
Theoretical Studies Aimed at Finding FLT3 Inhibitors and a Promising Compound and Molecular Pattern with Dual Aurora B/FLT3 Activity


 ,
,  and
and
Abstract
:1. Introduction
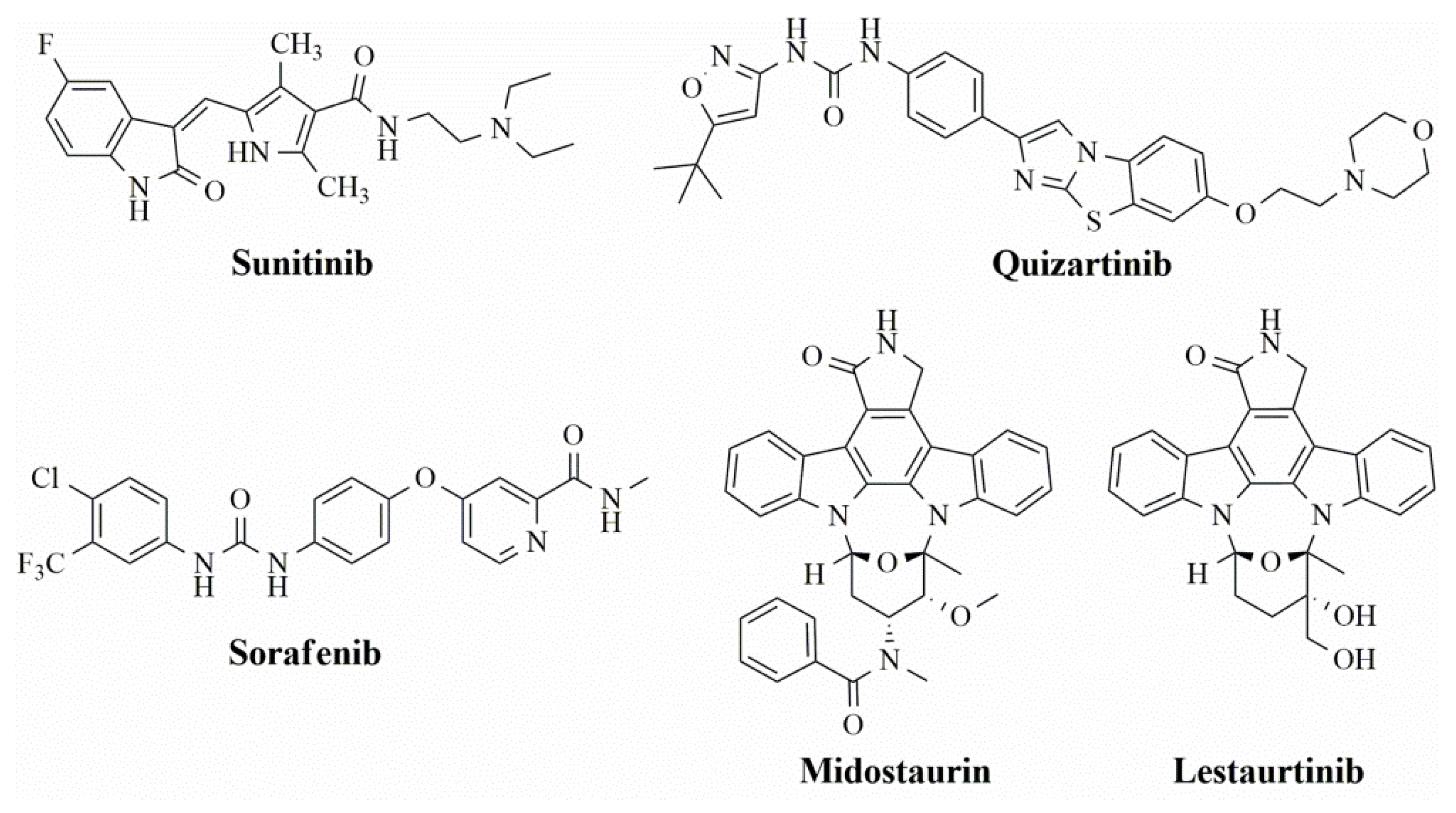
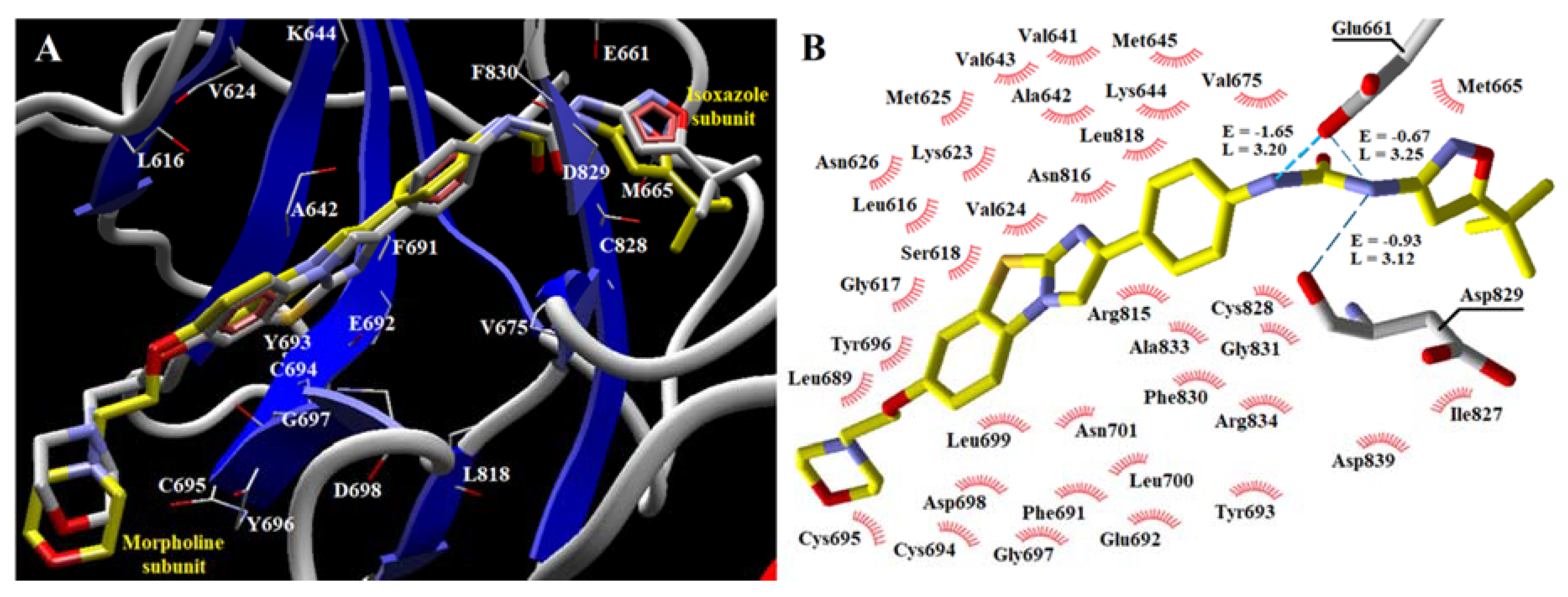
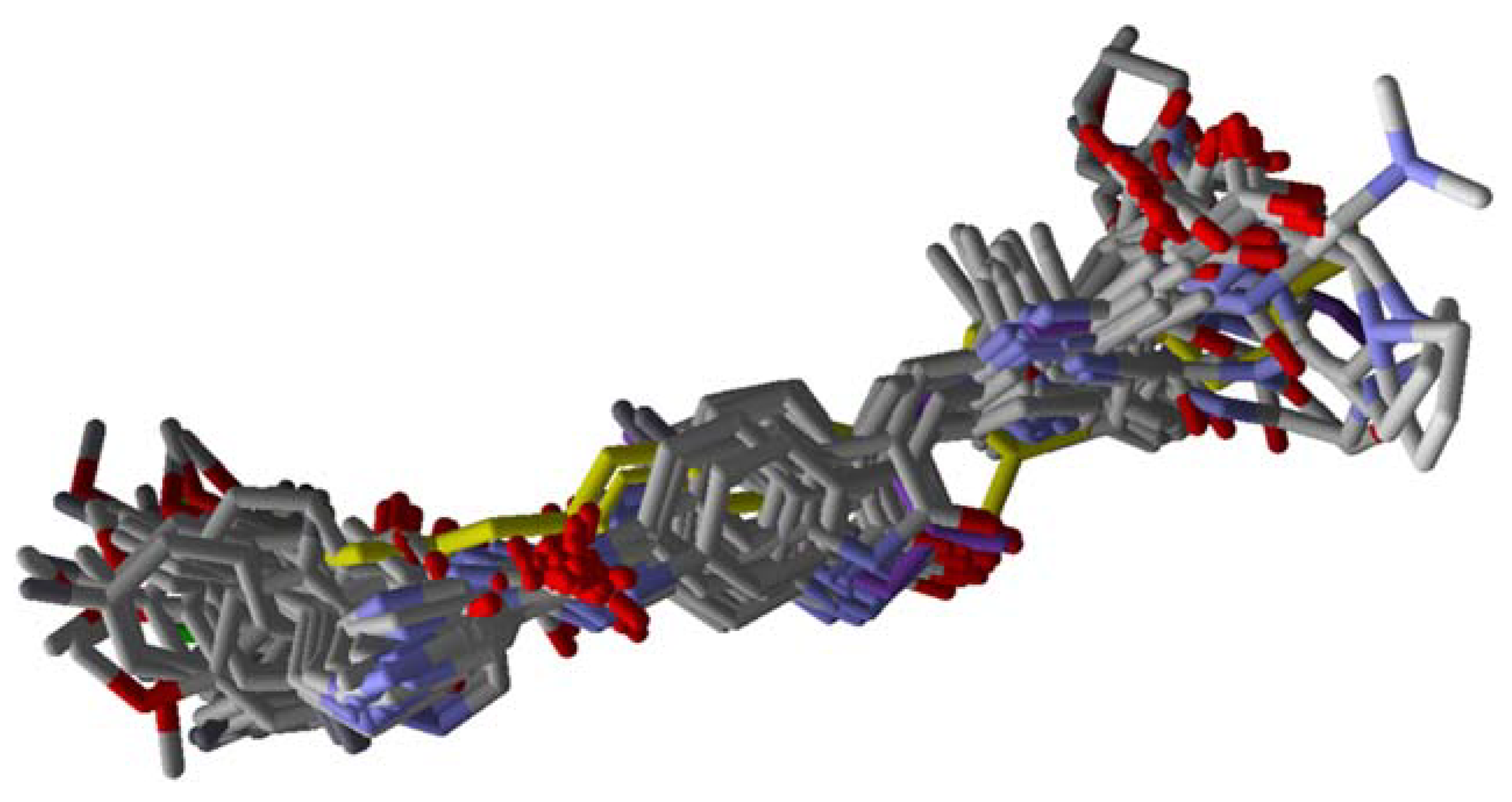
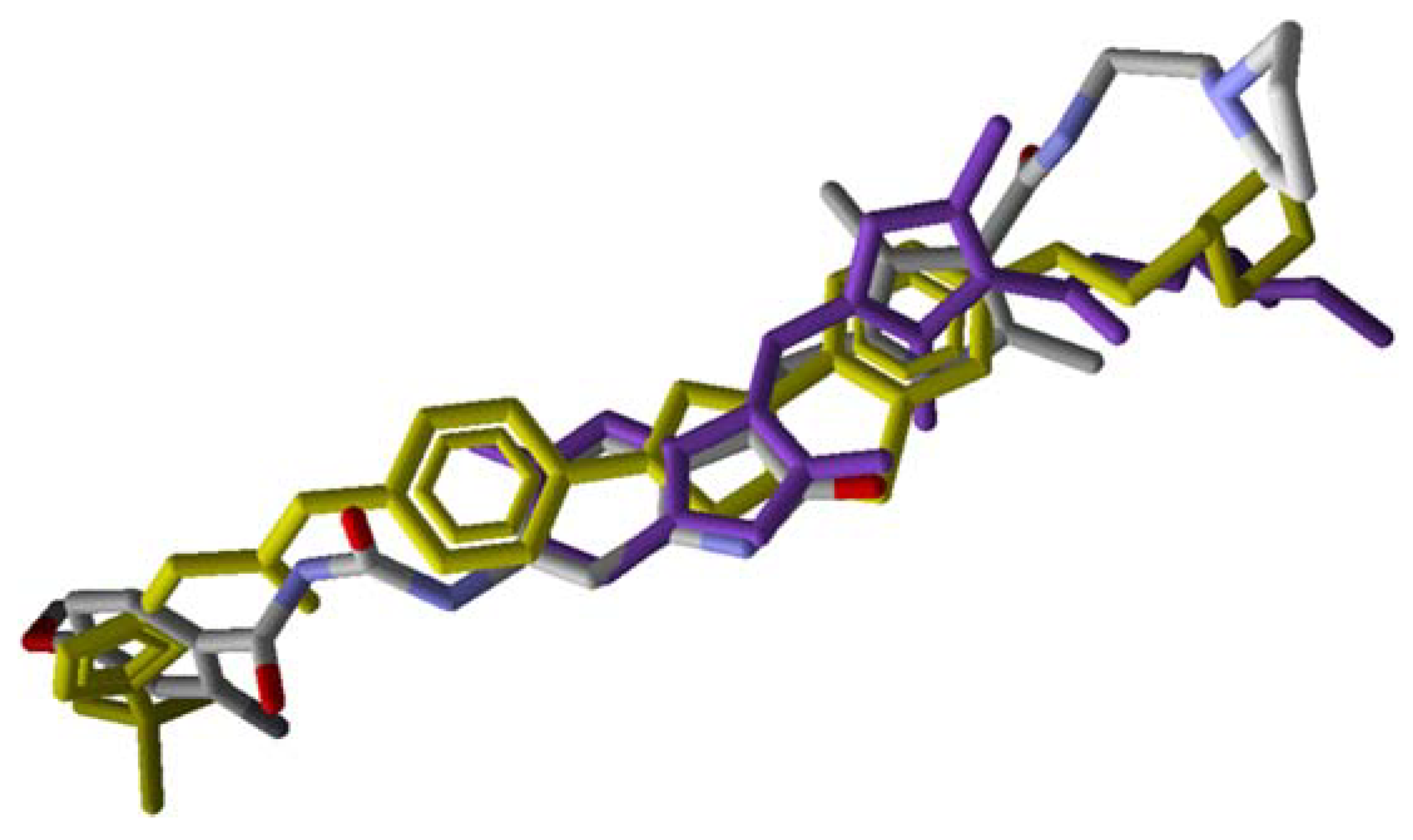
