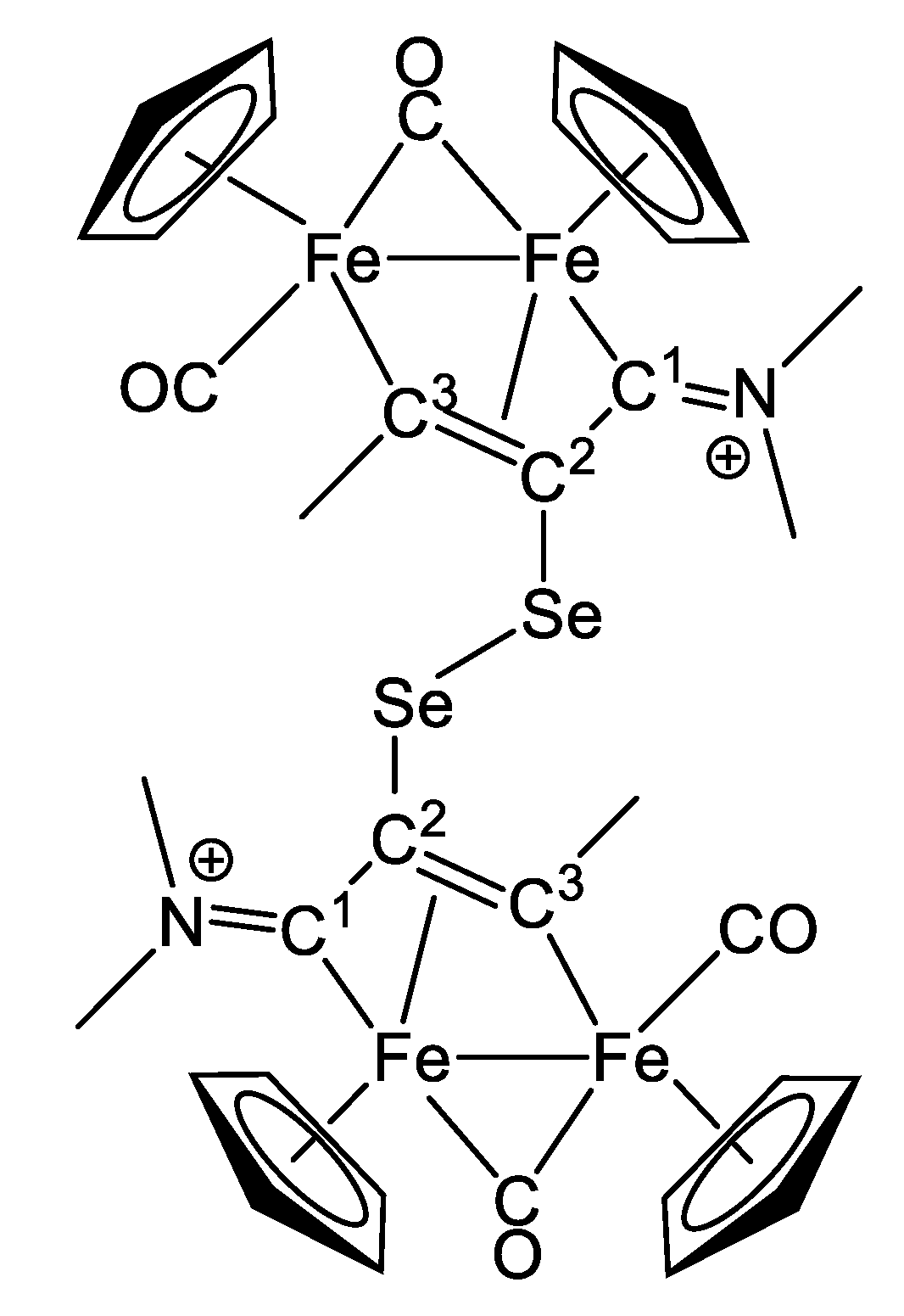
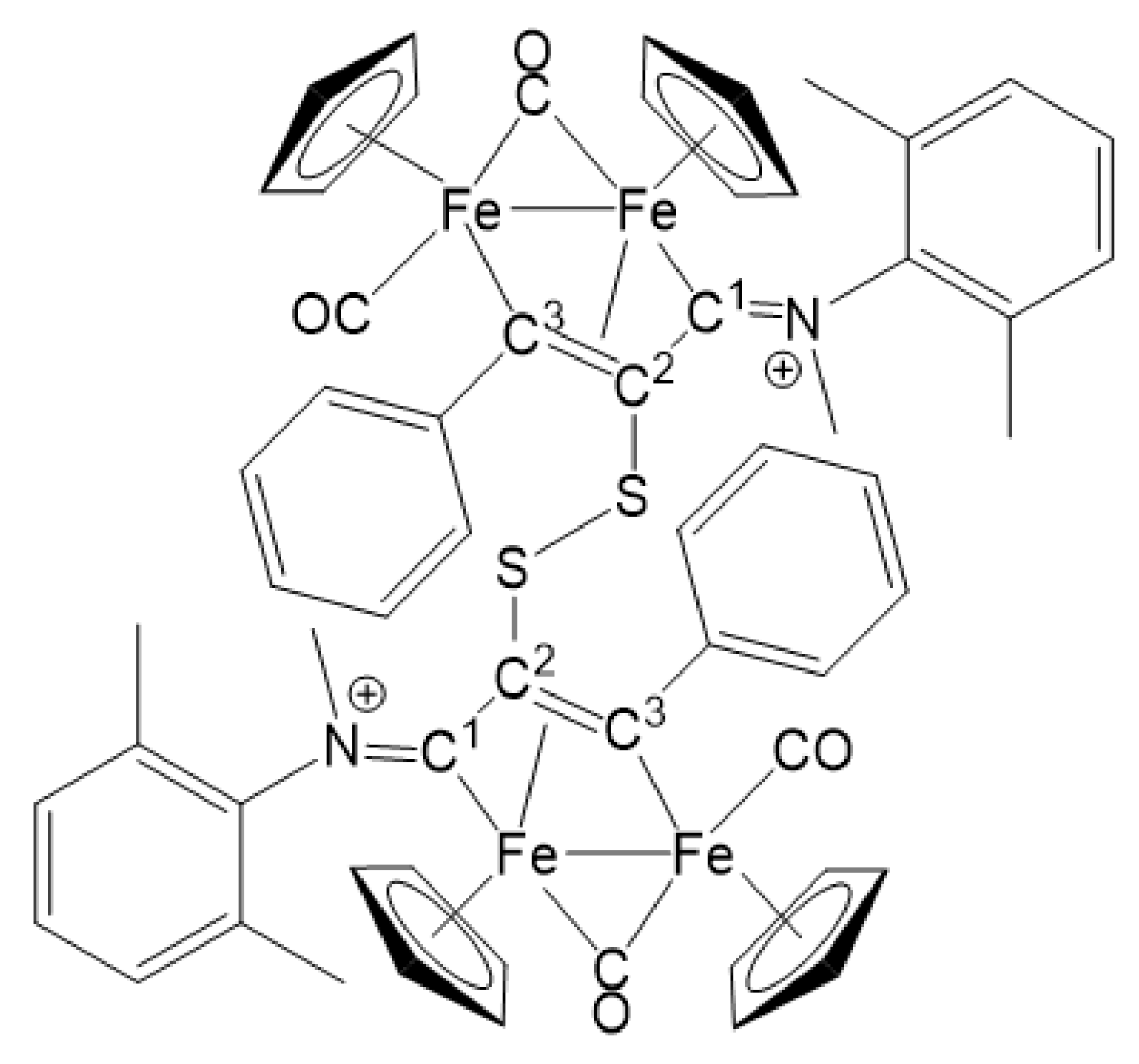
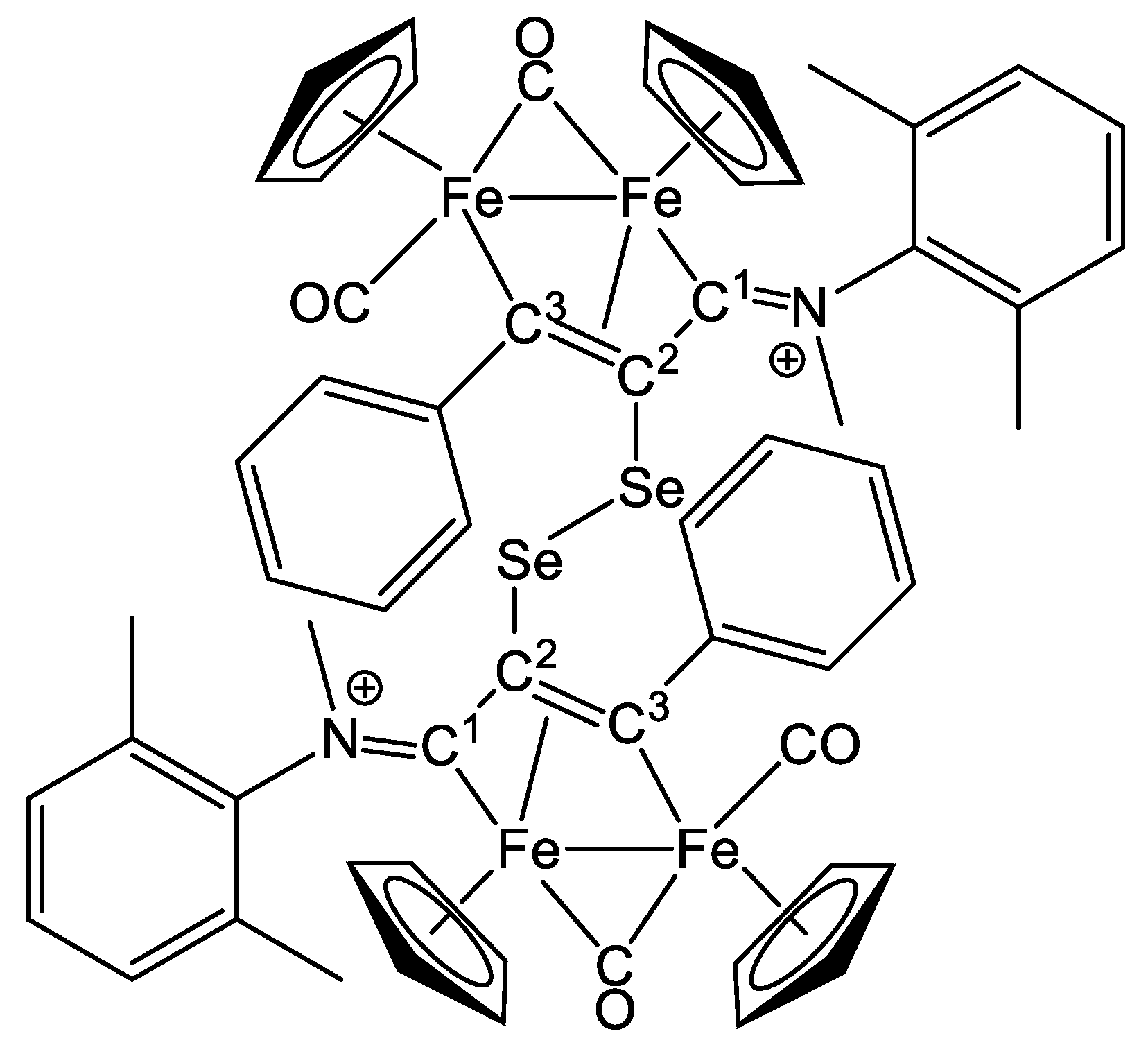
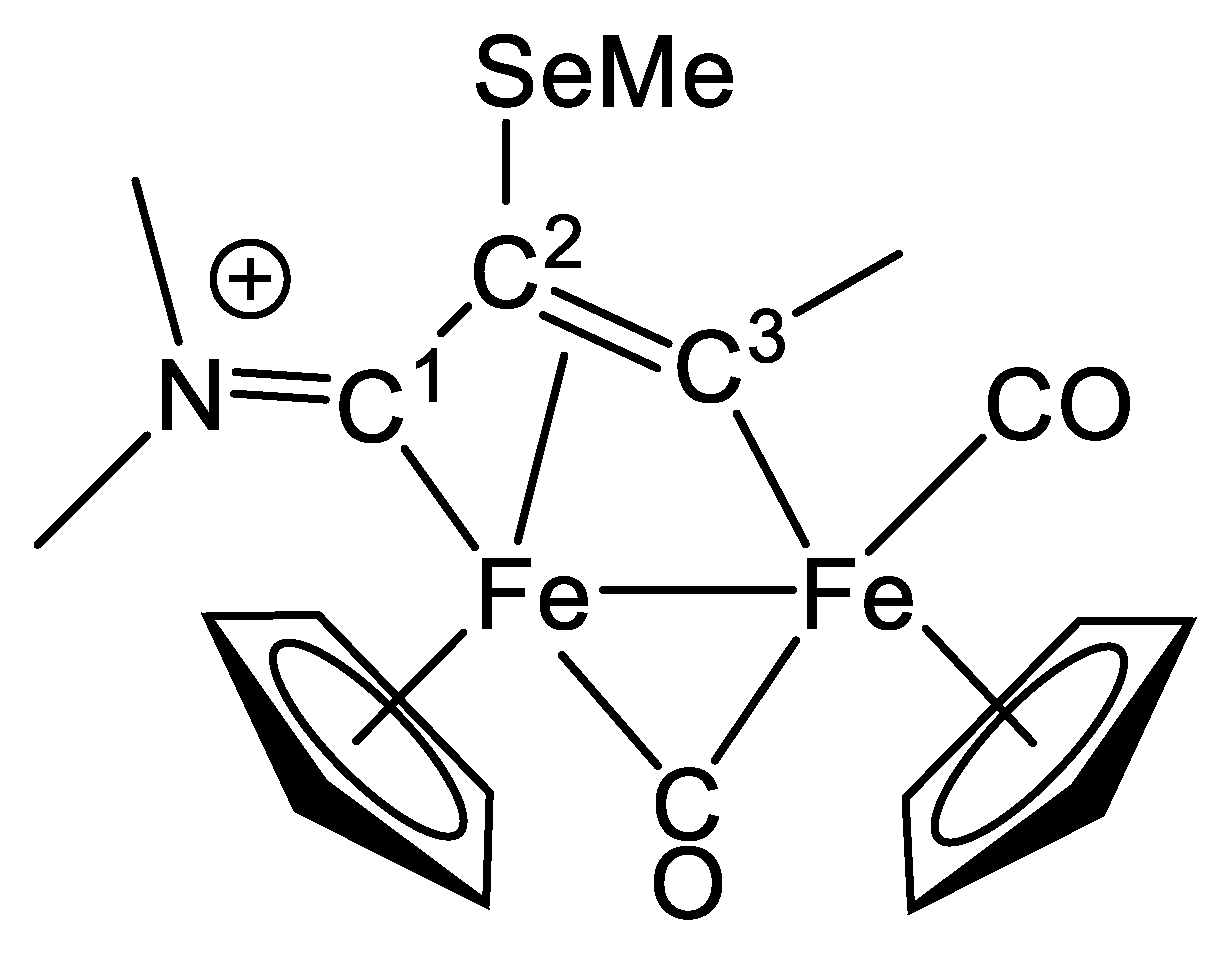
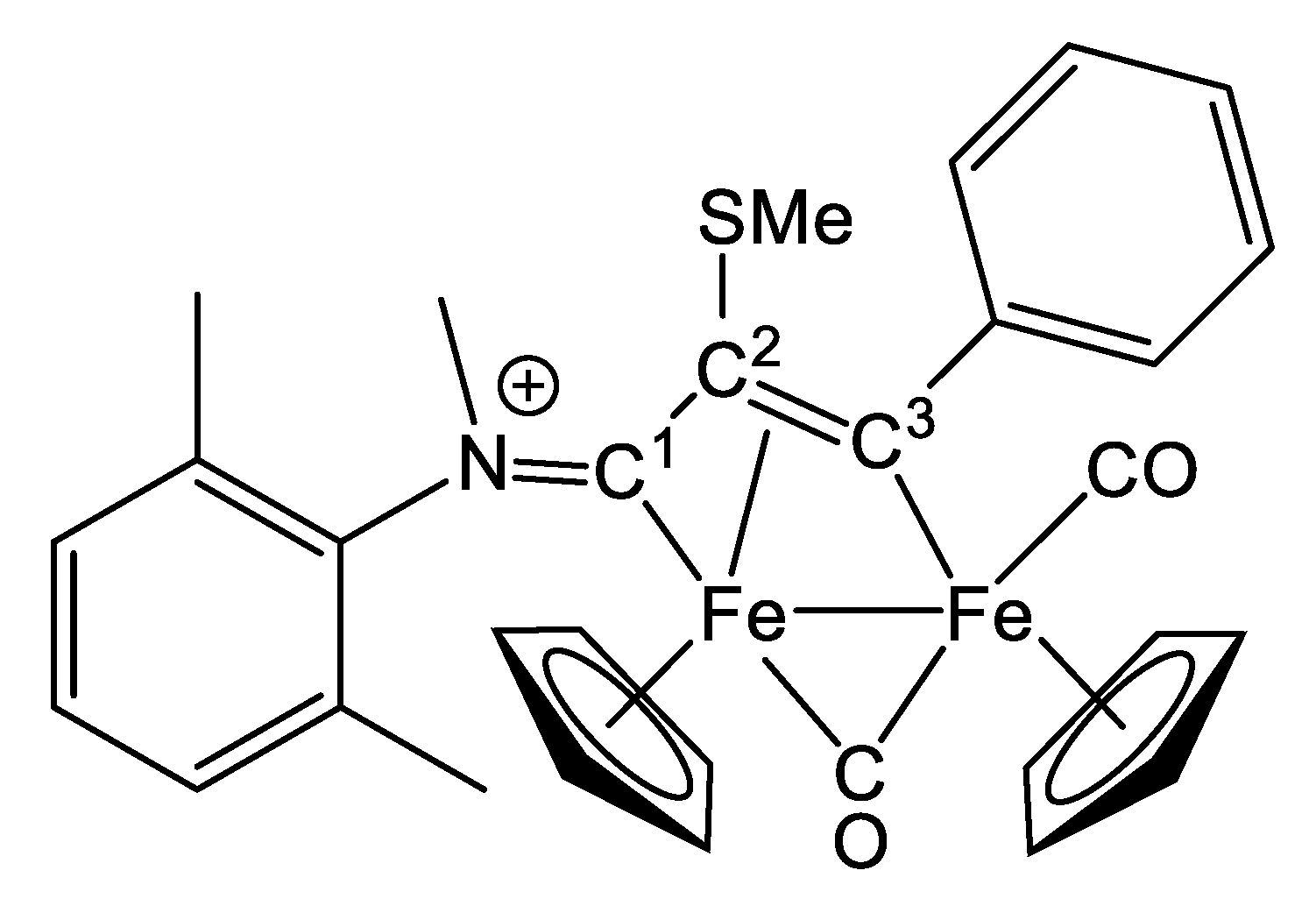
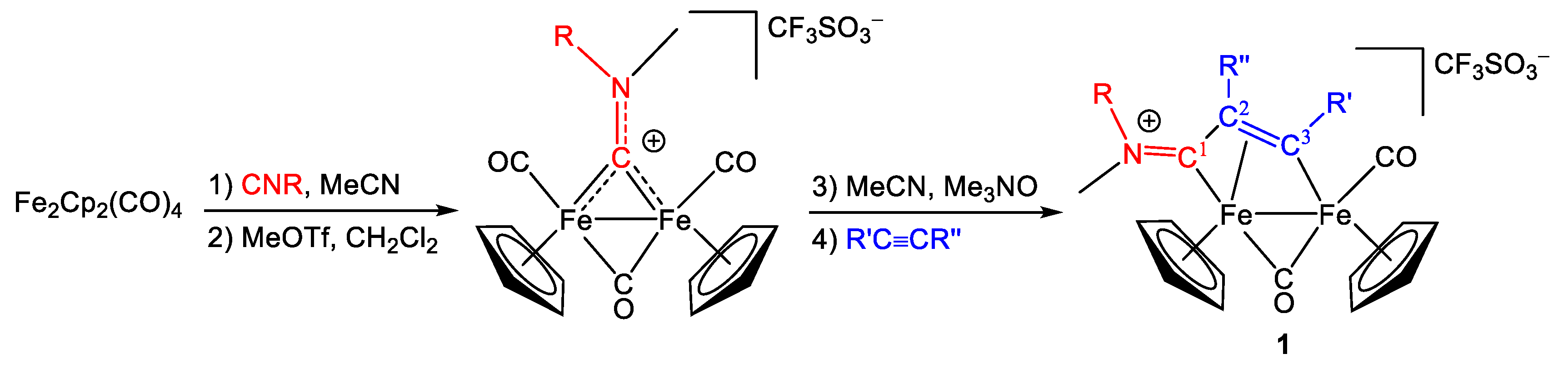
2.1. Synthesis and Characterization of Compounds, and DFT Analysis
Synthesis and Characterization of Compounds
2.2. Electrochemistry
2.3. Cytotoxicity Studies and Stability in Aqueous Media
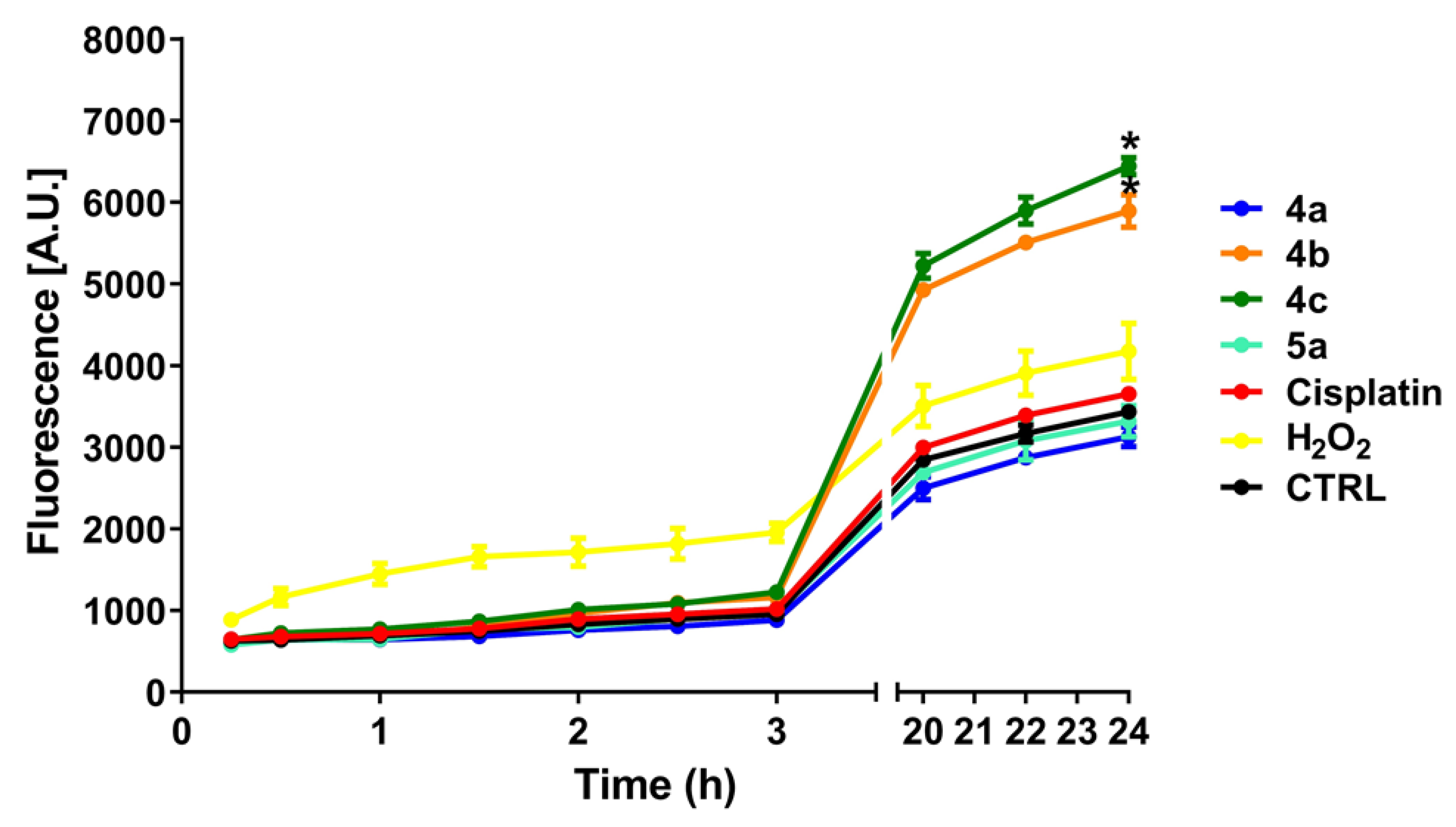
2.4. ROS Production and NADH Oxidation
3. Experimental
3.1. Synthetic Procedures and Compound Characterization
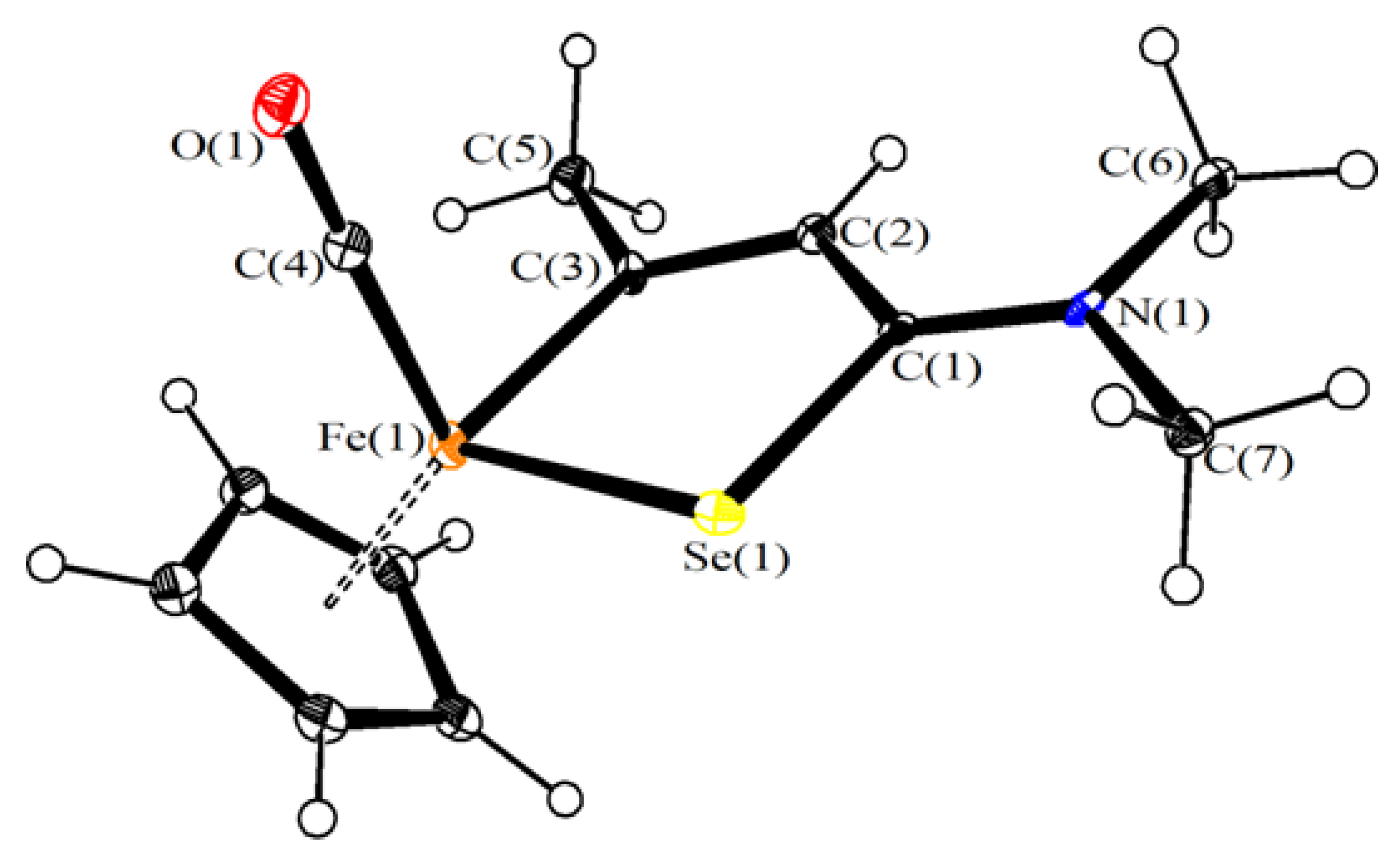
3.2. X-Ray Crystallography
3.3. Computational Studies
3.4. Stability in Aqueous Solutions
3.5. Electrochemistry
3.6. Cell Culture and Cytotoxicity Studies
3.7. ROS Production Assessment
3.8. Catalytic NADH Oxidation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Apps, M.G.; Choi, E.H.Y.; Wheate, N.J. The state-of-play and future of platinum drugs. Endocr. Relat. Cancer 2015, 22, R219–R233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, D. The mechanism of action of platinum anticancer agents—What do we really know about it? Dalton Trans. 2009, 10681–10689. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The Next Generation of Platinum Drugs: Targeted Pt(II) Agents, Nanoparticle Delivery, and Pt(IV) Prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raudenska, M.; Balvan, J.; Fojtu, M.; Gumulec, J.; Masarik, M. Unexpected therapeutic effects of cisplatin. Metallomics 2019, 11, 1182–1199. [Google Scholar] [CrossRef] [PubMed]
- Oun, R.; Moussa, Y.E.; Wheate, N.J. The side effects of platinum-based chemotherapy drugs: A review for chemists. Dalton Trans. 2018, 47, 6645–6653. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.; Mao, C.Q.; Chen, S.; Ma, G.; Wang, J.; Liu, Y. Combating the Drug Resistance of Cisplatin Using a Platinum Prodrug Based Delivery System. Angew. Chem. 2012, 124, 6846–6851. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of cytotoxic action and molecular basis of resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [Green Version]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Kartalou, M.; Essigmann, J.M. Mechanisms of resistance to cisplatin. Mutat. Res. 2001, 478, 23–43. [Google Scholar] [CrossRef]
