
Dimerization of Acetic Acid in the Gas Phase—NMR Experiments and Quantum-Chemical Calculations


Abstract
:
1. Introduction
2. Models and Methods
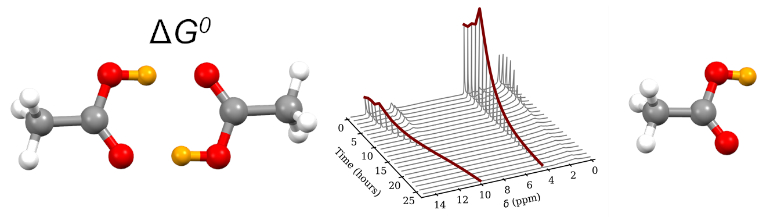
2.1. Theoretical Model
2.2. NMR Experiments
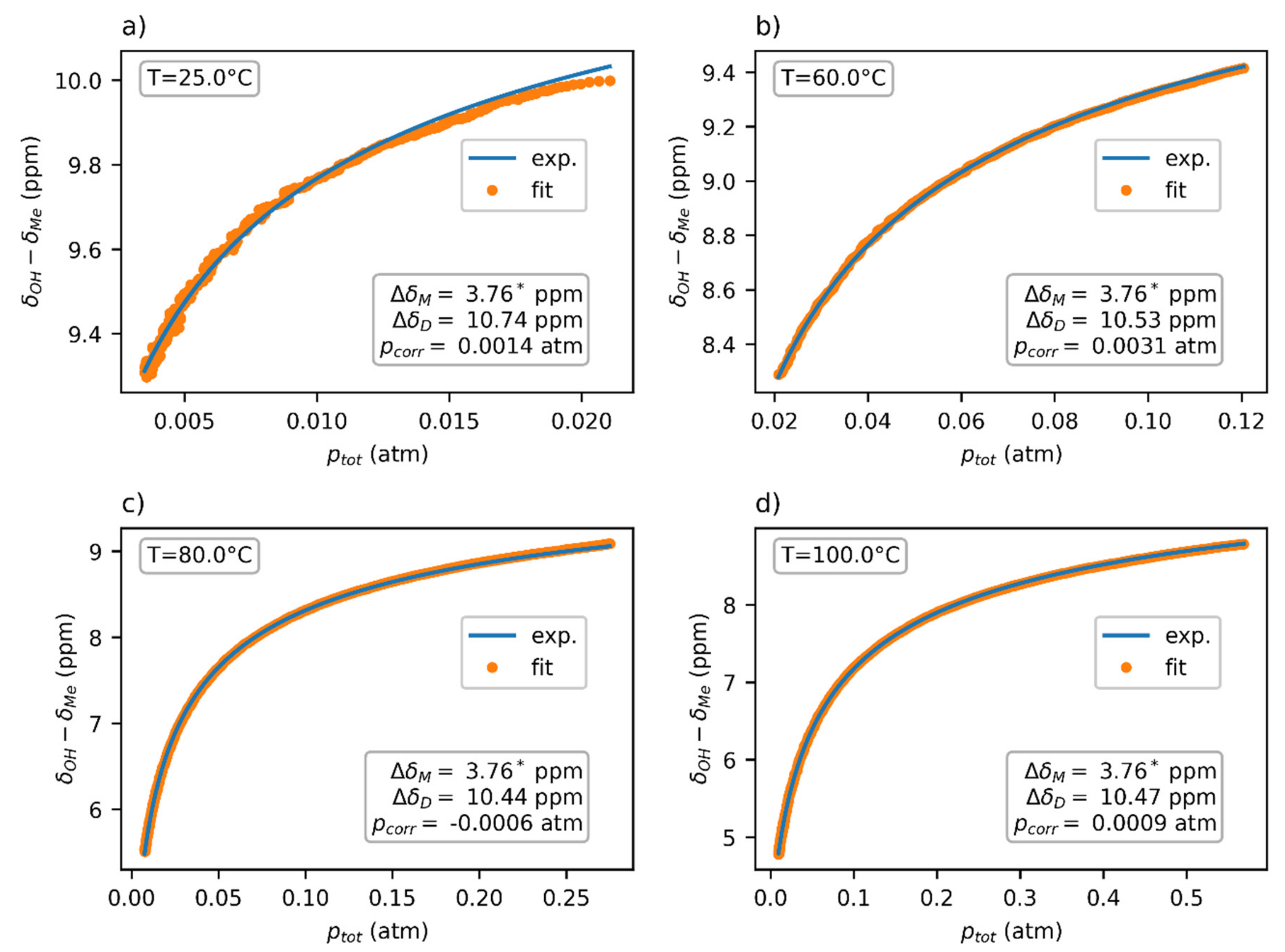
2.2.1. Pressure Estimation
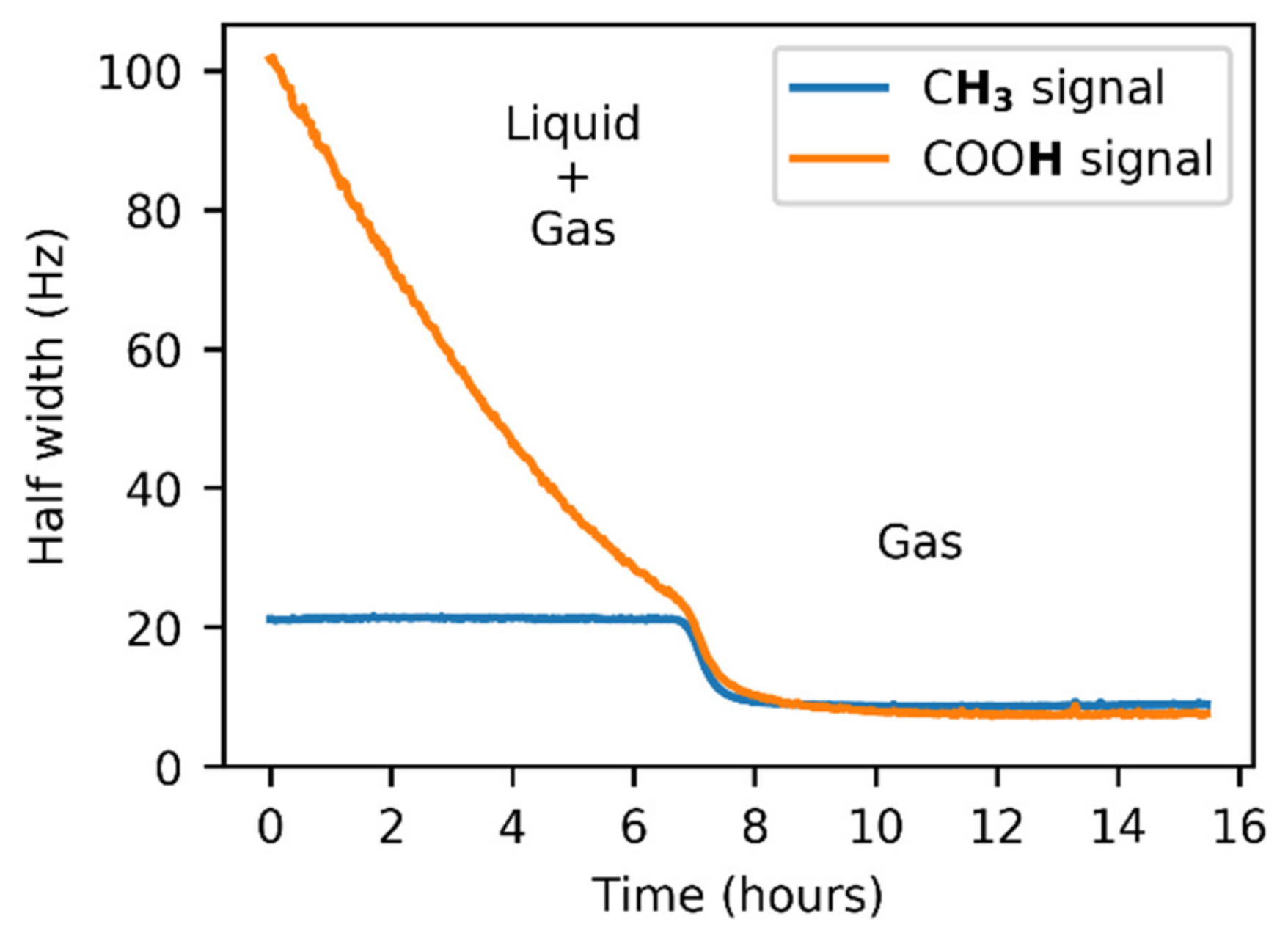
2.2.2. Data Processing
2.3. Computational Methods
3. Results and Discussion
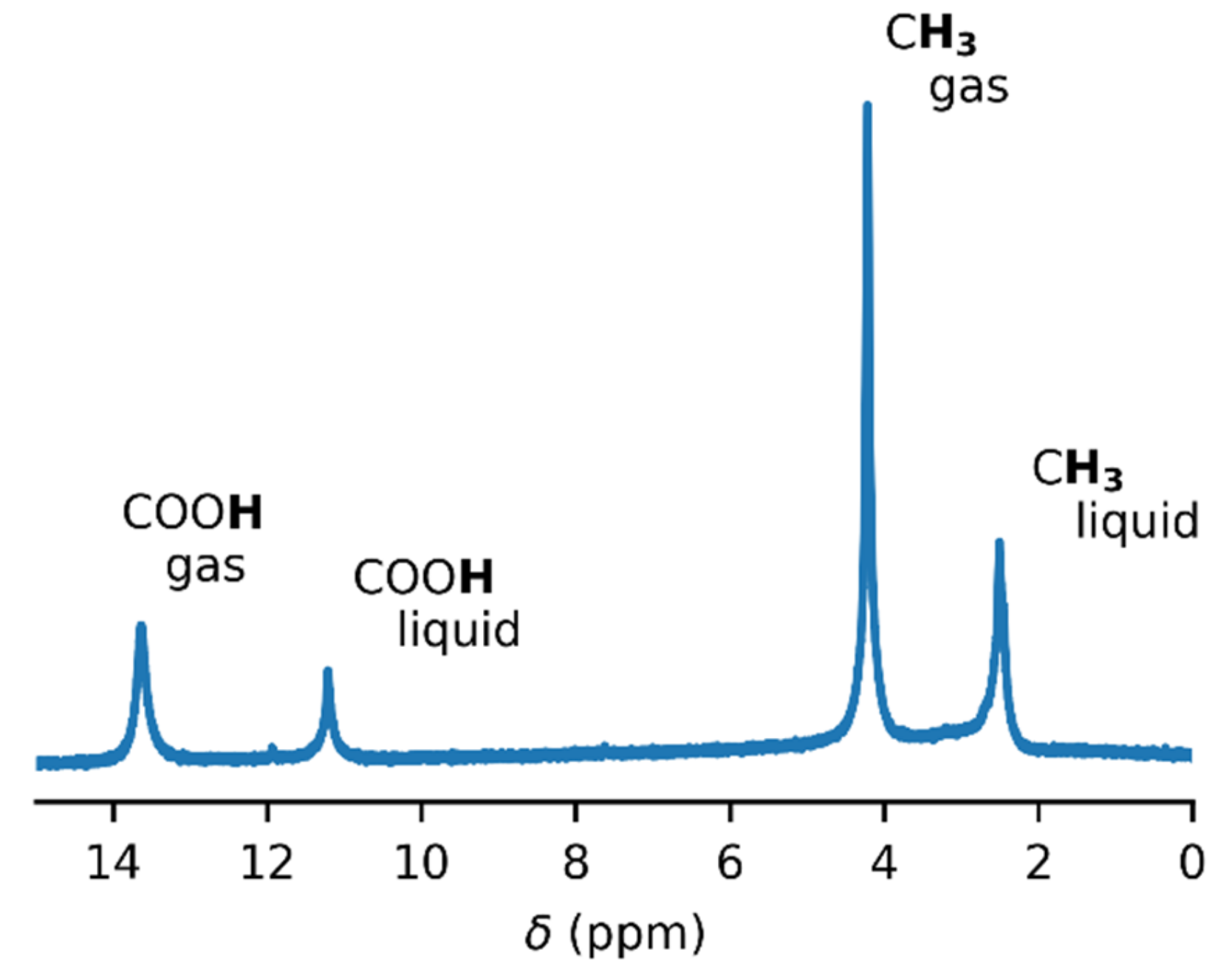
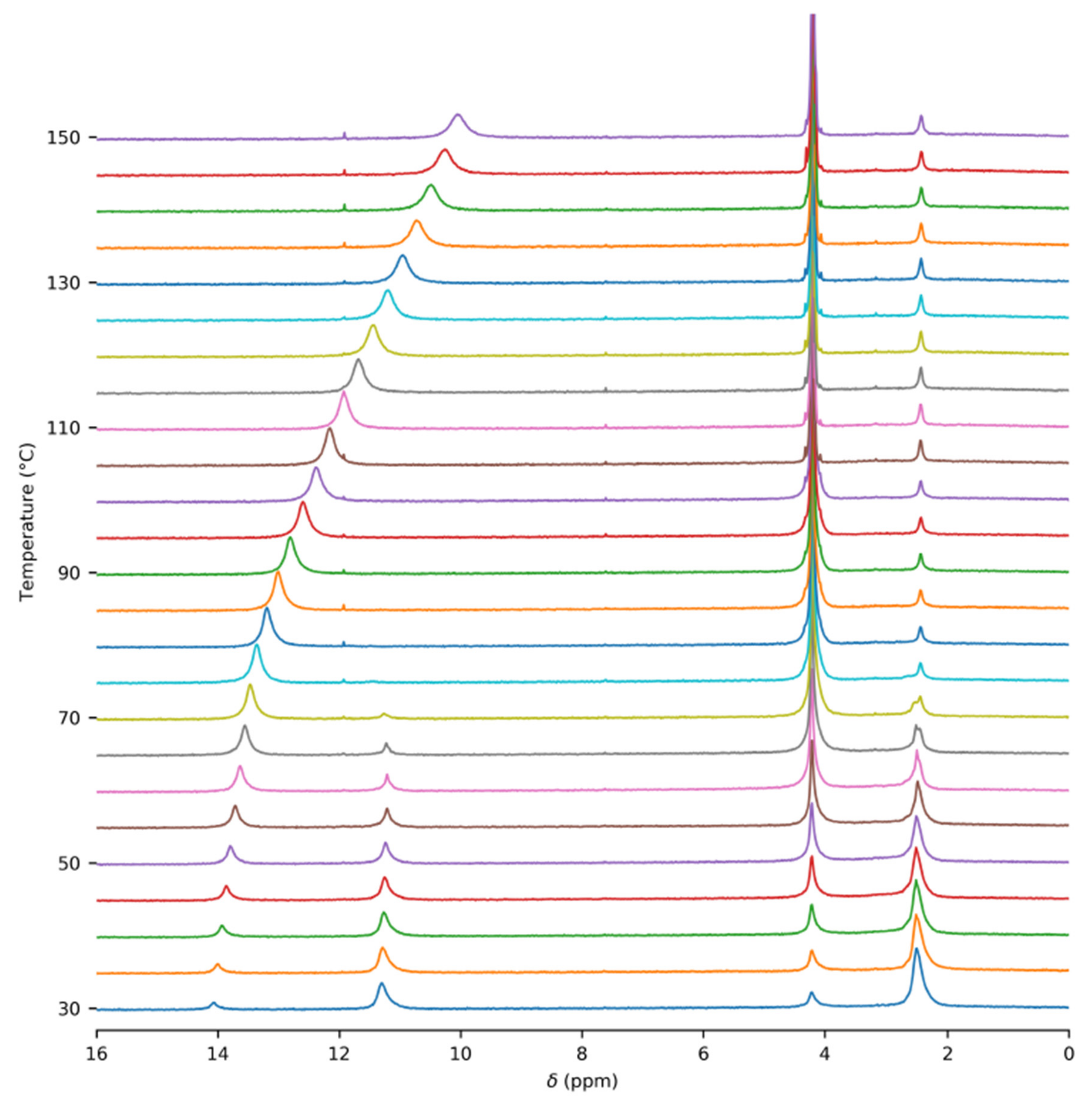
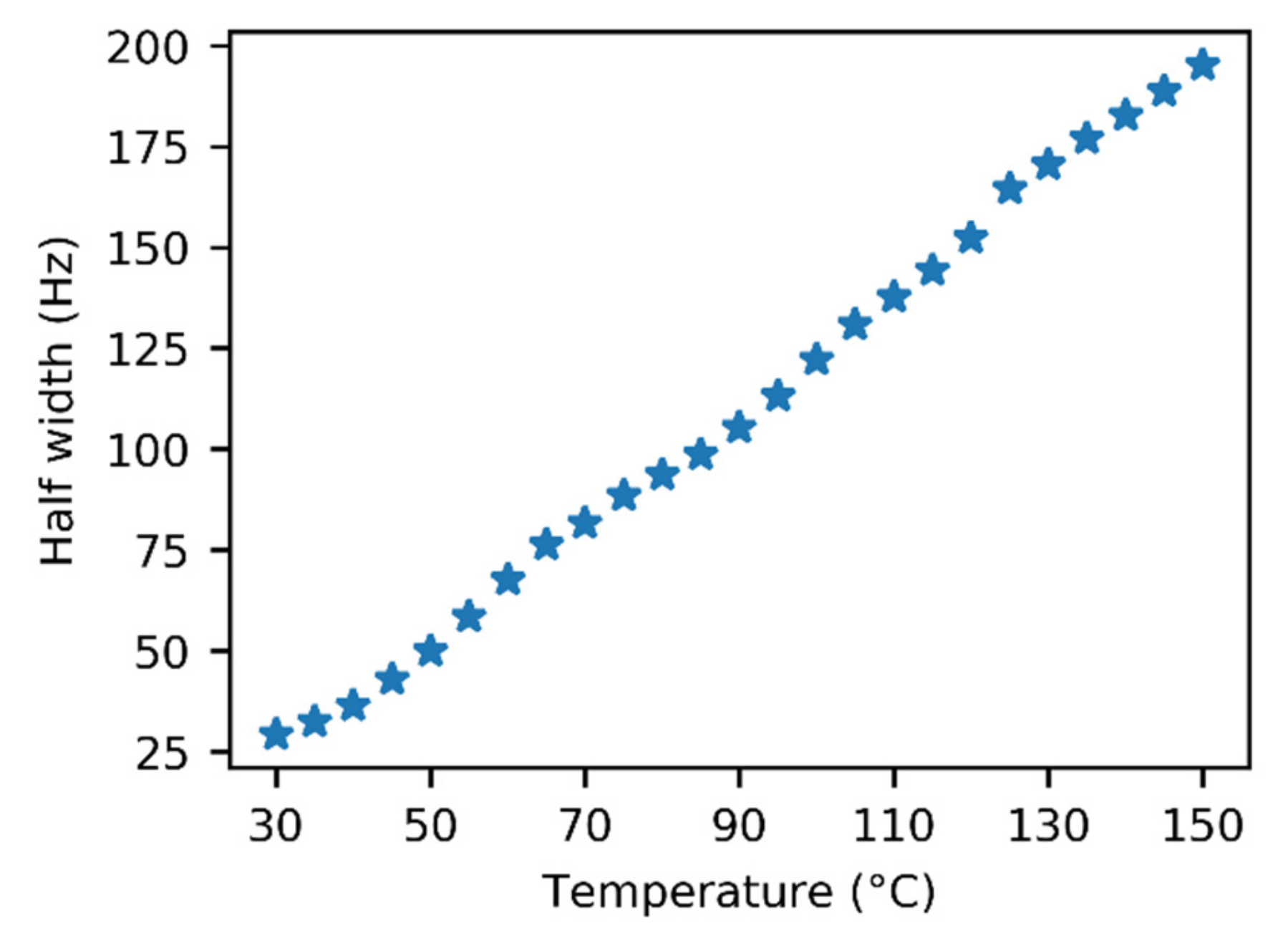
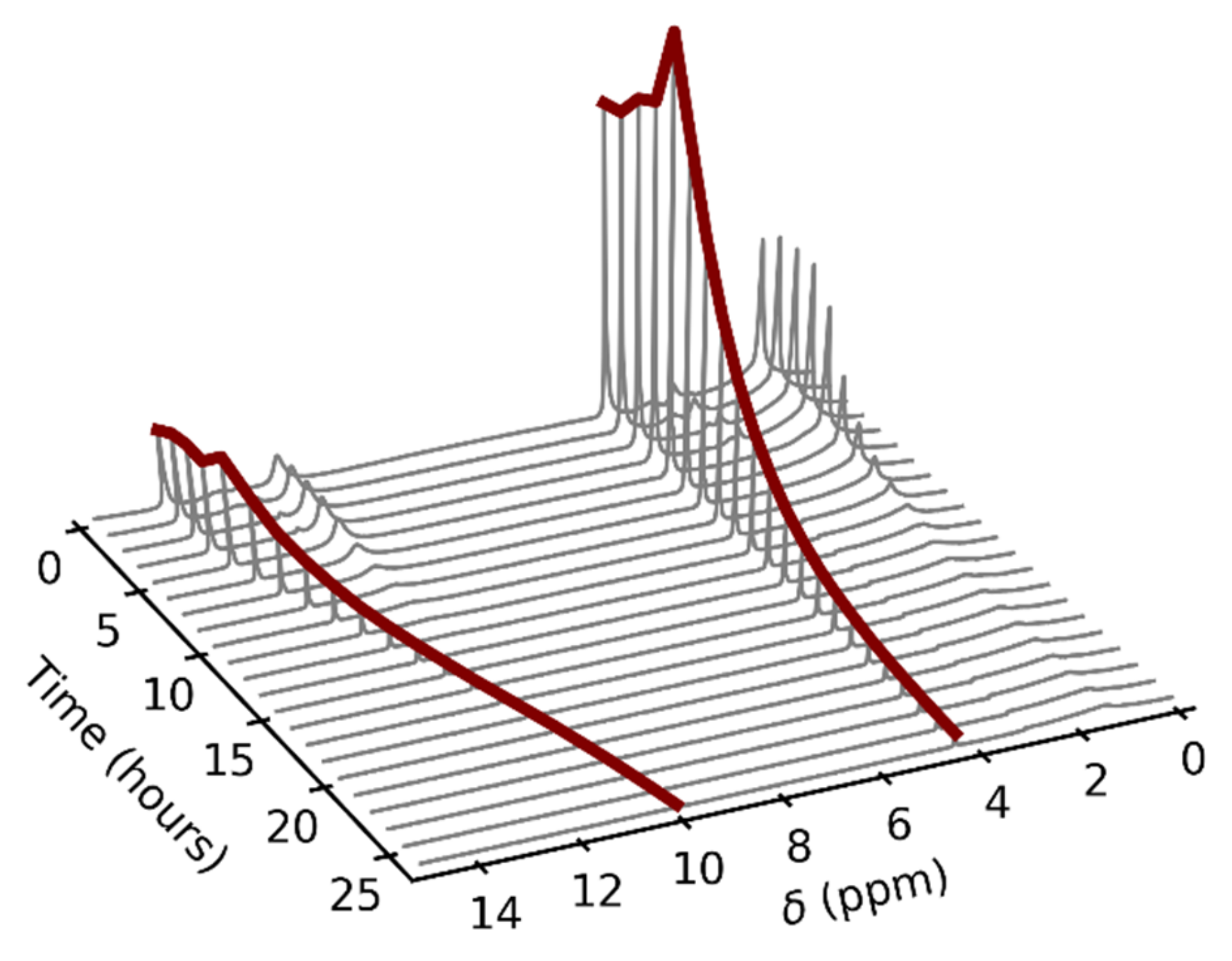
3.1. NMR Experiments
3.2. Computations
3.2.1. The Structure of the Acetic Acid Dimer
3.2.2. Dimerization Energy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Davies, J.A.; Hanson-Heine, M.W.D.; Besley, N.A.; Shirley, A.; Trowers, J.; Yang, S.F.; Ellis, A.M. Dimers of acetic acid in helium nanodroplets. Phys. Chem. Chem. Phys. 2019, 21, 13950–13958. [Google Scholar] [CrossRef] [Green Version]
- Nahringbauer, I. Hydrogen bond studies .39. Reinvestigation of crystal structure of acetic acid (at +5 degrees c and −190 degrees C). Acta Chem. Scand. 1970, 24, 453–462. [Google Scholar] [CrossRef]
- Frurip, D.J.; Curtiss, L.A.; Blander, M. Vapor-phase association in acetic and trifluoroacetic acids—thermal-conductivity measurements and molecular-orbital calculations. J. Am. Chem. Soc. 1980, 102, 2610–2616. [Google Scholar] [CrossRef]
- Emmeluth, C.; Suhm, M.A. A chemical approach towards the spectroscopy of carboxylic acid dimer isomerism. Phys. Chem. Chem. Phys. 2003, 5, 3094–3099. [Google Scholar] [CrossRef]
