
The Exchange of Cyclometalated Ligands

Contents
- Introduction
- The Exchange Discovery
- Scope: A Neat Synthetic Procedure
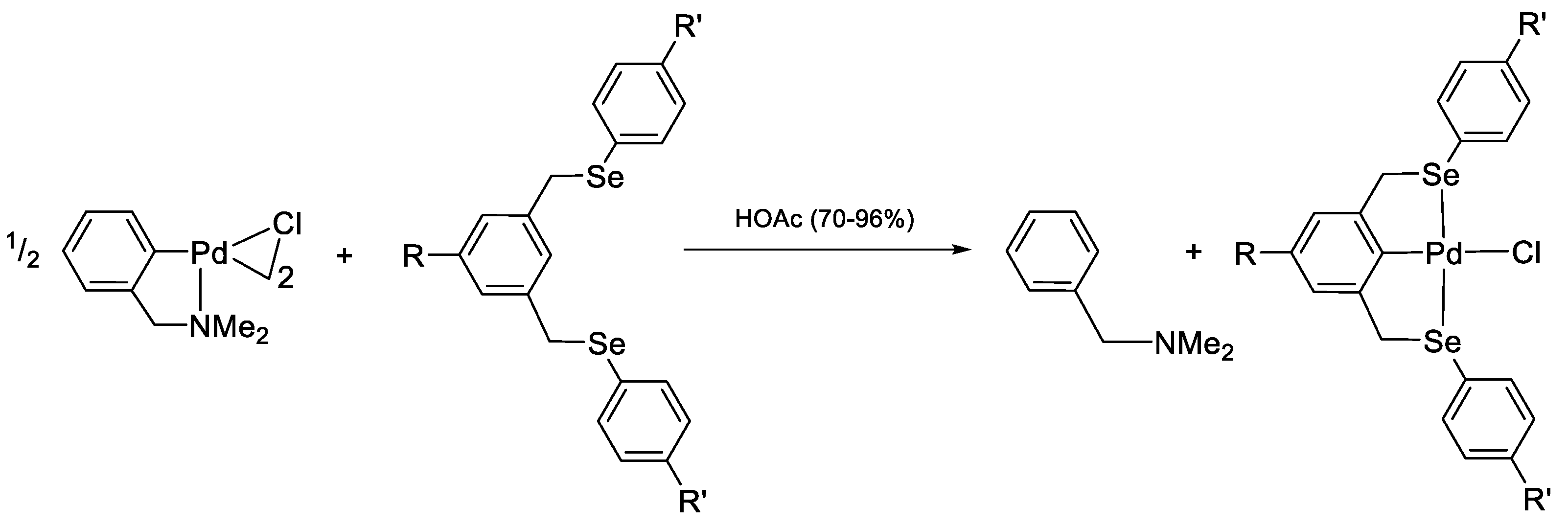
- Cyclopalladation of Phosphorous and Antimony Donor Ligands Including o-Carboranes
- Asymmetric Version of the Ligand Exchange
- The Mechanism of Palladium N/N Ligand Exchange6.1. Thermodynamics of Palladium(II) Exchange6.2. Kinetics and Mechanism of Palladium(II) Exchange6.3. “Mechanism” of Thermodynamic Control6.4. A Mechanistic Comment Regarding Section 5
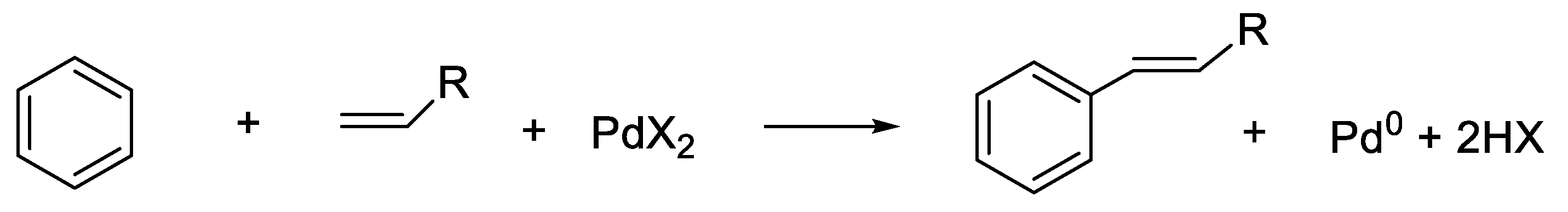
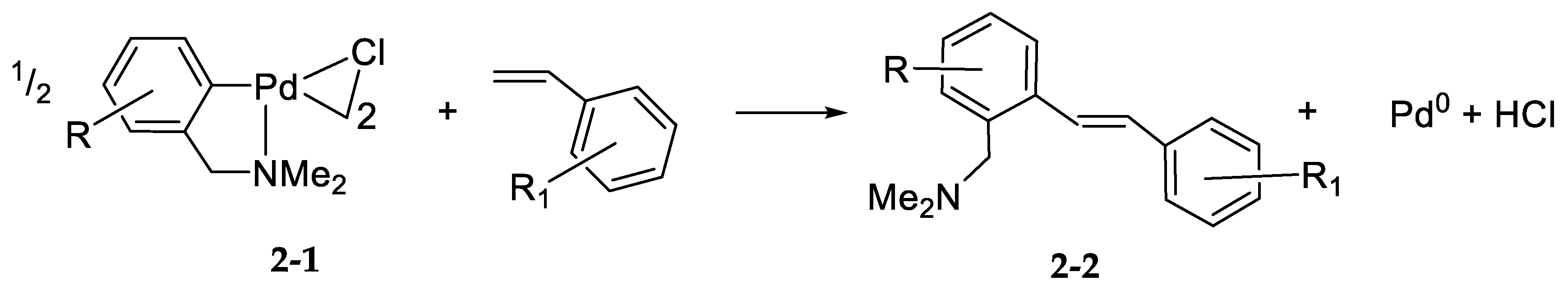