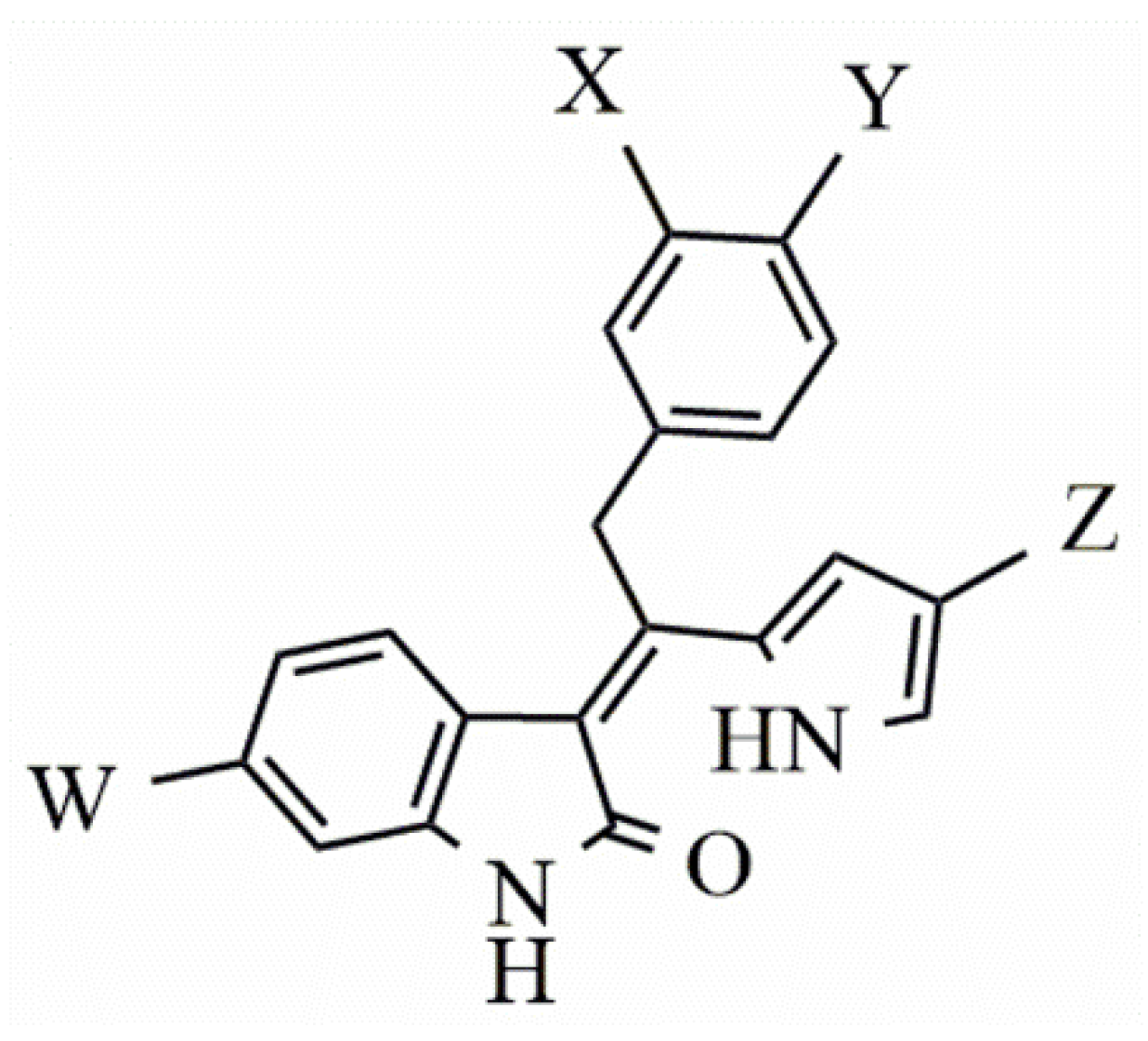
2. Results
3. Discussion
4. Materials and Methods
4.1. Data Set
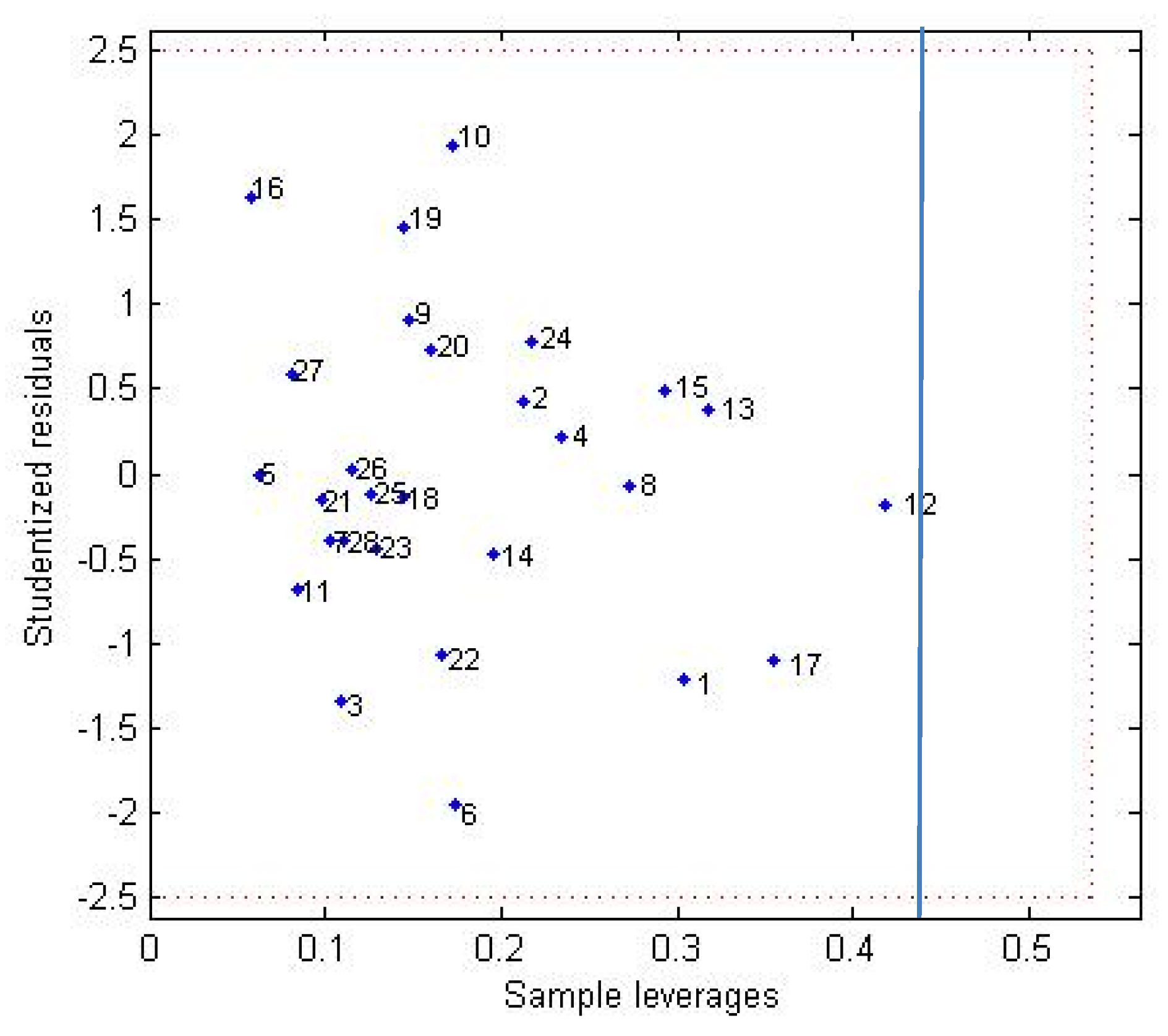
4.2. Predicted Model
- i)
- Coefficient of determination (R2): is the proportion of the variance in the dependent variable that is predictable from the independent variable.
- ii)
- Leave-one-out cross-validation (LOOcv) correlation coefficient (q2): estimating the performance of a predictive model.
- iii)
- Y-Randomization (R2rand): consists of the random exchange of the independent variable values. Thus, the R2rand value must be less than the correlation coefficient of the non-randomized models.
- iv)
- R2p: penalizes the model for the difference between R2 of randomized models and the R2 of the non-randomized model:
- v)
- Correlation coefficient of external validation set (R2pred): reflects the degree of correlation between the observed (YExp(test)) and predicted (YPred(test)) activity data of the test set:where is average value for the dependent variable for the training set.