- Zhang, P.; Sadler, P.J. Advances in the design of organometallic anticancer complexes. J. Organomet. Chem. 2017, 839, 5–14. [Google Scholar] [CrossRef]
- Barry, N.P.E.; Sadler, P.J. Exploration of the medical periodic table: Towards new targets. Chem. Commun. 2013, 49, 5016–5041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Štarha, P.; Trávníček, Z. Non-platinum complexes containing releasable biologically active ligands. Coord. Chem. Rev. 2019, 395, 130–145. [Google Scholar] [CrossRef]
- Santini, C.; Pellei, M.; Gandin, V.; Porchia, M.; Tisato, F.; Marzano, C. Advances in Copper Complexes as Anticancer Agents. Chem. Rev. 2014, 114, 815–862. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.N.; Huq, F. Comprehensive review on tumour active palladium compounds and structure–activity relationships. Coord. Chem. Rev. 2016, 316, 36–67. [Google Scholar] [CrossRef]
- Nazarov, A.A.; Hartinger, C.G.; Dyson, P.J. Opening the lid on piano-stool complexes: An account of ruthenium(II)-arene complexes with medicinal applications. J. Organomet. Chem. 2014, 751, 251–260. [Google Scholar] [CrossRef]
- Rilak Simovica, A.; Masnikosa, R.; Bratsos, I.; Alessio, E. Chemistry and reactivity of ruthenium(II) complexes: DNA/protein binding mode and anticancer activity are related to the complex structure. Coord. Chem. Rev. 2019, 398, 113011. [Google Scholar] [CrossRef]
- Zou, T.; Lum, C.T.; Lok, C.N.; Zhang, J.J.; Che, C.M. Chemical biology of anticancer gold(iii) and gold(i) complexes. Chem. Soc. Rev. 2015, 44, 8786–8801. [Google Scholar] [CrossRef] [PubMed]
- Lazarevi, T.; Rilak, A.; Bugarcic, Z.D. Platinum, palladium, gold and ruthenium complexes as anticancer agents: Current clinical uses, cytotoxicity studies and future perspectives. Eur. J. Med. Chem. 2017, 142, 8–31. [Google Scholar] [CrossRef] [PubMed]
- Bratsos, T.; Gianferrara, E.; Alessio, C.G.; Hartinger, M.A.; Jakupec, B.K. Keppler, Bioinorganic Medicinal Chemistry; Alessio, E., Ed.; Wiley-VCH: Weinheim, Germany, 2011; pp. 151–174. [Google Scholar]