- Processes Mechanistically Relevant to Palladium Ligand ExchangePalladation by η3-Allyl Palladium(II) Dimers
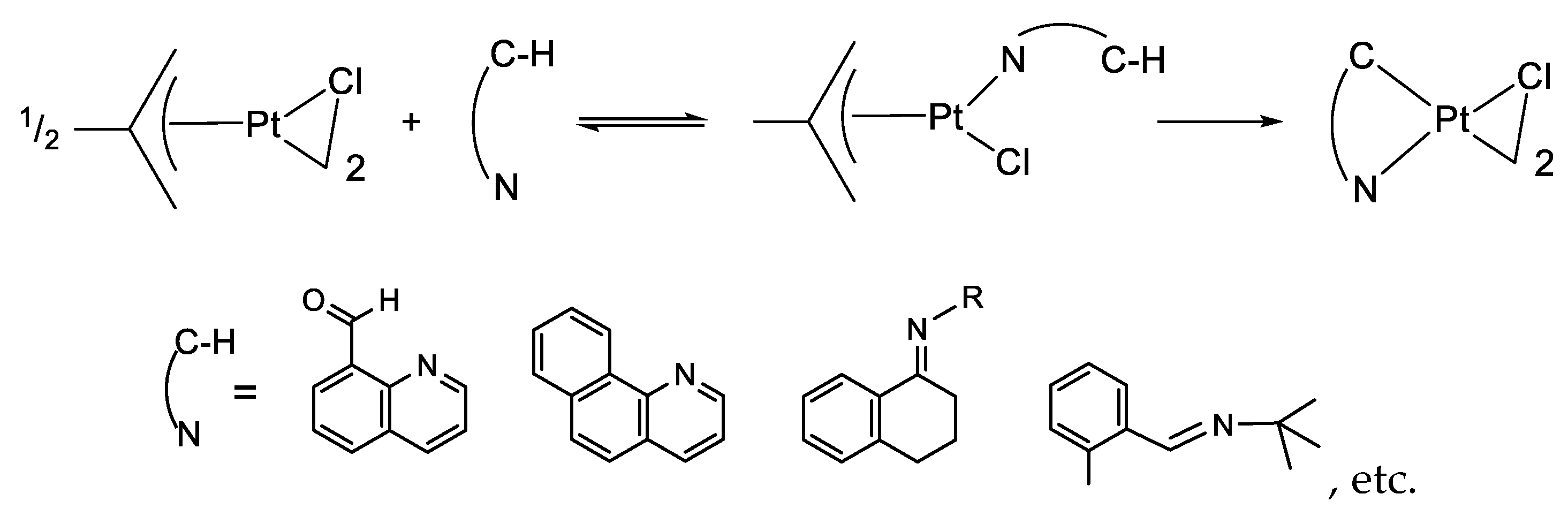
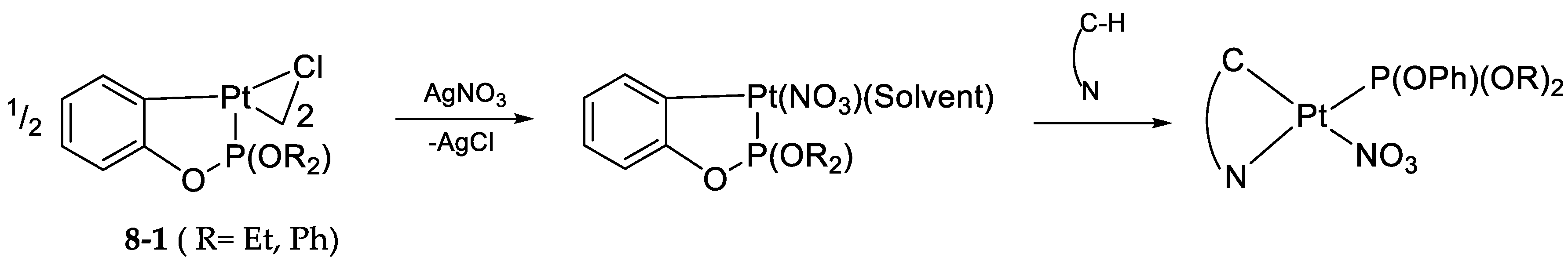
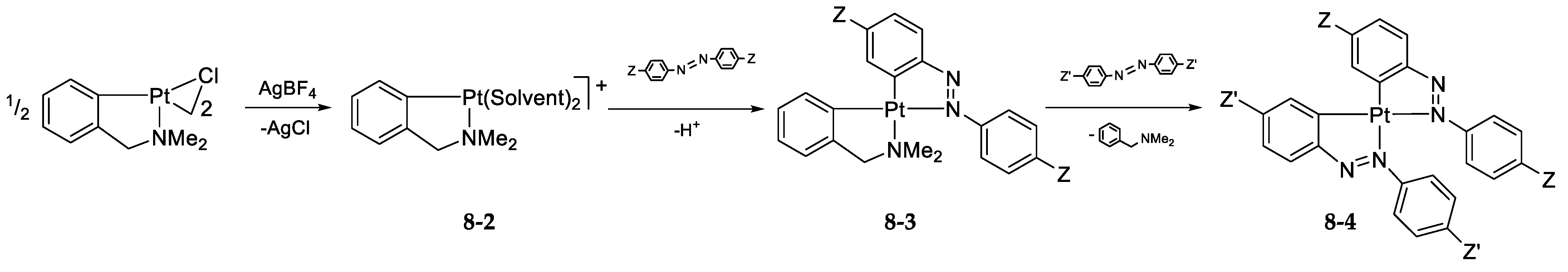
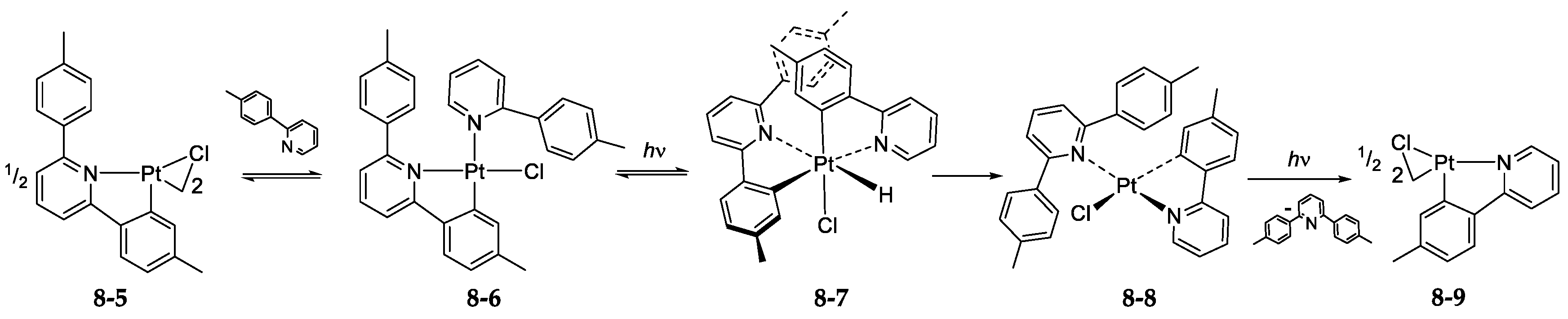
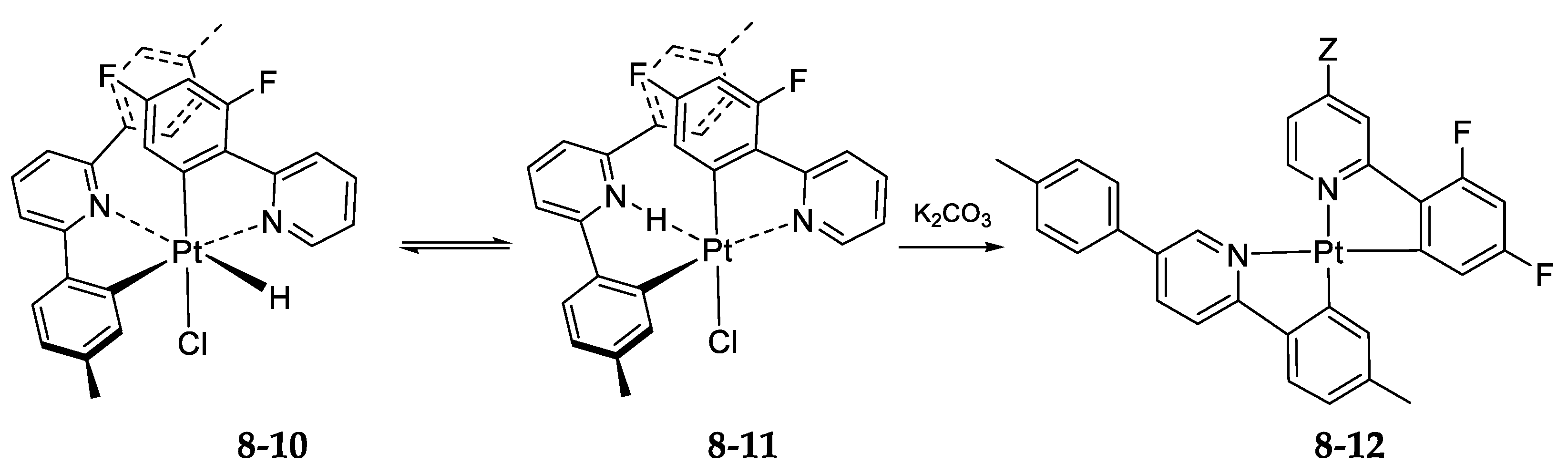
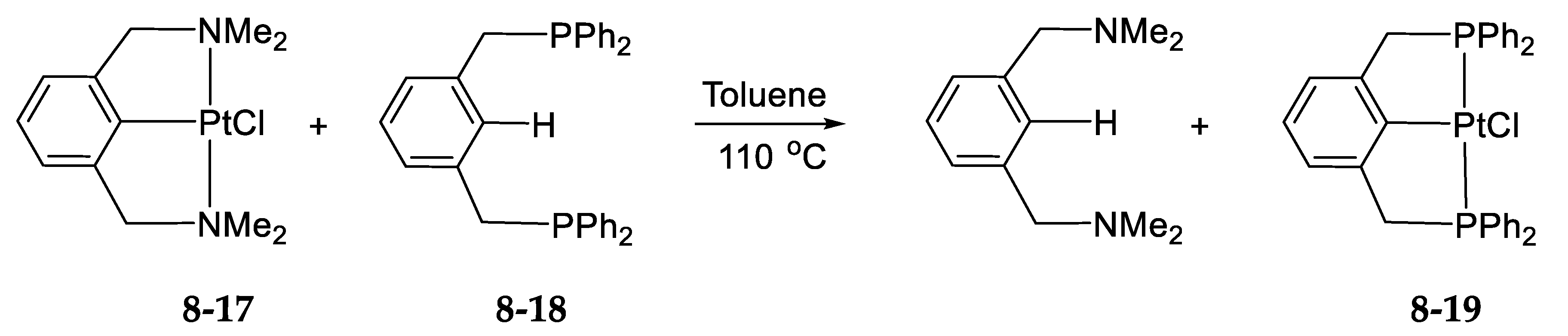
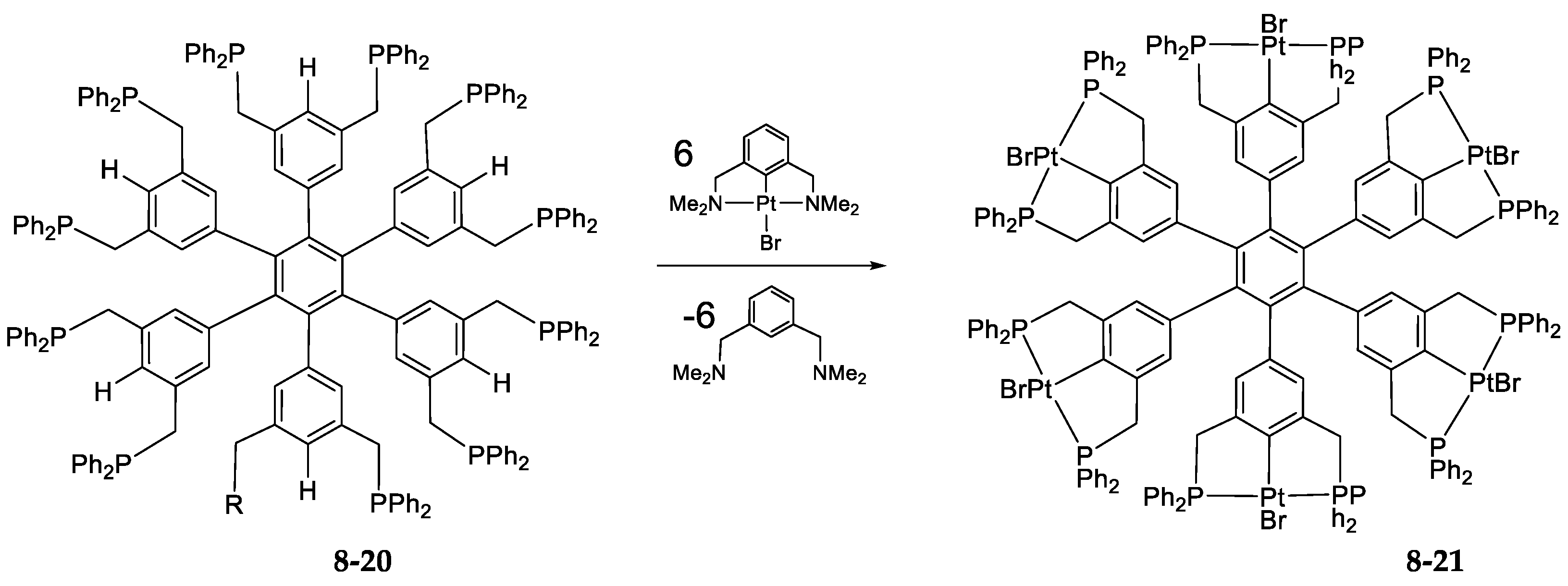
- Exchange in the Platinum Series8.1. η3-Allyls as Leaving Ligands8.2. True Exchange of Cycloplatinated Ligands8.3. Intramolecular Exchange of Cycloplatinated Ligands8.4. Pincer-Pincer Ligand Exchange in Cycloplatinated Complexes8.5. Exchange of Cyclometalated Ligands or Transcyclometalation?