- vi)
- Modified R2 (R2m(test)): equation determining the proximity between the observed and predicted values with the zero axis intersection:
4.3. Searching for More Potent Indolin−2-one FLT3 Inhibitors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Berenstein, R. Class III Receptor Tyrosine Kinases in Acute Leukemia—Biological Functions and Modern Laboratory Analysis. Biomark. Insights 2015, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayser, S.; Schlenk, R.F.; Londono, M.C.; Breitenbuecher, F.; Wittke, K.; Du, J.; Groner, S.; Späth, D.; Krauter, J.; Ganser, A.; et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood 2009, 114, 2386–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Port, M.; Böttcher, M.; Thol, F.; Ganser, A.; Schlenk, R.; Wasem, J.; Neumann, A.; Pouryamout, L. Prognostic significance of FLT3 internal tandem duplication, nucleophosmin 1, and CEBPA gene mutations for acute myeloid leukemia patients with normal karyotype and younger than 60 years: A systematic review and meta-analysis. Ann. Hematol. 2014, 93, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Pastore, D. FLT3 inhibitors in acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 133–144. [Google Scholar] [CrossRef]
- Bavetsias, V.; Crumpler, S.; Sun, C.; Avery, S.; Atrash, B.; Faisal, A.; Moore, A.S.; Kosmopoulou, M.; Brown, N.; Sheldrake, P.W.; et al. Optimization of Imidazo [4–b]pyridine-Based Kinase Inhibitors: Identification of a Dual FLT3/Aurora Kinase Inhibitor as an Orally Bioavailable Preclinical Development Candidate for the Treatment of Acute Myeloid Leukemia. J. Med. Chem. 2012, 55, 8721–8734. [Google Scholar] [CrossRef]
- Moore, A.S.; Faisal, A.; de Castro, G.D.; Bavetsias, V.; Sun, C.; Atrash, B.; Valenti, M.; de Haven, B.A.; Avery, S.; Mair, D.; et al. Selective FLT3 inhibition of FLT3-ITD+ acute myeloid leukaemia resulting in secondary D835Y mutation: A model for emerging clinical resistance patterns. Leukemia 2012, 26, 1462–1470. [Google Scholar] [CrossRef]
- Ma, F.; Liu, P.; Lei, M.; Liu, J.; Wang, H.; Zhao, S.; Hu, L. Design, synthesis and biological evaluation of indolin-2-one-based derivatives as potent, selective and efficacious inhibitors of FMS-like tyrosine kinase3 (FLT3). Eur. J. Med. Chem. 2017, 127, 72–86. [Google Scholar] [CrossRef]
- Zarrinkar, P.P.; Gunawardane, R.N.; Cramer, M.D.; Gardner, M.F.; Brigham, D.; Belli, B.; Karaman, M.W.; Pratz, K.W.; Pallares, G.; Chao, Q.; et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood 2009, 114, 2984–2992. [Google Scholar] [CrossRef]
- Nguyen, B.; Williams, A.B.; Young, D.J.; Ma, H.; Li, L.; Levis, M.; Brown, P.; Small, D. FLT3 activating mutations display differential sensitivity to multiple tyrosine kinase inhibitors. Oncotarget 2017, 8, 10931–10944. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, I.A.; Assis, T.M.; Rosa, I.A.; da Cunha, E.F.F. Indolin-2-one Derivatives: Theoretical Studies Aimed at Finding More Potent Aurora B Kinase Inhibitors. Lett. Drug Des. Discov. 2019, 16, 138–152. [Google Scholar] [CrossRef]
- Chern, J.W.; Jagtap, A.D.; Wang, H.C.; Chen, G.S. Indolin-2-one Derivatives as Protein Kinase Inhibitors. WO 2013/158373 A1, 24 October 2013. [Google Scholar]
- Zorn, J.A.; Wang, Q.; Fujimura, E.; Barros, T.; Kuriyan, J. Crystal structure of the FLT3 kinase domain bound to the inhibitor Quizartinib (AC220). PLoS ONE 2015, 10, e0121177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopfinger, A.J.; Wang, S.; Tokarski, J.S.; Jin, B.; Albuquerque, M.; Madhav, P.J.; Duraiswami, C. Construction of 3D-QSAR Models Using the 4D-QSAR Analysis Formalism. J. Am. Chem. Soc. 1997, 119, 10509–10524. [Google Scholar] [CrossRef]
- Gehlhaar, D.K.; Verkhivker, G.M.; Rejto, P.A.; Sherman, C.J.; Fogel, D.R.; Fogel, L.J.; Freer, S.T. Molecular Recognition of the Inhibitor AG-1343 by HIV-1 Protease: Conformationally Flexible Docking by Evolutionary Programming. Chem. Biol. 1995, 2, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, R.; Christensen, M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]