- Derissen, J.L. Reinvestigation of molecular structure of acetic acid monomer and dimer by gas electron diffraction. J. Mol. Struct. 1971, 7, 67–80. [Google Scholar] [CrossRef]
- Bertie, J.E.; Michaelian, K.H. The raman spectrum of gaseous acetic acid at 21 °C. J. Chem. Phys. 1982, 77, 5267–5271. [Google Scholar] [CrossRef]
- Togeas, J.B. Acetic acid vapor: 2. A statistical mechanical critique of vapor density experiments. J. Phys. Chem. A 2005, 109, 5438–5444. [Google Scholar] [CrossRef] [PubMed]
- Lumbrosobader, N.; Coupry, C.; Baron, D.; Clague, D.H. Dimerization of carboxylic acids: A vapor-phase NMR Study. J. Magn. Reson. 1975, 17, 386–392. [Google Scholar] [CrossRef]
- Lengvinaite, D.; Aidas, K.; Kimtys, L. Molecular aggregation in liquid acetic acid: Insight from molecular dynamics/quantum mechanics modelling of structural and NMR properties. Phys. Chem. Chem. Phys. 2019, 21, 14811–14820. [Google Scholar] [CrossRef] [PubMed]
- Bertagnolli, H. The structure of liquid acetic-acid—an interpretation of neutron-diffraction results by geometrical models. Chem. Phys. Lett. 1982, 93, 287–292. [Google Scholar] [CrossRef]
- Heisler, I.A.; Mazur, K.; Yamaguchi, S.; Tominaga, K.; Meech, S.R. Measuring acetic acid dimer modes by ultrafast time-domain Raman spectroscopy. Phys. Chem. Chem. Phys. 2011, 13, 15573–15579. [Google Scholar] [CrossRef] [PubMed]
- Lütgens, M.; Friedriszik, F.; Lochbrunner, S. Direct observation of the cyclic dimer in liquid acetic acid by probing the C=O vibration with ultrafast coherent Raman spectroscopy. Phys. Chem. Chem. Phys. 2014, 16, 18010–18016. [Google Scholar] [CrossRef] [PubMed]