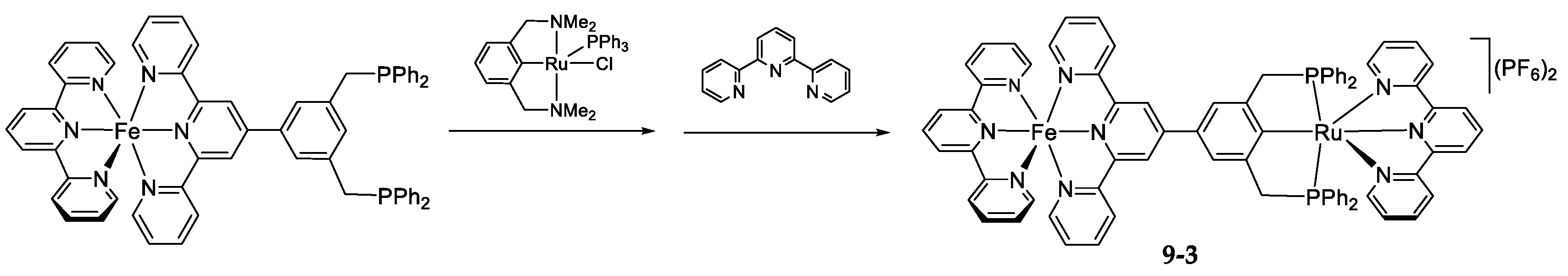
- The Ligand Exchange with Pincer Ruthenium Complexes
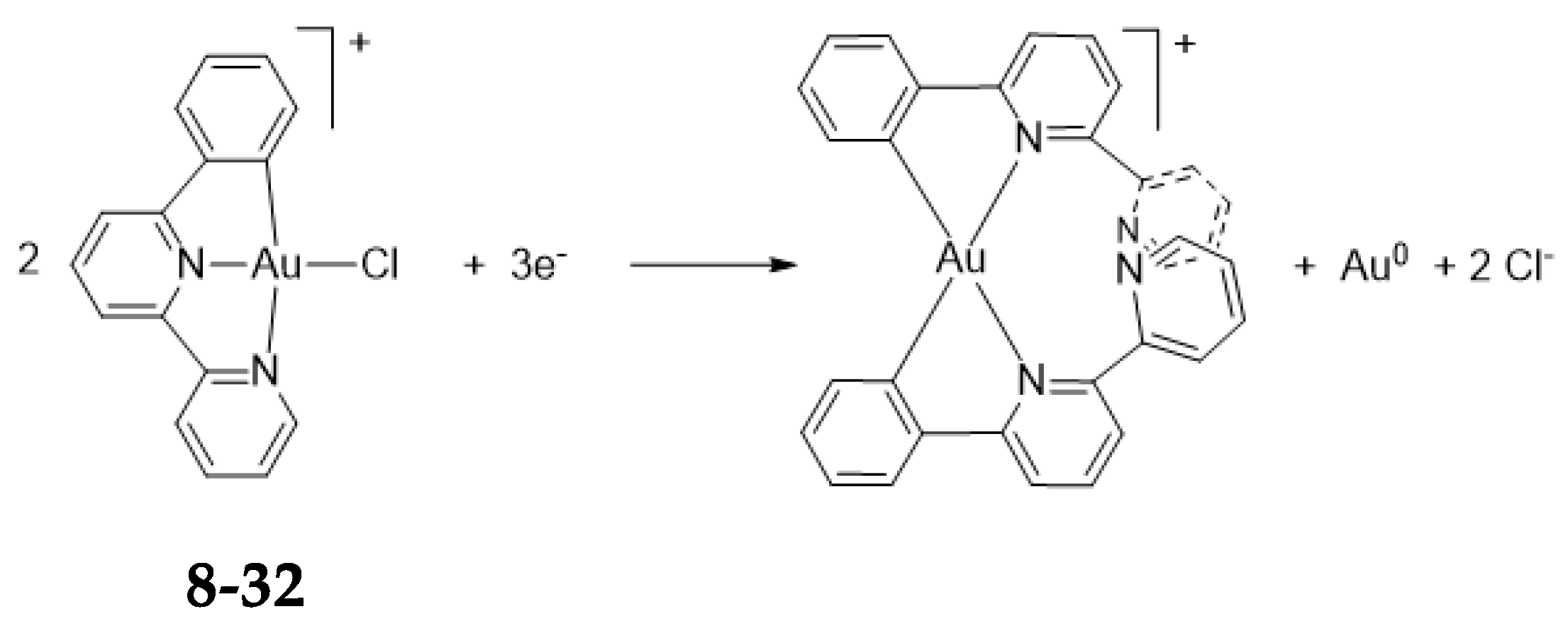
- Other Examples of Exchange of Cyclometalated Ligands10.1. Manganese(I)10.2. Rhodium(III)10.3. Iridium(III)
- Summary and Conclusions
1. Introduction
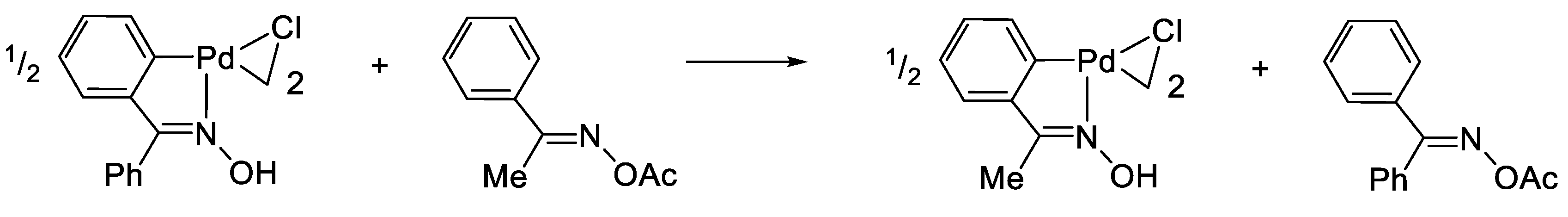
2. The Exchange Discovery