- Guimarães, A.P.; França, T.C.C.; Ramalho, T.C.; Rennó, M.N.; da Cunha, E.F.F.; Matos, K.S.; Mancini, D.T.; Kuca, K. Docking studies and effects of syn-anti isomery of oximes derived from pyridine imidazol bicycled systems as potential human acetylcholinesterase reactivators. J. Appl. Biomed. 2011, 9, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Ramalho, T.C.; França, T.C.C.; Renno, M.N.; Guimaraes, A.P.; da Cunha, E.F.F.; Kuca, K. Development of new acetylcholinesterase reactivators: Molecular modeling versus in vitro data. Chem. Biol. Interact. 2010, 185, 73–77. [Google Scholar] [CrossRef]
- Da Cunha, E.F.F.; Ramalho, T.C.; Maia, E.R.; de Alencastro, R.B. The search for new DHFR inhibitors: A review of patents, January 2001 February 2005. Expert Opin. Ther. Pat. 2005, 8, 967–986. [Google Scholar] [CrossRef]
- Ramalho, T.C.; da Cunha, E.F.F.; de Alencastro, R.B. A density functional study on the complexation of ethambutol with divalent cations. J. Mol. Struct. 2004, 676, 149–153. [Google Scholar] [CrossRef]
- Tavares, T.S.; da rocha, E.P.; Nogueira, F.G.E.; Torres, J.A.; Silva, M.C.; Kuca, K.; Ramalho, T.C. Δ-FeOOH as Support for Immobilization Peroxidase: Optimization via a Chemometric Approach. Molecules 2020, 25, 259. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.Y.S.; Leung, A.Y.H. Overcoming Resistance to FLT3 Inhibitors in the Treatment of FLT3-Mutated AML. Int. J. Mol. Sci. 2020, 21, 1537. [Google Scholar] [CrossRef] [Green Version]
- Bohmer, A.; Barz, S.; Schwab, K.; Kolve, U.; Gabel, A.; Kirkpatrick, J.; Ohlenshlager, O.; Gorlach, M.; Bohmer, F.D. Modulation of FLT3 signal transduction through cytoplasmic cysteine residues indicates the potential for redox regulation. Redox Biol. 2020, 28, 101325. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Chemical Structure | IC50 (nM) FLT3 | pIC50 FLT3 |
|---|---|---|---|
| 1 |  | 0.5 | 9.301 |
| 2 |  | 1.3 | 8.886 |
| 3 |  | 1.4 | 8.854 |
| 4* |  | 1.6 | 8.796 |
| 5* |  | 2.4 | 8.620 |
| 6* |  | 2.7 | 8.569 |
| 7 |  | 2.7 | 8.569 |
| 8 |  | 2.7 | 8.569 |
| 9 |  | 2.9 | 8.538 |
| 10 |  | 2.9 | 8.538 |
| 11 |  | 3.5 | 8.456 |
| 12 |  | 4.4 | 8.357 |
| 13* |  | 4.8 | 8.319 |
| 14* |  | 6.1 | 8.215 |
| 15 |  | 7.3 | 8.137 |
| 16* |  | 8.1 | 8.092 |
| 17 |  | 8.6 | 8.066 |
| 18 |  | 10.0 | 8.000 |
| 19* |  | 10.0 | 8.000 |
| 20 |  | 10.3 | 7.987 |
| 21 |  | 11.6 | 7.936 |
| 22* |  | 15.4 | 7.812 |
| 23 |  | 16.2 | 7.790 |
| 24 |  | 24.6 | 7.609 |
| 25 |  | 24.9 | 7.604 |
| 26 |  | 27.7 | 7.558 |
| 27 |  | 31.7 | 7.499 |
| 28 |  | 34.9 | 7.457 |
| 29 |  | 37.5 | 7.426 |
| 30 |  | 37.9 | 7.421 |
| 31* |  | 38.1 | 7.419 |
| 32 |  | 39.4 | 7.404 |
| 33 |  | 45.7 | 7.340 |
| 34* |  | 48.4 | 7.315 |
| 35 |  | 54.2 | 7.266 |
| 36 |  | 64.2 | 7.192 |
| 37 |  | 74.1 | 7.130 |
| 38* |  | 86.6 | 7.062 |
| 39* |  | 115.3 | 6.938 |
| 40 |  | 148.4 | 6.828 |
| Quizartinib |  | 4.2 a1 1.1 a2 | 8.377 8.959 |
| Sunitinib |  | 9.9 a1 34.0 a2 | 8.004 7.468 |
| Cpd/aa | Ala 642 | Asp 698 | Asp 829 | Cys 694 | Cys 695 | Cys 828 | Glu 661 | Glu 692 | Gly 697 | Leu 616 | Leu 818 | Lys 644 | Met 665 | Phe 691 | Phe 830 | Tyr 693 | Tyr 696 | Val 624 | Val 675 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −1.90 | −1.91 | −28.29 | −12.11 | −3.75 | −18.82 | −10.10 | −4.10 | −10.57 | −23.07 | −11.32 | −1.91 | 4.22 | −14.37 | −8.25 | −14.65 | −4.19 | −3.53 | −10.21 |
| 2 | −3.47 | −2.52 | −8.60 | −11.61 | −3.78 | −8.46 | 11.60 | −4.30 | −9.89 | −23.55 | −12.07 | −9.62 | 70.68 | −12.02 | −8.44 | −12.14 | −3.83 | −5.74 | −7.10 |
| 3 | −3.18 | −2.03 | −14.63 | −10.25 | −8.59 | −8.33 | −21.14 | −1.75 | −10.32 | −21.16 | −13.13 | −1.05 | 27.99 | −12.19 | −9.04 | −13.02 | −10.30 | −4.48 | −4.60 |
| 4 | −2.06 | −1.81 | −12.19 | −11.28 | −8.05 | −9.41 | −15.47 | −1.70 | −9.61 | −16.22 | −14.06 | −3.23 | 1.95 | −4.00 | −12.28 | −13.12 | −6.78 | −5.76 | −8.14 |
| 5 | −3.93 | −4.79 | −28.56 | −10.96 | −3.05 | −24.34 | −7.84 | −4.92 | −11.72 | −14.22 | −12.61 | −2.69 | 2.79 | −14.80 | −9.70 | −10.11 | −5.02 | −5.04 | −8.56 |
| 6 | −3.72 | −3.25 | −19.77 | −12.07 | −2.87 | −21.27 | −4.86 | −4.94 | −9.70 | −24.01 | −11.81 | −2.57 | 7.50 | −12.93 | −8.96 | −10.96 | −2.66 | −5.69 | −10.26 |