- Lord, R.; Zegke, M.; Henderson, I.R.; Pask, C.M.; Shepherd, H.J.; McGowan, P.C. β-Ketoiminato Iridium(III) organometallic complexes: Selective cytotoxicity towards colorectal cancer cells HCT116 p53-/-. Chem. Eur. J. 2019, 25, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Gallardo, J.; Elie, B.T.; Sadhukha, T.; Prabha, S.; Sanau, M.; Rotenberg, S.A.; Ramos, J.W.; Contel, M. Heterometallic titanium–gold complexes inhibit renal cancer cells in vitro and in vivo. Chem. Sci. 2015, 6, 5269–5283. [Google Scholar] [CrossRef] [Green Version]
- Patra, M.; Gasser, G. The medicinal chemistry of ferrocene and its derivatives. Nat. Rev. Chem. 2017, 1, 1–12. [Google Scholar] [CrossRef]
- Braga, S.S.; Silva, A.M.S. A New Age for Iron: Antitumoral Ferrocenes. Organometallics 2013, 32, 5626–5639. [Google Scholar] [CrossRef]
- Jaouen, G.; Vessiéres, A.; Top, S. Ferrocifen type anti cancer drugs. Chem. Soc. Rev. 2015, 44, 8802–8817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agonigi, G.; Biancalana, L.; Lupo, M.G.; Montopoli, M.; Ferri, N.; Zacchini, S.; Binacchi, F.; Biver, T.; Campanella, B.; Pampaloni, G.; et al. Exploring the anticancer potential of diiron bis-cyclopentadienyl complexes with bridging hydrocarbyl ligands: Behavior in aqueous media and in vitro cytotoxicity. Organometallics 2020, 39, 645–657. [Google Scholar] [CrossRef]
- Hirschhäuser, C.; Velcicky, J.; Schlawe, D.; Hessler, E.; Majdalani, A.; Neudçrfl, J.M.; Prokop, A.; Wieder, T.; Schmalz, H.G. Nucleoside analogues with a 1,3-diene-Fe(CO)3 substructure: Stereoselective synthesis, configurational assignment, and apoptosis-inducing activity. Chem. Eur. J. 2013, 19, 13017–13029. [Google Scholar] [CrossRef]
- Cingolani, A.; Zanotti, V.; Zacchini, S.; Massi, M.; Simpson, P.V.; Maheshkumar Desai, N.; Casari, I.; Falasca, M.; Rigamonti, R.; Mazzoni, R. Synthesis, reactivity and preliminary biological activity of iron(0) complexes with cyclopentadienone and amino-appended N-heterocyclic carbene ligands. Appl. Organomet. Chem. 2019, 33, e4779. [Google Scholar] [CrossRef]
- Prinz, C.; Vasyutina, E.; Lohmann, G.; Schrader, A.; Romanski, S.; Hirschhäuser, C.; Mayer, P.; Frias, C.; Herling, C.D.; Hallek, M.; et al. Organometallic nucleosides induce non-classical leukemic cell death that is mitochondrial-ROS dependent and facilitated by TCL1-oncogene burden Organometallic nucleosides induce non-classical leukemic cell death that is mitochondrial-ROS dependent and facilitated by TCL1-oncogene burden. Mol. Cancer 2015, 14, 114. [Google Scholar]