3. Scope: A Neat Synthetic Procedure
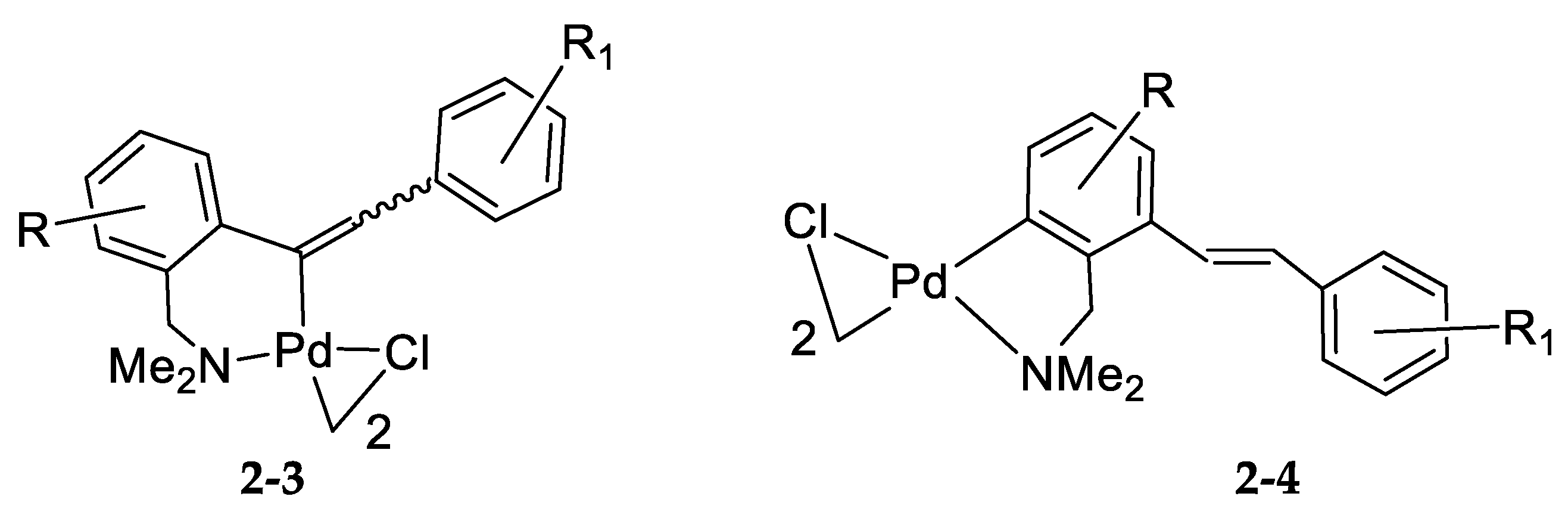
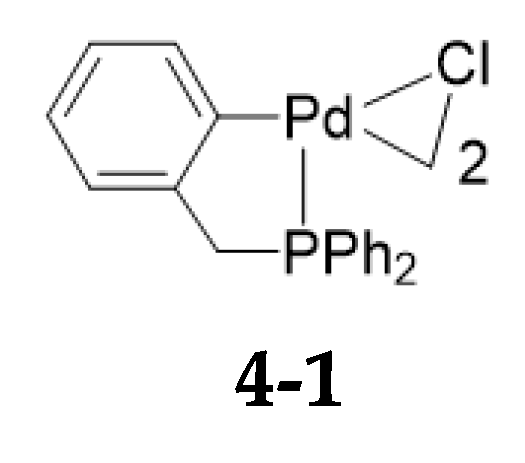
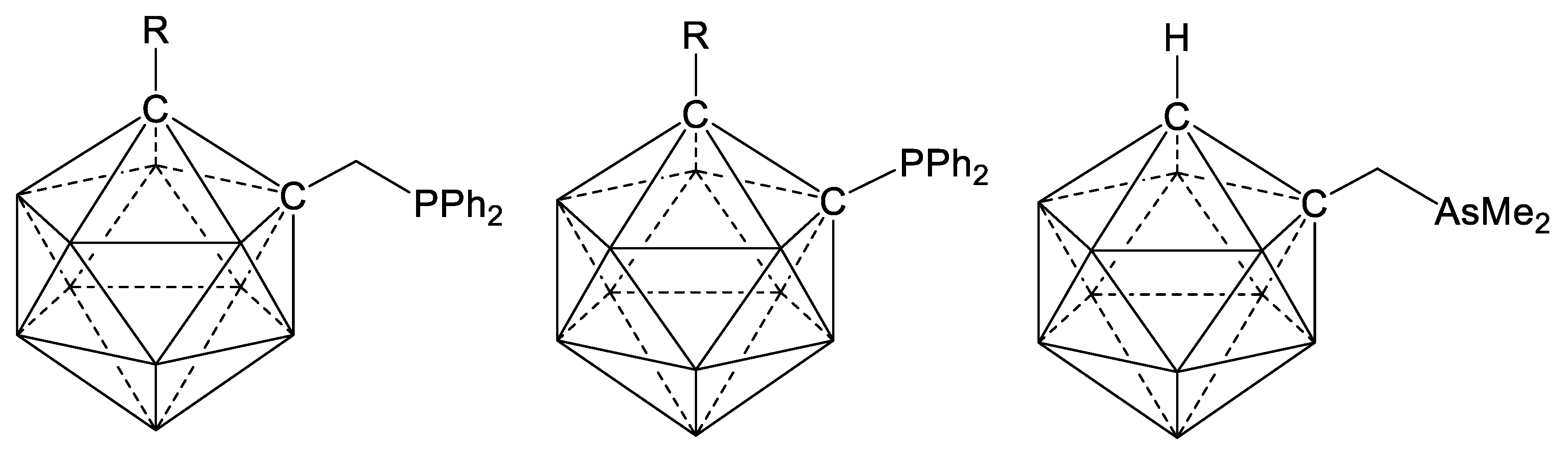
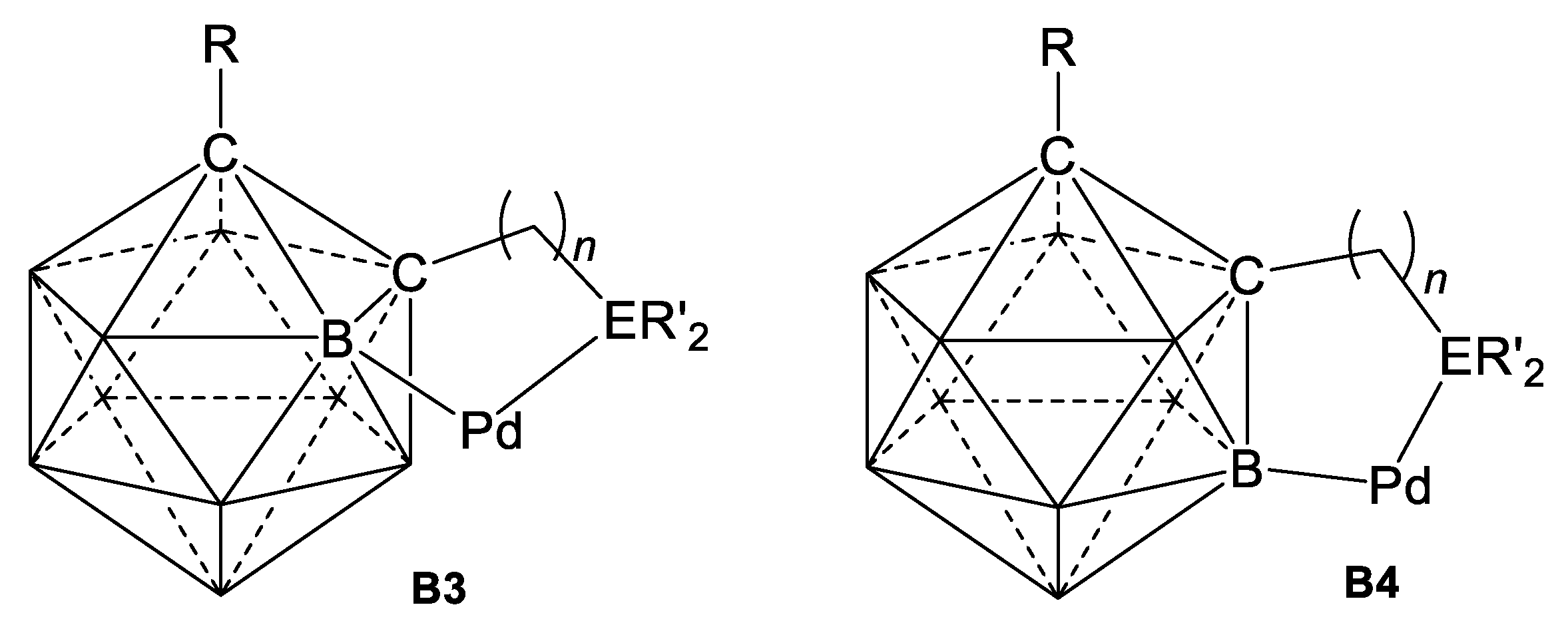
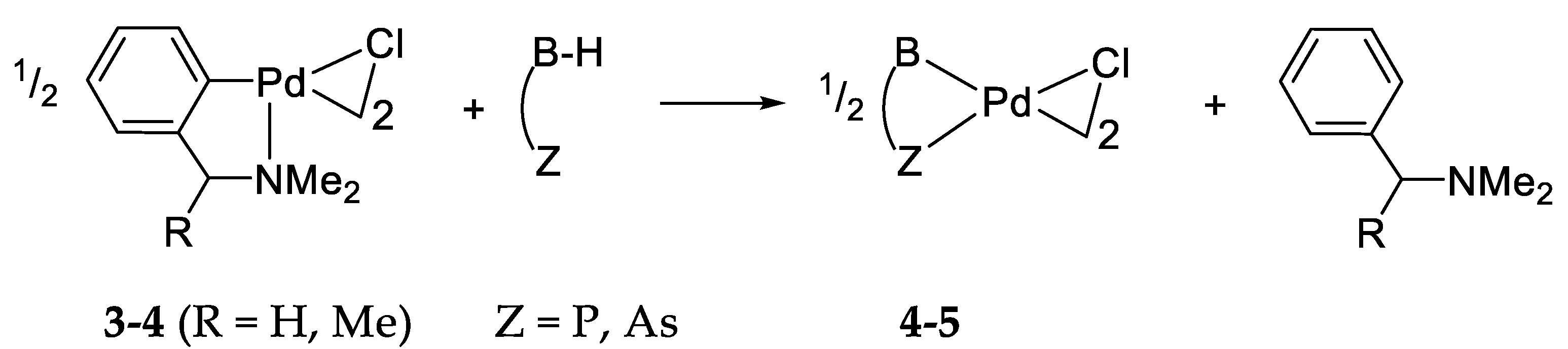
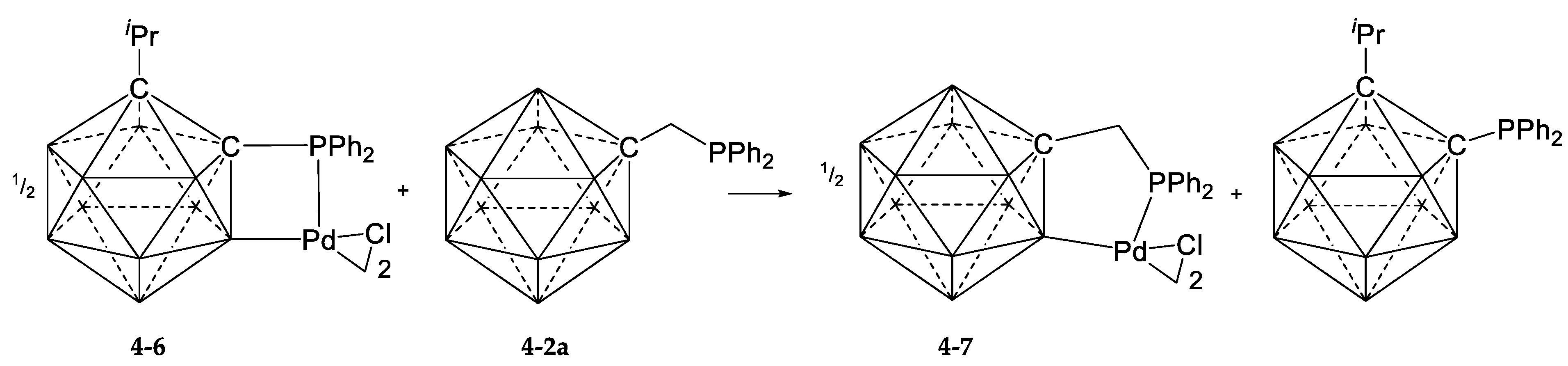
4. Cyclopalladation of Phosphorous and Antimony Donor Ligands Including o-Carboranes