| 7 | −3.58 | −5.82 | −9.43 | −12.55 | −3.54 | −10.52 | 0.52 | −2.20 | −10.90 | −17.27 | −11.75 | −7.18 | 77.63 | −8.56 | −7.08 | −10.75 | −6.27 | −5.58 | −4.75 |
| 8 | −4.03 | −5.13 | −15.80 | −12.87 | −2.02 | −23.44 | −2.34 | −5.47 | −8.53 | −20.14 | −12.68 | −1.61 | 9.50 | −14.83 | −8.84 | −9.66 | −2.13 | −5.05 | −7.23 |
| 9 | −5.14 | −2.17 | −11.28 | −8.87 | −3.82 | −6.94 | 3.47 | −3.14 | −10.13 | −18.14 | −9.58 | −6.53 | 65.61 | −12.54 | −7.25 | −11.71 | −4.78 | −8.17 | −5.69 |
| 10 | −2.30 | −1.62 | −12.27 | −11.99 | −3.68 | −12.31 | −9.30 | −5.20 | −9.17 | −21.41 | −11.99 | −4.35 | 38.66 | −3.53 | −7.94 | −12.27 | −3.17 | −3.58 | −8.62 |
| 11 | −2.39 | −1.70 | −13.05 | −12.58 | −9.24 | −10.14 | −16.87 | −2.00 | −9.45 | −11.96 | −14.02 | −2.97 | 3.85 | −2.70 | −11.56 | −15.15 | −7.67 | −5.44 | −8.52 |
| 12 | −3.22 | −9.77 | −17.90 | −10.84 | −4.04 | −11.92 | −25.33 | −3.83 | −15.35 | −15.56 | −12.73 | −4.41 | 15.24 | −14.71 | −9.85 | −10.81 | −7.82 | −5.72 | −6.56 |
| 13 | −1.79 | −1.59 | −13.55 | −10.83 | −6.14 | −11.91 | −8.55 | −2.41 | −9.81 | −15.30 | −13.45 | −5.53 | −4.10 | −2.98 | −10.12 | −15.00 | −6.59 | −5.87 | −7.99 |
| 14 | −2.15 | −1.80 | −25.45 | −12.12 | −3.52 | −19.77 | −8.72 | −5.03 | −9.32 | −21.43 | −11.89 | −1.71 | 3.03 | −16.56 | −8.81 | −12.82 | −3.25 | −3.42 | −7.48 |
| 15 | −2.66 | −2.27 | −10.52 | −12.48 | −4.21 | −10.40 | −1.77 | −3.69 | −10.22 | −24.67 | −12.05 | −6.20 | 54.98 | −1.02 | −7.62 | −11.22 | −4.02 | −3.64 | −8.03 |
| 16 | −2.23 | −1.92 | −13.13 | −13.07 | −3.91 | −10.60 | −1.06 | −4.51 | −10.82 | −17.85 | −11.47 | −5.12 | 39.50 | −7.34 | −7.64 | −11.77 | −4.50 | −3.50 | −9.17 |
| 17 | −3.33 | -0.91 | −15.54 | −14.26 | −2.48 | −12.12 | 8.11 | −4.44 | −6.15 | −12.24 | −11.87 | −7.22 | 34.52 | −3.82 | −6.98 | −12.19 | −1.25 | −5.27 | −7.94 |
| 18 | −3.48 | −5.04 | −20.27 | −11.13 | −2.16 | −12.32 | 44.19 | −1.95 | −9.89 | −22.54 | −10.40 | −2.13 | −2.50 | −18.51 | −11.80 | −7.48 | −2.21 | −4.43 | −7.12 |
| 19 | −2.15 | −1.32 | −25.70 | −14.60 | −2.07 | −18.54 | −9.08 | −6.09 | −6.94 | −17.34 | −11.95 | −2.47 | 2.22 | −16.83 | −7.05 | −12.42 | −1.64 | −4.73 | −9.06 |
| 20 | −2.94 | −1.97 | −12.01 | −12.66 | −6.93 | −11.42 | −17.81 | −4.69 | −14.44 | −16.58 | −10.88 | -0.31 | 33.24 | −20.81 | −8.59 | −11.82 | −14.06 | −2.89 | 5.41 |
| 21 | −1.80 | −1.48 | −10.81 | −11.82 | −7.05 | −11.32 | −12.80 | −1.94 | −6.00 | −19.99 | −13.32 | −4.11 | -0.97 | −3.64 | −11.52 | −6.08 | −3.90 | −6.15 | −7.82 |
| 22 | −1.70 | −1.31 | −12.20 | −14.35 | −1.81 | −23.16 | −2.52 | −6.30 | −4.04 | −10.48 | −11.75 | −2.02 | 8.27 | −13.68 | −6.37 | −10.84 | −1.64 | −5.55 | −6.99 |
| 23 | −2.59 | −1.66 | −18.36 | −11.64 | −3.54 | −12.63 | 32.16 | −3.09 | −9.04 | −13.48 | −12.99 | −7.69 | −2.75 | −19.03 | −8.37 | −14.20 | −3.84 | −3.67 | −8.31 |
| 24 | −4.49 | −2.12 | −13.80 | −9.24 | −3.64 | −11.07 | 7.10 | −3.21 | −10.41 | −18.62 | −11.74 | −9.56 | 27.49 | −10.24 | −8.61 | −11.81 | −4.27 | −6.65 | −7.45 |
| 25 | −4.44 | −1.16 | −23.81 | −11.81 | −3.31 | −11.65 | 2.30 | −2.16 | −4.03 | −10.48 | −12.26 | −8.10 | −1.23 | 11.81 | −8.98 | −11.76 | −2.85 | −6.58 | −8.21 |
| 26 | −3.46 | −1.01 | −17.06 | −12.26 | −2.70 | −15.76 | 5.74 | −1.79 | −5.98 | −10.60 | −12.48 | −3.40 | 50.76 | −13.89 | −7.20 | −10.54 | −2.08 | −6.03 | −1.60 |
| 27 | −3.04 | −1.25 | −21.42 | −10.60 | −3.94 | −11.96 | −21.37 | −4.68 | −8.62 | −15.67 | −11.82 | −4.61 | −1.05 | −14.40 | −9.63 | −16.59 | −3.29 | −6.16 | −7.27 |
| 28 | −1.88 | −1.12 | −16.49 | −8.57 | −6.61 | −10.78 | −7.95 | -0.93 | −7.19 | −21.41 | −10.72 | −6.68 | 4.37 | 0.54 | −9.77 | −12.12 | −4.89 | −8.58 | −7.27 |
| 29 | −2.58 | -0.85 | −19.75 | −12.76 | −3.21 | −10.32 | 12.68 | −1.89 | −6.22 | −13.12 | −11.82 | −4.08 | 29.51 | −12.09 | −6.24 | −12.57 | −2.05 | −6.41 | −4.56 |
| 30 | −1.89 | -0.79 | −14.09 | −13.14 | −2.10 | −11.77 | −6.60 | −5.49 | −5.51 | −5.58 | −12.82 | −4.48 | −4.64 | −8.71 | −11.04 | −11.76 | −1.12 | −4.29 | −7.49 |
| 31 | −3.00 | -0.67 | −20.12 | −13.76 | −2.70 | −11.78 | −4.00 | −4.92 | −5.82 | −10.84 | −11.69 | −6.47 | −2.56 | 5.56 | −7.77 | −12.88 | −1.14 | −5.24 | −8.90 |