- Pathania, S.; Narang, R.K.; Rawal, R.K. Role of sulphur-heterocycles in medicinal chemistry: An update. Eur. J. Med. Chem. 2019, 180, 486–508. [Google Scholar] [CrossRef]
- Gandin, V.; Fernandes, A.P. Organoselenium Compounds in Biology and Medicine: Synthesis, Biological and Therapeutic Treatments; Jain, V.K., Priyadarsini, K.I., Eds.; The Royal Society of Chemistry: London, UK, 2018. [Google Scholar]
- Gandin, V.; Khalkar, P.; Braude, J.; Fernandes, A.P. Organic selenium compounds as potential chemotherapeutic agents for improved cancer treatment. Free Radic. Biol. Med. 2018, 127, 80–97. [Google Scholar] [CrossRef]
- Fernandes, A.P.; Gandin, V. Selenium compounds as therapeutic agents in cancer. Biochim. Biophys. Acta 2015, 1850, 1642–1660. [Google Scholar] [CrossRef]
- Tan, H.W.; Mo, H.Y.; Lau, A.T.Y.; Xu, Y.M. Selenium Species: Current Status and Potentials in Cancer Prevention and Therapy. Int. J. Mol. Sci. 2019, 20, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spengler, G.; Gajdács, M.; Marć, M.A.; Domínguez-Álvarez, E.; Sanmartín, C. Organoselenium Compounds as Novel Adjuvants of Chemotherapy Drugs—A Promising Approach to Fight Cancer Drug Resistance. Molecules 2019, 24, 336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amouri, H.; Moussa, J.; Renfrew, A.K.; Dyson, P.J.; Rager, M.N.; Chamoreau, L.M. Discovery, structure, and anticancer activity of an iridium complex of diselenobenzoquinone. Angew. Chem. Int. Ed. 2010, 49, 7530–7533. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Su, Q. Molecular mechanisms for the anti-cancer effects of diallyl disulfide. Food Chem. Toxic. 2013, 57, 362–370. [Google Scholar] [CrossRef]
- Cytarska, J.; Skowerski, K.; Jaworski, S.; Misiura, K.; Filip-Psurska, B.; Wietrzyk, J. The Disulfide Analogues of Isophosphoramide Mustard for Anticancer Therapy. Lett. Drug Des. Discov. 2015, 12, 172–179. [Google Scholar] [CrossRef]
- Álvarez-Pérez, M.; Ali, W.; Marć, M.A.; Handzlik, J.; Domínguez-Álvarez, E. Selenides and Diselenides: A Review of Their Anticancer and Chemopreventive Activity. Molecules 2018, 23, 628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocco, D.; Batchelor, L.K.; Agonigi, G.; Braccini, S.; Chiellini, F.; Schoch, S.; Biver, T.; Funaioli, T.; Zacchini, S.; Biancalana, L.; et al. Anticancer Potential of Diiron Vinyliminium Complexes. Chem. Eur. J. 2019, 25, 14801–14816. [Google Scholar] [CrossRef]