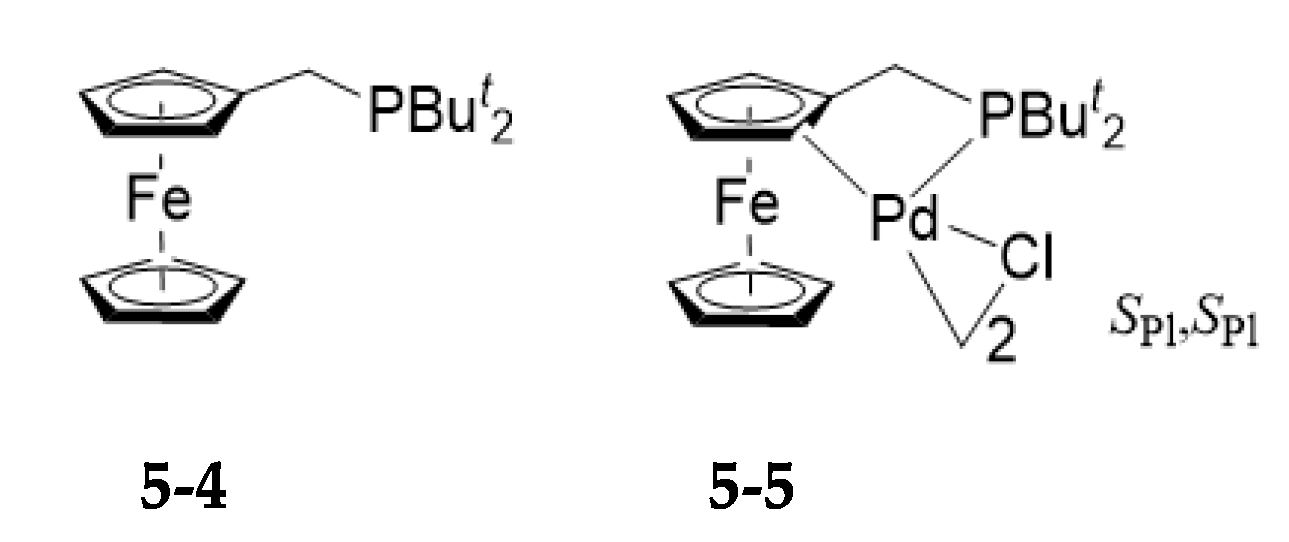
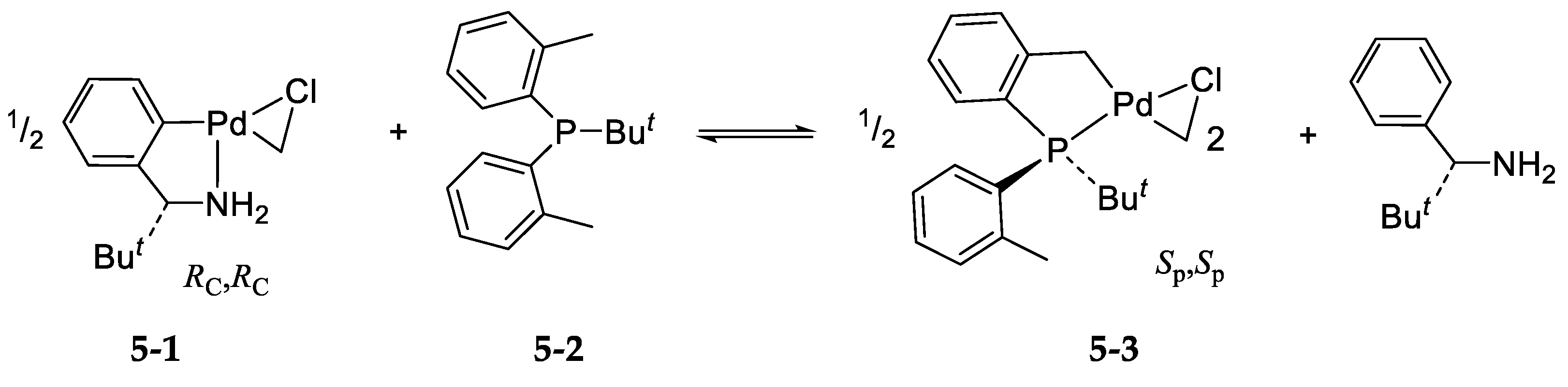
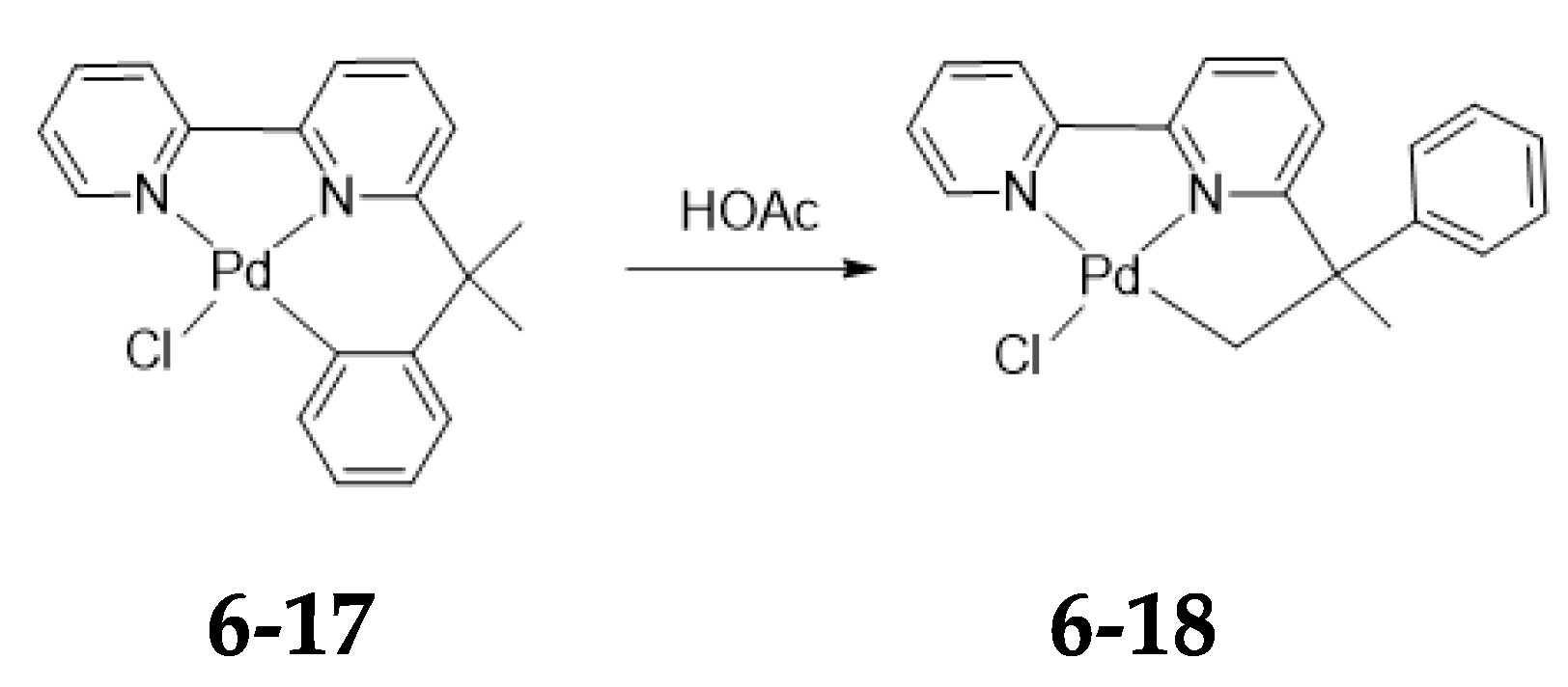
5. Asymmetric Version of the Ligand Exchange
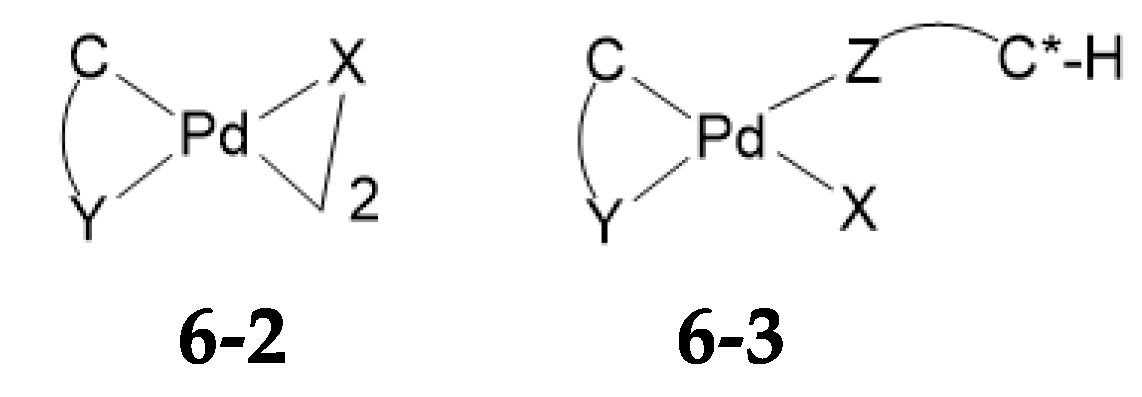
6. The Mechanism of Palladium N/N Ligand Exchange
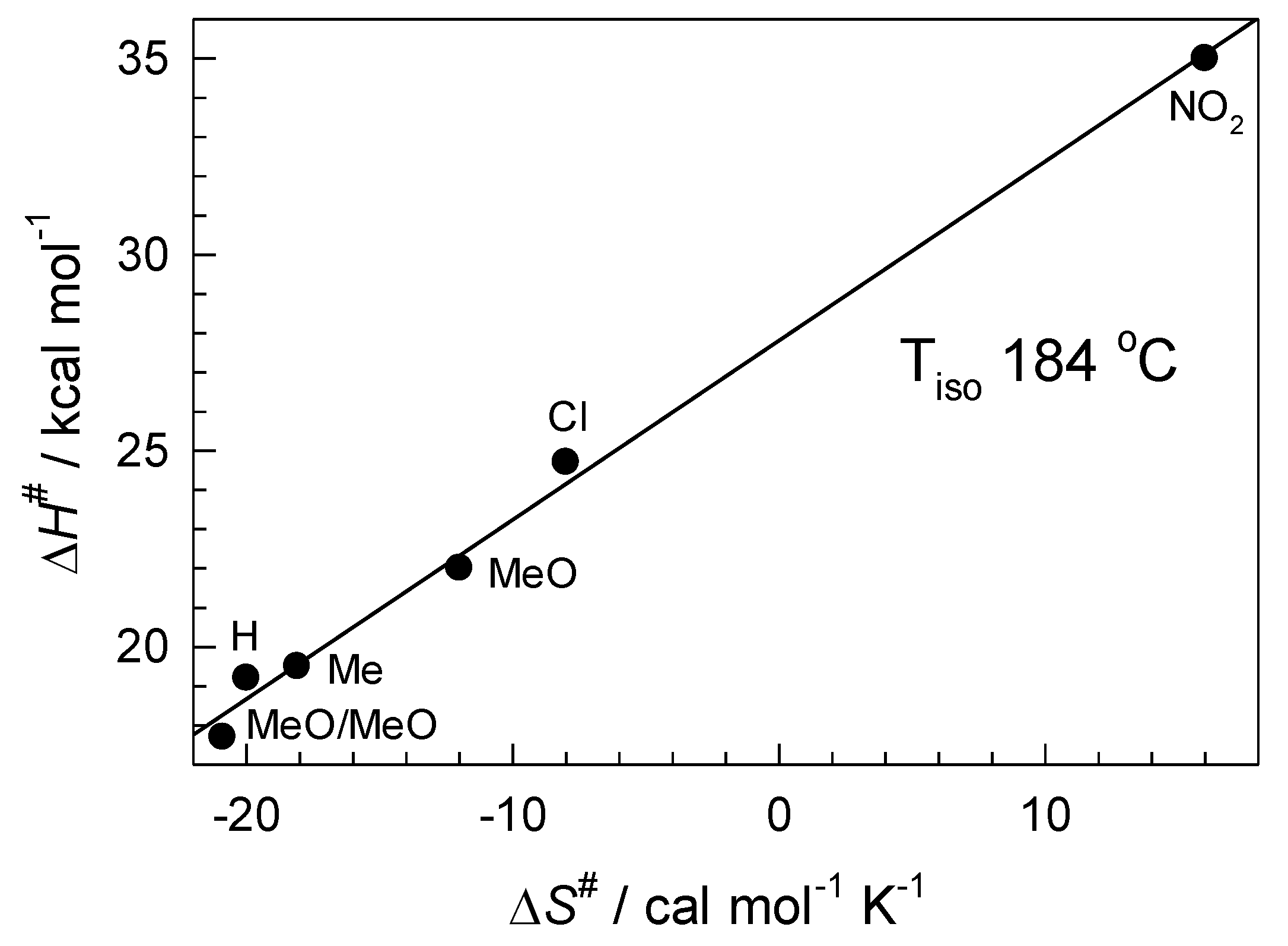
6.1. Thermodynamics of Palladium(II) Exchange