- Takamuku, T.; Kyoshoin, Y.; Noguchi, H.; Kusano, S.; Yamaguchi, T. Liquid structure of acetic acid-water and trifluoroacetic acid-water mixtures studied by large-angle x-ray scattering and NMR. J. Phys. Chem. B 2007, 111, 9270–9280. [Google Scholar] [CrossRef] [PubMed]
- Flakus, H.T.; Hachula, B. The source of similarity of the IR spectra of acetic acid in the liquid and solid-state phases. Vib. Spectrosc. 2011, 56, 170–176. [Google Scholar] [CrossRef]
- Nakabayashi, T.; Kosugi, K.; Nishi, N. Liquid structure of acetic acid studied by Raman spectroscopy and ab initio molecular orbital calculations. J. Phys. Chem. A 1999, 103, 8595–8603. [Google Scholar] [CrossRef]
- Wu, J.P. Gaussian analysis of Raman spectroscopy of acetic acid reveals a significant amount of monomers that effectively cooperate with hydrogen bonded linear chains. Phys. Chem. Chem. Phys. 2014, 16, 22458–22461. [Google Scholar] [CrossRef]
- Fathi, S.; Bouazizi, S.; Trabelsi, S.; Gonzalez, M.A.; Bahri, M.; Nasr, S.; Bellissent-Funel, M.C. Structural investigation of liquid acetic acid by neutron scattering, DFT calculations and molecular dynamics simulations. Complementarity to x-ray scattering results. J. Mol. Liq. 2014, 196, 69–76. [Google Scholar] [CrossRef]
- Pašalić, H.; Tunega, D.; Aquino, A.J.A.; Haberhauer, G.; Gerzabek, M.H.; Lischka, H. The stability of the acetic acid dimer in microhydrated environments and in aqueous solution. Phys. Chem. Chem. Phys. 2012, 14, 4162–4170. [Google Scholar] [CrossRef]