- Agonigi, G.; Bortoluzzi, M.; Marchetti, F.; Pampaloni, G.; Zacchini, S.; Zanotti, V. Regioselective Nucleophilic Additions to Diiron Carbonyl Complexes Containing a Bridging Aminocarbyne Ligand: A Synthetic, Crystallographic and DFT Study. Eur. J. Inorg. Chem. 2018, 8, 960–971. [Google Scholar] [CrossRef]
- Albano, V.G.; Busetto, L.; Marchetti, F.; Monari, M.; Zacchini, S.; Zanotti, V. Diiron μ-Vinyliminium Complexes from Acetylene Insertion into a Metal−Aminocarbyne Bond. Organometallics 2003, 22, 1326–1331. [Google Scholar] [CrossRef]
- Ciancaleoni, G.; Zacchini, S.; Zanotti, V.; Marchetti, F. DFT Mechanistic Insights into the Alkyne Insertion Reaction Affording Diiron μ-Vinyliminium Complexes and New Functionalization Pathways. Organometallics 2018, 37, 3718–3731. [Google Scholar] [CrossRef]
- Marchetti, F. Constructing Organometallic Architectures from Aminoalkylidyne Diiron Complexes. Eur. J. Inorg. Chem. 2018, 3987–4003. [Google Scholar] [CrossRef] [Green Version]
- Mazzoni, R.; Salmi, M.; Zanotti, V. C–C Bond Formation in Diiron Complexes. Chem. Eur. J. 2012, 18, 10174–10194. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, F.; Zacchini, S.; Zanotti, V. Amination of Bridging Vinyliminium Ligands in Diiron Complexes: C–N Bond Forming Reactions for Amidine-Alkylidene Species. Organometallics 2018, 37, 107–115. [Google Scholar] [CrossRef]
- Schoch, S.; Batchelor, L.K.; Funaioli, T.; Ciancaleoni, G.; Zacchini, S.; Braccini, S.; Chiellini, F.; Biver, T.; Pampaloni, G.; Dyson, P.J.; et al. Diiron Complexes with a Bridging Functionalized Allylidene Ligand: Synthesis, Structural Aspects, and Cytotoxicity. Organometallics 2020, 39, 361–373. [Google Scholar] [CrossRef]
- Agonigi, G.; Ciancaleoni, G.; Funaioli, T.; Zacchini, S.; Pineider, F.; Pinzino, C.; Pampaloni, G.; Zanotti, V.; Marchetti, F. Controlled Dissociation of Iron and Cyclopentadienyl from a Diiron Complex with a Bridging C3 Ligand Triggered by One-Electron Reduction. Inorg. Chem. 2018, 57, 15172–15186. [Google Scholar] [CrossRef]
- Busetto, L.; Marchetti, F.; Zacchini, S.; Zanotti, V. Unprecedented Zwitterionic Iminium−Chalcogenide Bridging Ligands in Diiron Complexes. Organometallics 2006, 25, 4808–4816. [Google Scholar] [CrossRef]