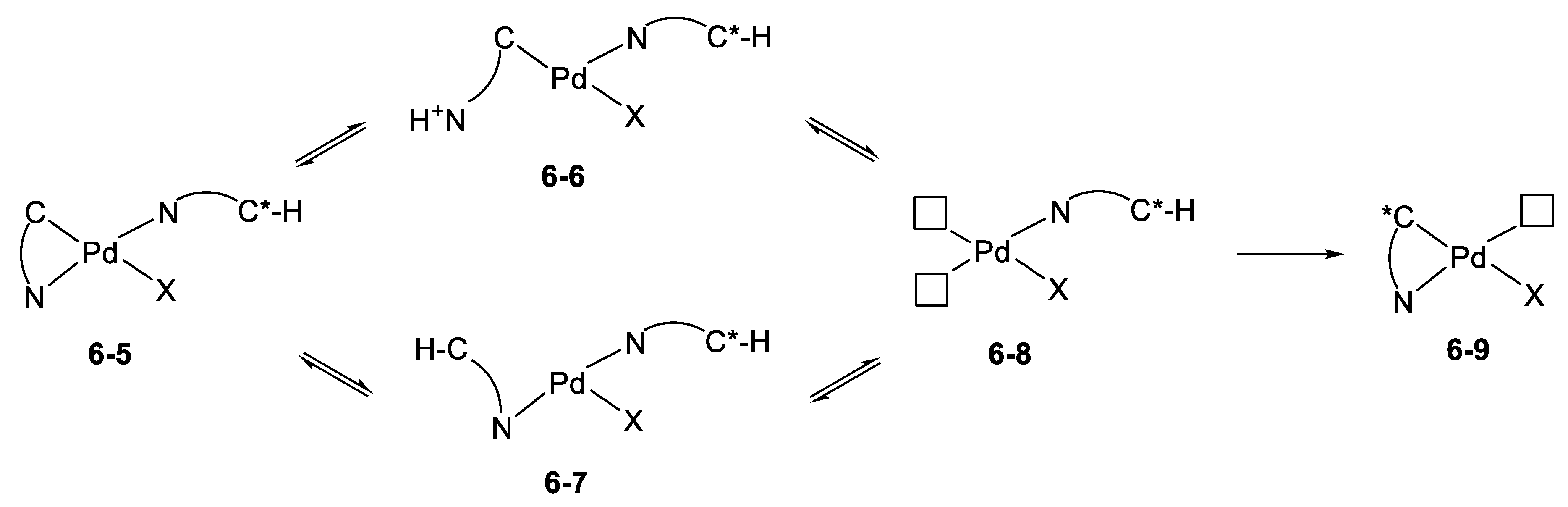
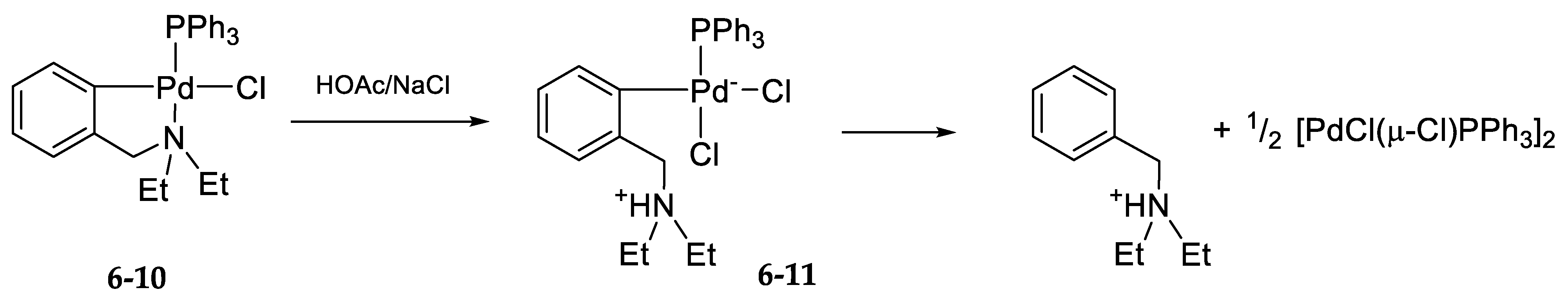
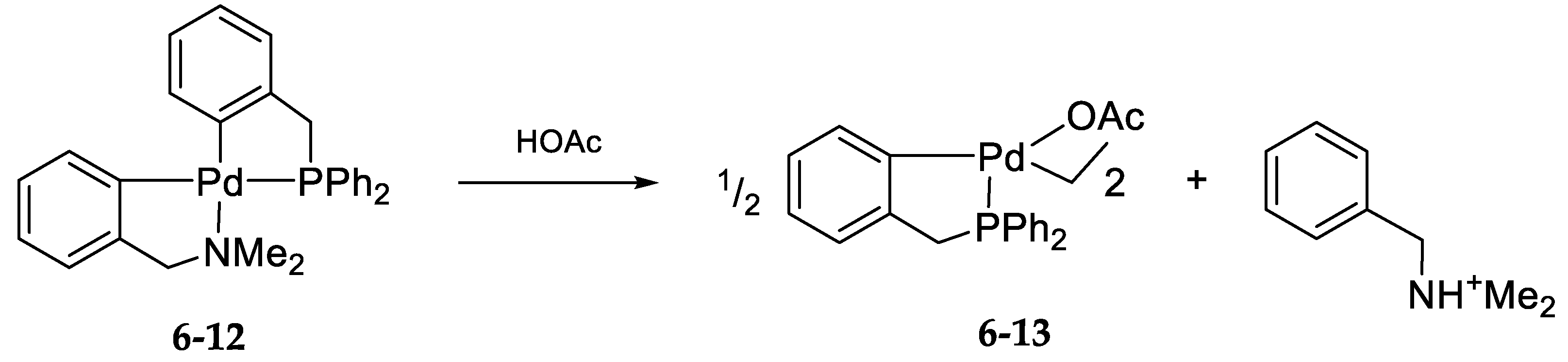
6.2. Kinetics and Mechanism of Palladium(II) Exchange
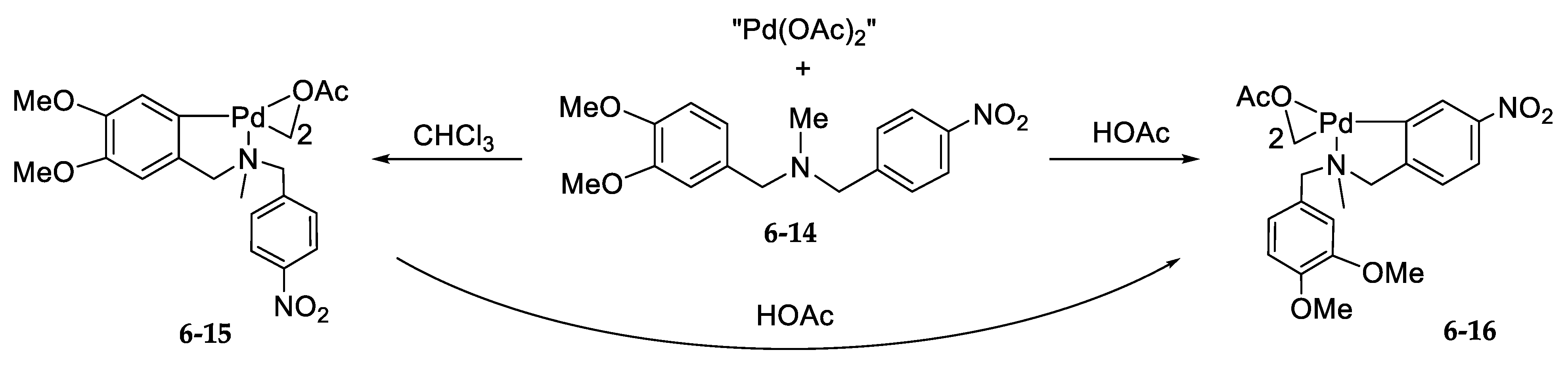
6.3. “Mechanism” of Thermodynamic Control
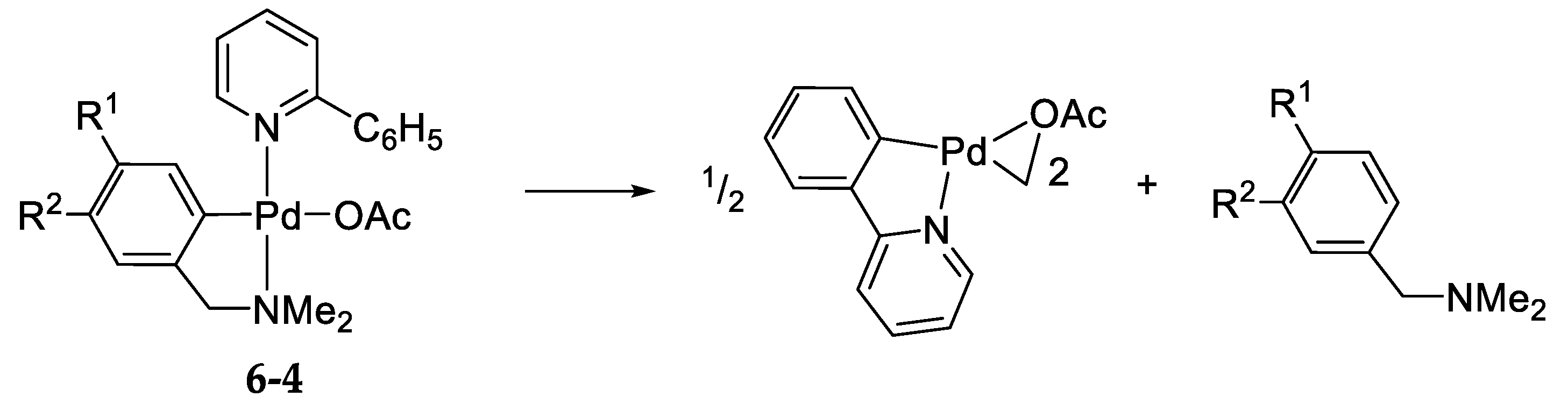
6.4. A Mechanistic Comment Regarding Section 5