- Lim, V.T.; Bayly, C.I.; Fusti-Molnar, L.; Mobley, D.L. Assessing the conformational equilibrium of carboxylic acid via quantum mechanical and molecular dynamics studies on acetic acid. J. Chem. Inf. Model. 2019, 59, 1957–1964. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.H.; Chen, L.H.; Yang, H.M.; Ma, J. Theoretical study of acetic acid association based on hydrogen bonding mechanism. J. Phys. Chem. A 2017, 121, 4560–4568. [Google Scholar] [CrossRef]
- Chocholoušová, J.; Vacek, J.; Hobza, P. Acetic acid dimer in the gas phase, nonpolar solvent, microhydrated environment, and dilute and concentrated acetic acid: Ab initio quantum chemical and molecular dynamics simulations. J. Phys. Chem. A 2003, 107, 3086–3092. [Google Scholar] [CrossRef]
- Colominas, C.; Teixido, J.; Cemeli, J.; Luque, F.J.; Orozco, M. Dimerization of carboxylic acids: Reliability of theoretical calculations and the effect of solvent. J. Phys. Chem. B 1998, 102, 2269–2276. [Google Scholar] [CrossRef]
- Řezáč, J.; Riley, K.E.; Hobza, P. S66: A Well-balanced database of benchmark interaction energies relevant to biomolecular structures. J. Chem. Theory Comput. 2011, 7, 2427–2438. [Google Scholar] [CrossRef] [PubMed]
- Řezáč, J.; Hobza, P. Advanced corrections of hydrogen bonding and dispersion for semiempirical quantum mechanical methods. J. Chem. Theory Comput. 2012, 8, 141–151. [Google Scholar] [CrossRef]
- Brauer, B.; Kesharwani, M.K.; Martin, J.M.L. Some observations on counterpoise corrections for explicitly correlated calculations on noncovalent interactions. J. Chem. Theory Comput. 2014, 10, 3791–3799. [Google Scholar] [CrossRef]
- Miller, C.E.; Francisco, J.S. A quadratic configuration interaction study of the proton affinity of acetic acid. Chem. Phys. Lett. 2002, 364, 427–431. [Google Scholar] [CrossRef]