- Busetto, L.; Dionisio, M.; Marchetti, F.; Mazzoni, R.; Salmi, M.; Zacchini, S.; Zanotti, V. Zwitterionic diiron vinyliminium complexes: Alkylation, metalation and oxidative coupling at the S and Se functionalities. J. Organomet. Chem. 2008, 693, 2383–2391. [Google Scholar] [CrossRef]
- Busetto, L.; Marchetti, F.; Mazzoni, R.; Salmi, M.; Zacchini, S.; Zanotti, V. SPh functionalized bridging-vinyliminium diiron and diruthenium complexes. J. Organomet. Chem. 2008, 693, 3191–3196. [Google Scholar] [CrossRef]
- Rocco, D.; Batchelor, L.K.; Ferretti, E.; Zacchini, S.; Pampaloni, G.; Dyson, P.J.; Marchetti, F. Piano Stool Aminoalkylidene-Ferracyclopentenone Complexes from Bimetallic Precursors: Synthesis and Cytotoxicity Data. ChemPlusChem 2020, 85, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Shao, L.; Geib, S.J.; Cooper, N.J. Electrophilic Addition of the Carbene Ligand in [Fe(CO)2(η5-C5H5){CMe(OMe)}]+ to the Reductively Activated Benzene Ligand in [Mn(CO)3(η4-C6H6)]−. Organometallics 2003, 22, 4361–4363. [Google Scholar] [CrossRef]
- Hurley, A.L.; Welker, M.E.; Day, C.S. On the Configuration Resulting from Oxidative Addition of RX to Pd(PPh3)4 and the Mechanism of the cis-to-trans Isomerization of [PdRX(PPh3)2] Complexes (R = Aryl, X = Halide). Organometallics 1998, 17, 2832–2838. [Google Scholar] [CrossRef]
- Busetto, L.; Marchetti, F.; Zacchini, S.; Zanotti, V.; Zoli, E. Nitrile ligands activation in dinuclear aminocarbyne complexes. J. Organomet. Chem. 2005, 690, 1959–1970. [Google Scholar] [CrossRef]
- Busetto, L.; Marchetti, F.; Salmi, M.; Zacchini, S.; Zanotti, V. Coupling of Allenes with Alkylidyne Ligands in Diiron Complexes: Synthesis of Novel Bridging Thio- and Aminobutadienylidene Complexes. Eur. J. Inorg. Chem. 2008, 15, 2437–2447. [Google Scholar] [CrossRef]
- El-khateeb, M. Preparation, characterization and structure determination of CpFe(CO)(EPh3)SeCO-het complexes. J. Mol. Struct. 2016, 1123, 300–304. [Google Scholar] [CrossRef]
- El-khateeb, M.; Harb, M.; Mansour, A.; Yousuf, S. Photochemical substitution of a single CO ligand of CpFe(CO)2SeC(Y)Y′Ar [(Y)Y′=(O)O,(S)O and (S)S] by EPh3 (E = P, As, Sb). Inorg. Chim. Acta 2019, 486, 694–697. [Google Scholar] [CrossRef]