7. Processes Mechanistically Relevant to Palladium Ligand Exchange
Palladation by η3-Allyl Palladium(II) Dimers
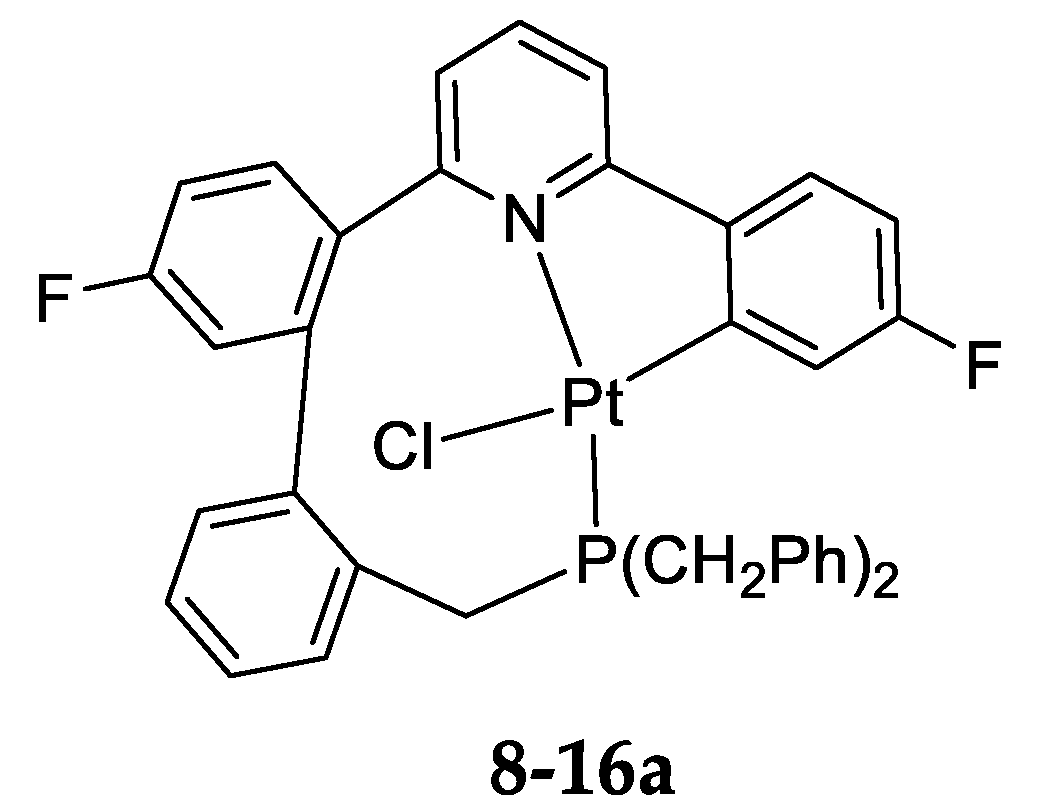
8. Exchange in the Platinum Series
8.1. η3-Allyls as Leaving Ligands
8.2. True Exchange of Cycloplatinated Ligands
8.3. Intramolecular Exchange of Cycloplatinated Ligands
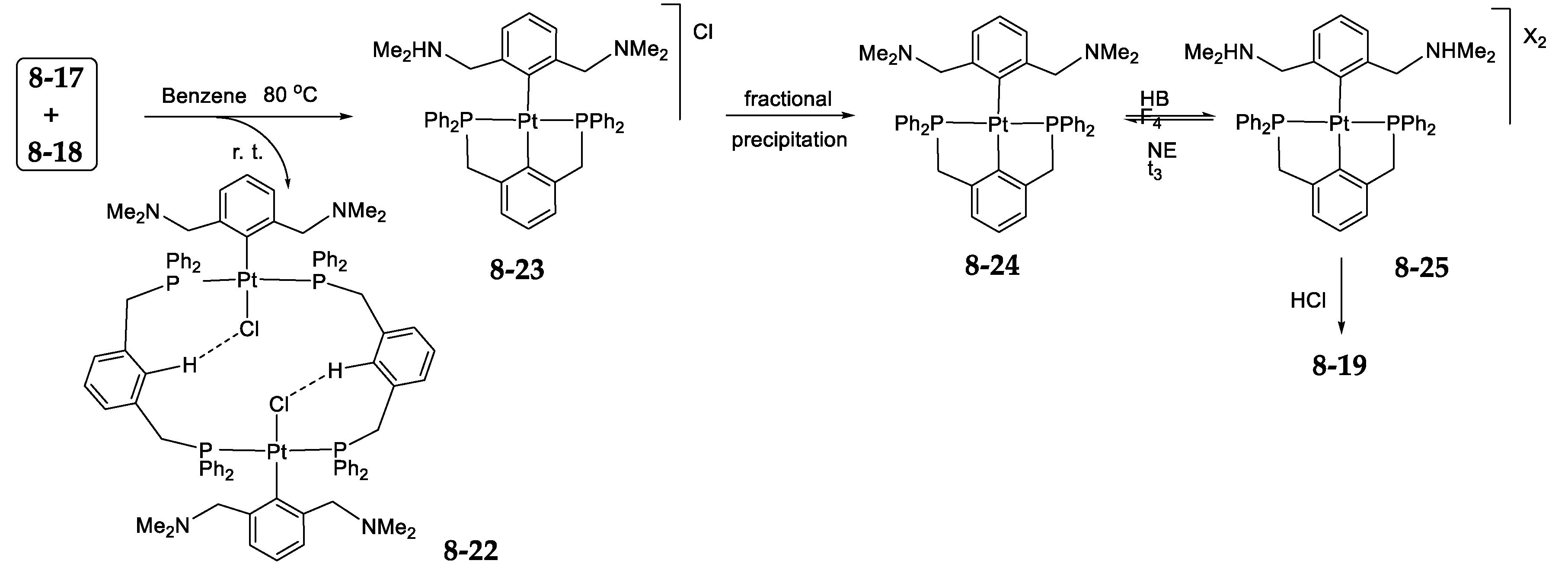