- Saunders, C.M.; Tantillo, D.J. Application of computational chemical shift prediction techniques to the cereoanhydride structure problem—carboxylate complications. Mar. Drugs 2017, 15, 171. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, W.W.; Fischer, D.; Irmer, G. Vibrational spectroscopic studies and DFT calculations on NaCH3CO2(aq) and CH3COOH(aq). Dalton Trans. 2014, 43, 3174–3185. [Google Scholar] [CrossRef]
- de Nevers, N. Air pollution control engineering; Waveland Pr Inc.: Long Grove, IL, USA, 2010. [Google Scholar]
- Dean, J.A. Lange’s Handbook of Chemistry, 15th ed.; McGraw-Hill, Inc.: New York, NY, 1999. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry 3. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H. Gaussian-basis sets for use in correlated molecular calculations.1. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron-affinities of the 1st-row atoms revisited–systematic basis-sets and wave-functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Boys, S.F.; Bernardi, F. Calculation of small molecular interactions by differences of separate total energies—some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an approximation treatment for many-electron systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, R.J.; Purvis, G.D. Many-body perturbation-theory, coupled-pair many-electron theory, and importance of quadruple excitations for correlation problem. Int. J. Quantum Chem. 1978, 14, 561–581. [Google Scholar] [CrossRef]
- Čížek, J. On the use of the cluster expansion and the technique of diagrams in calculations of correlation effects in atoms and molecules. In Advances in Chemical Physics; LeFebvre, R., Moser, C., Eds.; John Wiley & Sons, Ltd.: London, UK, 1969; Volume 14, pp. 35–89. [Google Scholar]