- Marchetti, F.; Zacchini, S.; Zanotti, V. C–S and C–Se Bond Formation at Bridging Vinyliminium Ligands in Diiron Complexes. Eur. J. Inorg. Chem. 2013, 29, 5145–5152. [Google Scholar] [CrossRef]
- Hecht, C.; Herdtweck, E.; Rohrmann, J.; Herrmann, W.A.; Beck, W.; Fritz, P.M. Mehrfachbindungen zwischen Hauptgruppenelementen und Übergangsmetallen: XLVI. Organoeisen-Komplexe mit Selen-Brücken. J. Organomet. Chem. 1987, 330, 389–396. [Google Scholar] [CrossRef]
- Liaw, W.F.; Horng, Y.C.; Ou, D.S.; Ching, C.Y.; Lee, G.H.; Peng, S.M. Distorted Square Planar Ni(II)−Chalcogenolate Carbonyl Complexes [Ni(CO)(SPh)n(SePh)3-n]− (n = 0, 1, 2): Relevance to the Nickel Site in CO Dehydrogenases and [NiFeSe] Hydrogenase. J. Am. Chem. Soc. 1997, 119, 9299–9300. [Google Scholar] [CrossRef]
- Ruiz, J.; Ceroni, M.; Quinzani, O.V.; Riera, V.; Piro, O.E. Reversible S−S Bond Breaking and Bond Formation in Disulfide-Containing Dinuclear Complexes of MnI. Angew. Chem. Int. Ed. 2001, 40, 220–222. [Google Scholar] [CrossRef]
- Murray, B.S.; Babak, M.V.; Hartinger, C.G.; Dyson, P.J. The development of RAPTA compounds for the treatment of tumors. Coord. Chem. Rev. 2016, 306, 86–114. [Google Scholar] [CrossRef]
- Mazzoni, R.; Gabiccini, A.; Cesari, C.; Zanotti, V.; Gualandi, I.; Tonelli, D. Diiron Complexes Bearing Bridging Hydrocarbyl Ligands as Electrocatalysts for Proton Reduction. Organometallics 2015, 34, 3228–3235. [Google Scholar] [CrossRef]
- Du, Q.; Guo, L.; Tian, M.; Ge, X.; Yang, Y.; Jian, X.; Xu, Z.; Tian, Z.; Liu, Z. Potent Half-Sandwich Iridium(III) and Ruthenium(II) Anticancer Complexes Containing a P^O-Chelated Ligand. Organometallics 2018, 37, 2880–2889. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Rad. Biol. Med. 2011, 30, 1191–1212. [Google Scholar] [CrossRef]
- Fu, Y.; Romero, M.J.; Habtemariam, A.; Snowden, M.E.; Song, L.; Clarkson, G.J.; Qamar, B.; Pizarro, A.M.; Unwin, P.R.; Sadler, P.J. The contrasting chemical reactivity of potent isoelectronic iminopyridine and azopyridine osmium(ii) arene anticancer complexes. Chem. Sci. 2012, 3, 2485–2493. [Google Scholar] [CrossRef]
- Menges, F. “Spectragryph—Optical Spectroscopy Software”, Version 1.2.5, @ 2016–2017. Available online: http://www.effemm2.de/spectragryph (accessed on 2 April 2020).