| 32 | −3.59 | −2.33 | −17.04 | −10.53 | −4.09 | −15.48 | −15.18 | −1.60 | −10.97 | −18.08 | −11.57 | −1.90 | 6.44 | −16.41 | −8.20 | −9.10 | −5.18 | −7.44 | 4.76 |
| 33 | −2.96 | −2.01 | −14.00 | −11.59 | −3.68 | −12.27 | −21.55 | −3.87 | −10.00 | −19.56 | −12.71 | −3.81 | 21.37 | −16.47 | −8.23 | −12.49 | −4.03 | −4.28 | −5.11 |
| 34 | −2.04 | −1.24 | −14.85 | −14.31 | −2.46 | −12.11 | 6.84 | −5.96 | −5.13 | −8.97 | −12.32 | −5.81 | 27.07 | −12.89 | −6.44 | −11.51 | −1.79 | −4.35 | −8.87 |
| 35 | −2.42 | −1.55 | −25.07 | −11.15 | −9.38 | −18.03 | −8.01 | −2.73 | -0.36 | −13.27 | −12.19 | −1.68 | −2.38 | −15.94 | −7.62 | −16.61 | −6.66 | −4.76 | −4.90 |
| 36 | −2.07 | −1.14 | −14.32 | −14.53 | −1.87 | −11.28 | 10.54 | −5.39 | −5.15 | −8.90 | −12.44 | −7.02 | 2.20 | −10.78 | −7.14 | −10.95 | −1.30 | −4.78 | −8.37 |
| 37 | −2.23 | −1.22 | −17.23 | −14.97 | −1.41 | −20.78 | −5.56 | −6.80 | −5.08 | −10.34 | −12.09 | −2.18 | 4.18 | −17.93 | −6.69 | −10.49 | −1.07 | −4.24 | -0.69 |
| 38 | −2.24 | −1.51 | −14.39 | −10.47 | −3.64 | −11.66 | −8.32 | −5.08 | −8.20 | −11.91 | −12.40 | −4.53 | −4.61 | −5.64 | −9.53 | −11.34 | −2.65 | −3.94 | −7.51 |
| 39 | −3.53 | −2.64 | −19.41 | −8.51 | −2.35 | −17.95 | −11.74 | −1.44 | −9.49 | −17.55 | −10.16 | −1.61 | 8.11 | −16.42 | −13.80 | −6.79 | −3.21 | −8.19 | 2.66 |
| 40 | −2.77 | -0.98 | −22.08 | −12.72 | −3.36 | −7.88 | 4.48 | −1.86 | −6.34 | −12.98 | −12.08 | −4.22 | −1.32 | −10.22 | −5.81 | −12.47 | −2.26 | −6.27 | −7.50 |
| Quiz | −4.48 | −1.55 | −18.63 | −8.59 | −7.63 | −19.56 | −11.13 | −1.64 | −10.93 | −15.17 | −9.37 | −4.92 | 2.19 | −9.99 | −12.34 | −10.03 | −13.10 | −7.47 | −6.01 |
| Sunit | −2.11 | −1.65 | −2.84 | −10.80 | −8.63 | −0.98 | - | −3.42 | −9.61 | −19.49 | −12.80 | - | - | −4.01 | −10.82 | −16.27 | −19.13 | −5.61 | −3.63 |
| Parameters | Accept Values | Obtained Values |
|---|---|---|
| R2 | >0.8 | 0.80 |
| RMSEc | - | 0.29 |
| q2 | >0.5 | 0.60 |
| RMSEcv | - | 0.46 |
| LV | - | 5 |
| R2rand | < r2 | 0.22 |
| RMSE y-rand | - | 0.60 |
| R2pred | >0.8 | 0.80 |
| RMSEpred | - | 0.31 |
| r2m(test) | >0.5 | 0.68 |
| R2p | >0.5 | 0.61 |
| Compd | Chemical Structure | pIC50 pred | MolDock Score | Inter Energy | H Bond Energy |
|---|---|---|---|---|---|
| IAF70 |  | 10.06 | −236.96 | −249.26 | −9.37 |
| IAF72 |  | 10.25 | −238.17 | −251.73 | −6.74 |
| IAF75 |  | 10.24 | −257.27 | −254.38 | −13.95 |
| IAF80 |  | 10.22 | −255.22 | −252.98 | −11.75 |
| IAF84 |  | 10.13 | −261.42 | −258.18 | −11.16 |
| IAF88 |  | 10.12 | −254.29 | −250.82 | −8.30 |
| Compd | Chemical Structure | Enzyme | pIC50 pred | MolDock Score | Inter Energy | H Bond Energy |
|---|---|---|---|---|---|---|
| IAF79 |  | Aurora B | 11.39 b | −236.71 b | −240.59 b | −15.61 b |
| FLT3 | 9.83 | −248.06 | −246.77 | −8.35 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, Í.A.; Braga Resende, D.; Ramalho, T.C.; Kuca, K.; da Cunha, E.F.F. Theoretical Studies Aimed at Finding FLT3 Inhibitors and a Promising Compound and Molecular Pattern with Dual Aurora B/FLT3 Activity. Molecules 2020, 25, 1726. https://doi.org/10.3390/molecules25071726
Fernandes ÍA, Braga Resende D, Ramalho TC, Kuca K, da Cunha EFF. Theoretical Studies Aimed at Finding FLT3 Inhibitors and a Promising Compound and Molecular Pattern with Dual Aurora B/FLT3 Activity. Molecules. 2020; 25(7):1726. https://doi.org/10.3390/molecules25071726
Chicago/Turabian StyleFernandes, Ítalo Antônio, Déborah Braga Resende, Teodorico Castro Ramalho, Kamil Kuca, and Elaine Fontes Ferreira da Cunha. 2020. "Theoretical Studies Aimed at Finding FLT3 Inhibitors and a Promising Compound and Molecular Pattern with Dual Aurora B/FLT3 Activity" Molecules 25, no. 7: 1726. https://doi.org/10.3390/molecules25071726
APA StyleFernandes, Í. A., Braga Resende, D., Ramalho, T. C., Kuca, K., & da Cunha, E. F. F. (2020). Theoretical Studies Aimed at Finding FLT3 Inhibitors and a Promising Compound and Molecular Pattern with Dual Aurora B/FLT3 Activity. Molecules, 25(7), 1726. https://doi.org/10.3390/molecules25071726