- Purvis, G.D.; Bartlett, R.J. A full coupled-cluster singles and doubles model—the inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Scuseria, G.E.; Janssen, C.L.; Schaefer, H.F. An efficient reformulation of the closed-shell coupled cluster single and double excitation (Ccsd) equations. J. Chem. Phys. 1988, 89, 7382–7387. [Google Scholar] [CrossRef]
- Helgaker, T.; Klopper, W.; Koch, H.; Noga, J. Basis-set convergence of correlated calculations on water. J. Chem. Phys. 1997, 106, 9639–9646. [Google Scholar] [CrossRef]
- Hobza, P. Theoretical studies of hydrogen bonding. In Annual Reports Section C (Physical Chemistry); Webb, G.A., Ed.; Royal Society of Chemistry: Cambridge, UK, 2004; Volume 100, pp. 3–27. [Google Scholar]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical-shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Jensen, F. Basis set convergence of nuclear magnetic shielding constants calculated by density functional methods. J. Chem. Theory Comput. 2008, 4, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, J.D. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Stapleton, J.J.; Suchy, D.L.; Banerjee, J.; Mueller, K.T.; Pantano, C.G. adsorption reactions of carboxylic acid functional groups on sodium aluminoborosilicate glass fiber surfaces. Acs Appl. Mater. Interfaces 2010, 2, 3303–3309. [Google Scholar] [CrossRef]
- Alvarez-Idaboy, J.R.; Galano, A. Counterpoise corrected interaction energies are not systematically better than uncorrected ones: Comparison with CCSD(T) CBS extrapolated values. Theor. Chem. Acc. 2010, 126, 75–85. [Google Scholar] [CrossRef]