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
- Willker, W.; Leibfritz, D.; Kerssebaum, R.; Bermel, W. Gradient selection in inverse heteronuclear correlation spectroscopy. Magn. Reson. Chem. 1993, 31, 287–292. [Google Scholar] [CrossRef]
- Sheldrick, G.M. TWINABS; Version 2012/1; University of Göttingen: Göttingen, Germany, 2012. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Minenkov, Y.; Singstad, A.; Occhipinti, G.; Jensen, V.R. The accuracy of DFT-optimized geometries of functional transition metal compounds: A validation study of catalysts for olefin metathesis and other reactions in the homogeneous phase. Dalton Trans. 2012, 41, 5526–5541. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Gerber, I.C.; Ángyán, J.G. Hybrid functional with separated range. Chem. Phys. Lett. 2005, 415, 100–105. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Lin, C.Y.; George, M.W.; Gill, P.M.W. EDF2: A Density Functional for Predicting Molecular Vibrational Frequencies. Aust. J. Chem. 2004, 57, 365–370. [Google Scholar] [CrossRef] [Green Version]
- Henre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XIV. An Extended Gaussian-Type Basis for Molecular Orbital Studies of Organic Molecules. Inclusion of Second Row Elements. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; Wiley: Chichester, UK, 2007. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Spartan16. Build 2.0.3; Wavefunction Inc.: Irvine, CA, USA, 2016. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Rosenkranz, A.R.; Schmaldienst, S.; Stuhlmeier, K.M.; Chen, W.; Knapp, W.; Zlabinger, G.J. A microplate assay for the detection of oxidative products using 2’,7’-dichlorofluorescin-diacetate. J. Immunol. Methods 1992, 156, 39–45. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds cited in this work are available from the authors. |















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule 1 | Molecule 2 | |
|---|---|---|
| Fe(1)-Se(1) | 2.391(8) | 2.357(9) |
| Fe(1)-Cp | 2.07(5)–2.17(5) | 2.09(5)–2.10(5) |
| Fe(1)-C(4) | 1.74(4) | 1.83(4) |
| Fe(1)-C(3) | 1.96(4) | 1.92(5) |
| C(4)-O(1) | 1.12(6) | 1.09(5) |
| Se(1)-C(1) | 1.90(4) | 1.88(5) |
| C(1)-C(2) | 1.46(6) | 1.43(6) |
| C(2)-C(3) | 1.39(6) | 1.39(6) |
| C(3)-C(5) | 1.50(6) | 1.43(7) |
| C(1)-N(1) | 1.28(6) | 1.27(6) |
| N(1)-C(6) | 1.53(6) | 1.50(6) |
| N(1)-C(7) | 1.52(5) | 1.52(6) |
| Se(1)-Fe(1)-C(3) | 84.9(13) | 86.6(14) |
| Fe(1)-C(4)-O(1) | 178(4) | 171(4) |
| Fe(1)-Se(1)-C(1) | 96.5(13) | 98.3(14) |
| Se(1)-C(1)-C(2) | 115(3) | 108(3) |
| C(1)-C(2)-C(3) | 118(4) | 127(4) |
| C(2)-C(3)-Fe(1) | 126(3) | 119(4) |
| Sum at N(1) | 360(6) | 360(6) |
| Sum at C(1) | 360(6) | 359(6) |
| Sum at C(3) | 360(6) | 360(6) |
| Compound | Oxidation [V] | Reduction [V] | ΔEp (red) [mV] |
|---|---|---|---|
| 1a | +0.65 a | −1.37 | 108 |
| 1c | +0.73 a | −1.35 a | - |
| 2c | +0.12 a | −1.7 a | - |
| 4c | - | −0.78 a | - |
| 5a | −0.44 a ÷ +0.66 a | −1.29 | 87 |
| Compnd. | A2780 | A2780cisR | HEK-293 |
|---|---|---|---|
| 1aa | 35 ± 3 | 86 ± 7 | >200 |
| 1ba | 0.50 ± 0.06 | 1.2 ± 0.2 | 2.4 ± 0.2 |
| 1ca | 11.6 ± 0.6 | 21.2 ± 1.6 | 13.4 ± 1.0 |
| 3 | 16.1 ± 1.3 | 20 ± 2 | 19 ± 2 |
| 4a | >200 | >200 | >200 |
| 4b | 0.6 ± 0.1 | 1.2 ± 0.6 | 0.72 ± 0.04 |
| 4c | 5.7 ± 0.8 | 12.8 ± 0.7 | 9.1 ± 0.7 |
| 4d | 1.4 ± 0.2 | 2.8 ± 0.3 | 2.2 ± 0.6 |
| 5a | 15.6 ± 0.8 | 28 ± 2 | 26 ± 3 |
| 5b | 0.5 ± 0.2 | 1.4 ± 0.2 | 0.7 ± 0.1 |
| 6 | 3.7 ± 0.4 | 14 ± 2 | 6.7 ± 0.6 |
| cisplatin | 2.7 ± 0.1 | 26 ± 3 | 10.0 ± 0.7 |
| RAPTA-C | >200 | >200 | >200 |
| Compound | TON |
| 4a | 3.8 |
| 4c | 4.1 |
| 4d | 3.7 |
| 5a | 1.8 |
| 6 | 1.6 |
| 1a | 3.7 |
| 1c | 3.5 |
| FeSO4 [a] | 2.3 |
| 3 | |
|---|---|
| Formula | C12H15FeNOSe |
| FW | 324.06 |
| T, K | 100(2) |
| λ, Å | 0.71073 |
| Crystal system | Monoclinic |
| Space group | Pc |
| a, Å | 13.454(3) |
| b, Å | 7.675(2) |
| c, Å | 12.285(2) |
| β,° | 99.07(3) |
| Cell Volume, Å3 | 1252.5(5) |
| Z | 4 |
| Dc, g∙cm−3 | 1.719 |
| μ, mm−1 | 4.088 |
| F(000) | 648 |
| Crystal size, mm | 0.21 × 0.19 × 0.15 |
| θ limits,° | 1.533–24.999 |
| Reflections collected | 11149 |
| Independent reflections | 2119 [Rint = 0.0687] |
| Data/restraints/parameters | 2119/350/290 |
| Goodness on fit on F2 | 1.116 |
| R1 (I > 2σ(I)) | 0.1175 |
| wR2 (all data) | 0.2914 |
| Largest diff. peak and hole, e Å−3 | 1.622/–1.857 |
| Comp. | Stability in DMSO-d6/D2O (v/v) + NaCl (0.05 M) a | Stability in DMSO/RPMI-1640 (v/v) b |
|---|---|---|
| 4a | <15% degradation (3:2) | 4a + other species c (1:3) |
| 4b | ca. 50% degradation (1:1) | 4b (1:3) |
| 4c | <15% degradation (2:1) | 2c + 4c (1:4) |
| 4d | <15% degradation (1:1) | 4d (1:2) |
| 5a | 5a (1:1) | |
| 6 | 6 (1:1) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agonigi, G.; Batchelor, L.K.; Ferretti, E.; Schoch, S.; Bortoluzzi, M.; Braccini, S.; Chiellini, F.; Biancalana, L.; Zacchini, S.; Pampaloni, G.; et al. Mono-, Di- and Tetra-iron Complexes with Selenium or Sulphur Functionalized Vinyliminium Ligands: Synthesis, Structural Characterization and Antiproliferative Activity. Molecules 2020, 25, 1656. https://doi.org/10.3390/molecules25071656
Agonigi G, Batchelor LK, Ferretti E, Schoch S, Bortoluzzi M, Braccini S, Chiellini F, Biancalana L, Zacchini S, Pampaloni G, et al. Mono-, Di- and Tetra-iron Complexes with Selenium or Sulphur Functionalized Vinyliminium Ligands: Synthesis, Structural Characterization and Antiproliferative Activity. Molecules. 2020; 25(7):1656. https://doi.org/10.3390/molecules25071656
Chicago/Turabian StyleAgonigi, Gabriele, Lucinda K. Batchelor, Eleonora Ferretti, Silvia Schoch, Marco Bortoluzzi, Simona Braccini, Federica Chiellini, Lorenzo Biancalana, Stefano Zacchini, Guido Pampaloni, and et al. 2020. "Mono-, Di- and Tetra-iron Complexes with Selenium or Sulphur Functionalized Vinyliminium Ligands: Synthesis, Structural Characterization and Antiproliferative Activity" Molecules 25, no. 7: 1656. https://doi.org/10.3390/molecules25071656
APA StyleAgonigi, G., Batchelor, L. K., Ferretti, E., Schoch, S., Bortoluzzi, M., Braccini, S., Chiellini, F., Biancalana, L., Zacchini, S., Pampaloni, G., Sarkar, B., Dyson, P. J., & Marchetti, F. (2020). Mono-, Di- and Tetra-iron Complexes with Selenium or Sulphur Functionalized Vinyliminium Ligands: Synthesis, Structural Characterization and Antiproliferative Activity. Molecules, 25(7), 1656. https://doi.org/10.3390/molecules25071656