- Sheng, X.W.; Mentel, L.; Gritsenko, O.V.; Baerends, E.J. Counterpoise correction is not useful for short and van der waals distances but may be useful at long range. J. Comput. Chem. 2011, 32, 2896–2901. [Google Scholar] [CrossRef]
- Mentel, L.M.; Baerends, E.J. Can the counterpoise correction for basis set superposition effect be justified? J. Chem. Theory Comput. 2014, 10, 252–267. [Google Scholar] [CrossRef]
- Ruud, K.; Åstrand, P.O.; Taylor, P.R. Zero-point vibrational effects on proton shieldings: Functional-group contributions from ab initio calculations. J. Am. Chem. Soc. 2001, 123, 4826–4833. [Google Scholar] [CrossRef]
- Dračínský, M.; Hodgkinson, P. Effects of quantum nuclear delocalisation on NMR parameters from path integral molecular dynamics. Chem. Eur. J. 2014, 20, 2201–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dračínský, M.; Bouř, P.; Hodgkinson, P. Temperature dependence of NMR parameters calculated from path integral molecular dynamics simulations. J. Chem. Theory Comput. 2016, 12, 968–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, R.; Socha, O.; Slavíček, P.; Šála, M.; Hodgkinson, P.; Dračínský, M. Proton transfer in guanine-cytosine base pair analogues studied by NMR spectroscopy and PIMD simulations. Faraday Discuss. 2018, 212, 331–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance/Angle | MP2 | B3LYP | B3LYP and GD3 |
|---|---|---|---|
| O–H | 0.998 | 0.002 | 0.003 |
| O–H···O | 2.650 | 0.012 | 0.005 |
| C1,C1’ | 3.832 | 0.015 | 0.005 |
| C1–C2 | 1.495 | 0.006 | 0.006 |
| C=O | 1.226 | −0.003 | −0.003 |
| C–O | 1.319 | 0.000 | −0.001 |
| ∢ O–C–O | 123.90 | −0.16 | 0.00 |
| Method | Basis Set | ∆H | ∆S | ∆G0 |
|---|---|---|---|---|
| B3LYP | Aug-cc-pVQZ | −14.27 | −37.43 | −3.11 |
| B3LYP and GD3 | Aug-cc-pVQZ | −16.77 | −37.25 | −5.67 |
| MP2 | Aug-cc-pVTZ | −15.93 | −37.12 | −4.86 |
| MP2 | Aug-cc-pVQZ | −15.57 * | −4.51 * | |
| MP2 | Aug-cc-pV5Z | −15.34 * | −4.28 * | |
| MP2 | CBS [a] | −15.31 * | −4.24 * | |
| CCSD(T) | Aug-cc-pVDZ | −15.83 * | −4.76 * | |
| CCSD(T) | Aug-cc-pVTZ | −16.08 * | −5.02 * | |
| CCSD(T) | CBS [b] | −15.46 * | −4.40 * | |
| exptl. | −15.38 | −36.6 | −4.48 |
| Method | Monomer | Dimer |
|---|---|---|
| MP2 | 4.01 | 11.77 |
| B3LYP | 3.90 | 11.81 |
| B3LYP and GD3 | 3.86 | 11.91 |
| Vibrational correction [a] | −0.08 | −0.05 |
| Experiment | 3.76 | 10.55 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Socha, O.; Dračínský, M. Dimerization of Acetic Acid in the Gas Phase—NMR Experiments and Quantum-Chemical Calculations. Molecules 2020, 25, 2150. https://doi.org/10.3390/molecules25092150
Socha O, Dračínský M. Dimerization of Acetic Acid in the Gas Phase—NMR Experiments and Quantum-Chemical Calculations. Molecules. 2020; 25(9):2150. https://doi.org/10.3390/molecules25092150
Chicago/Turabian StyleSocha, Ondřej, and Martin Dračínský. 2020. "Dimerization of Acetic Acid in the Gas Phase—NMR Experiments and Quantum-Chemical Calculations" Molecules 25, no. 9: 2150. https://doi.org/10.3390/molecules25092150
APA StyleSocha, O., & Dračínský, M. (2020). Dimerization of Acetic Acid in the Gas Phase—NMR Experiments and Quantum-Chemical Calculations. Molecules, 25(9), 2150. https://doi.org/10.3390/molecules25092150