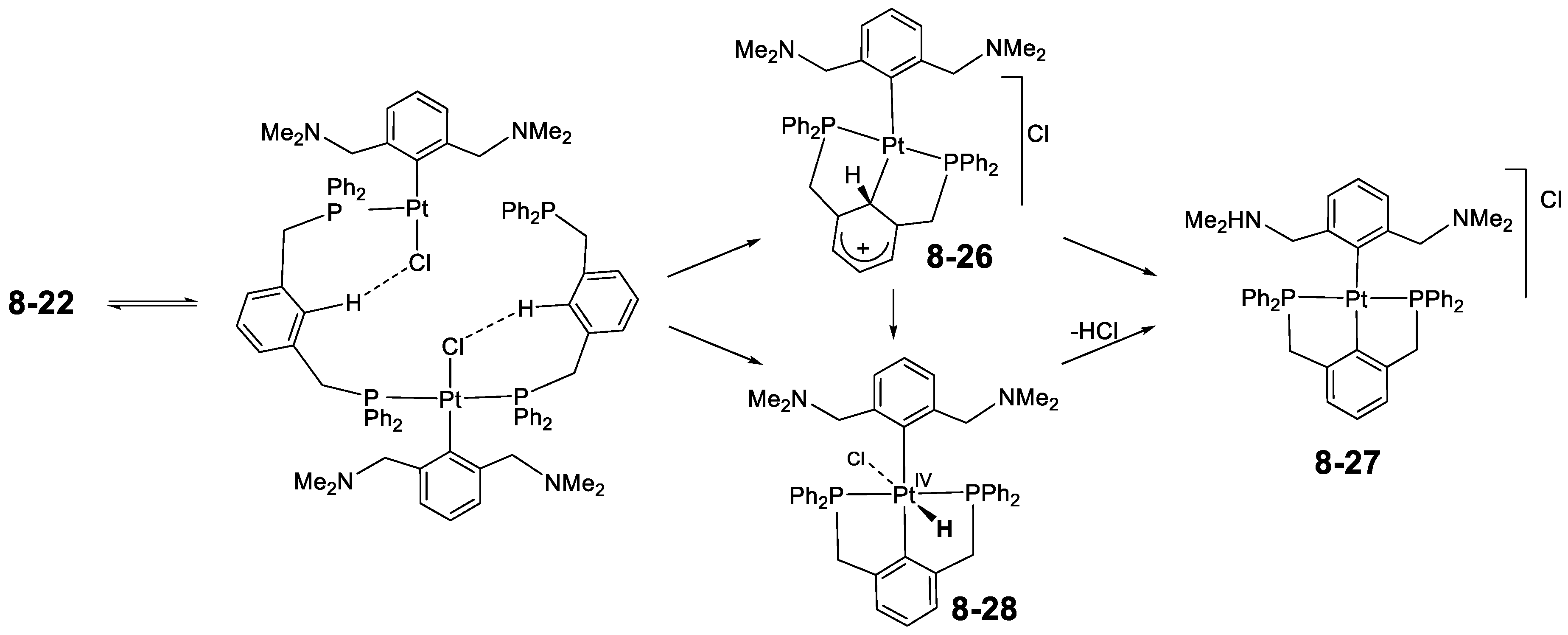
8.4. Pincer-Pincer Ligand Exchange in Cycloplatinated Complexes
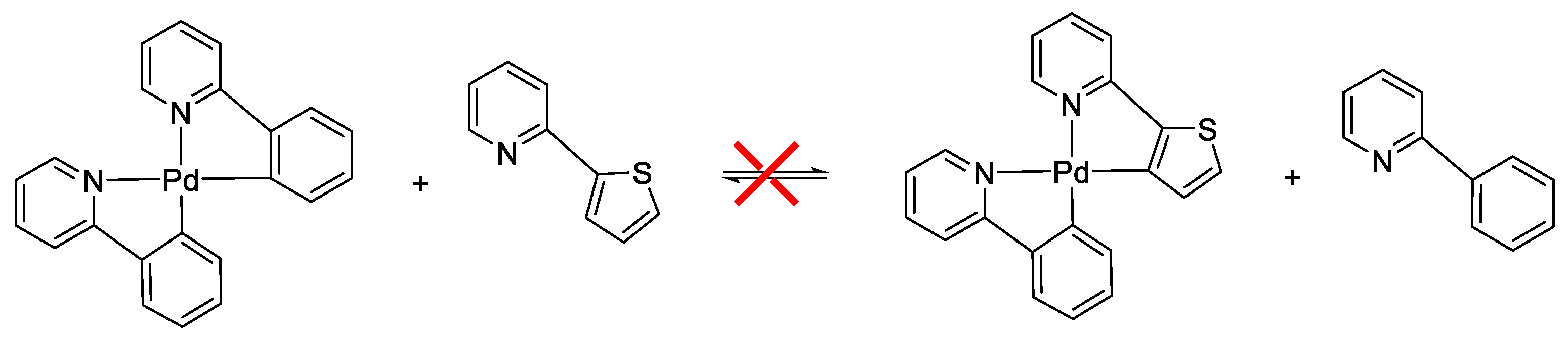
8.5. Exchange of Cyclometalated Ligands or Transcyclometalation?
9. The Ligand Exchange with Pincer Ruthenium Complexes
10. Other Examples of Exchange of Cyclometalated Ligands
10.1. Manganese(I)
10.2. Rhodium(III)
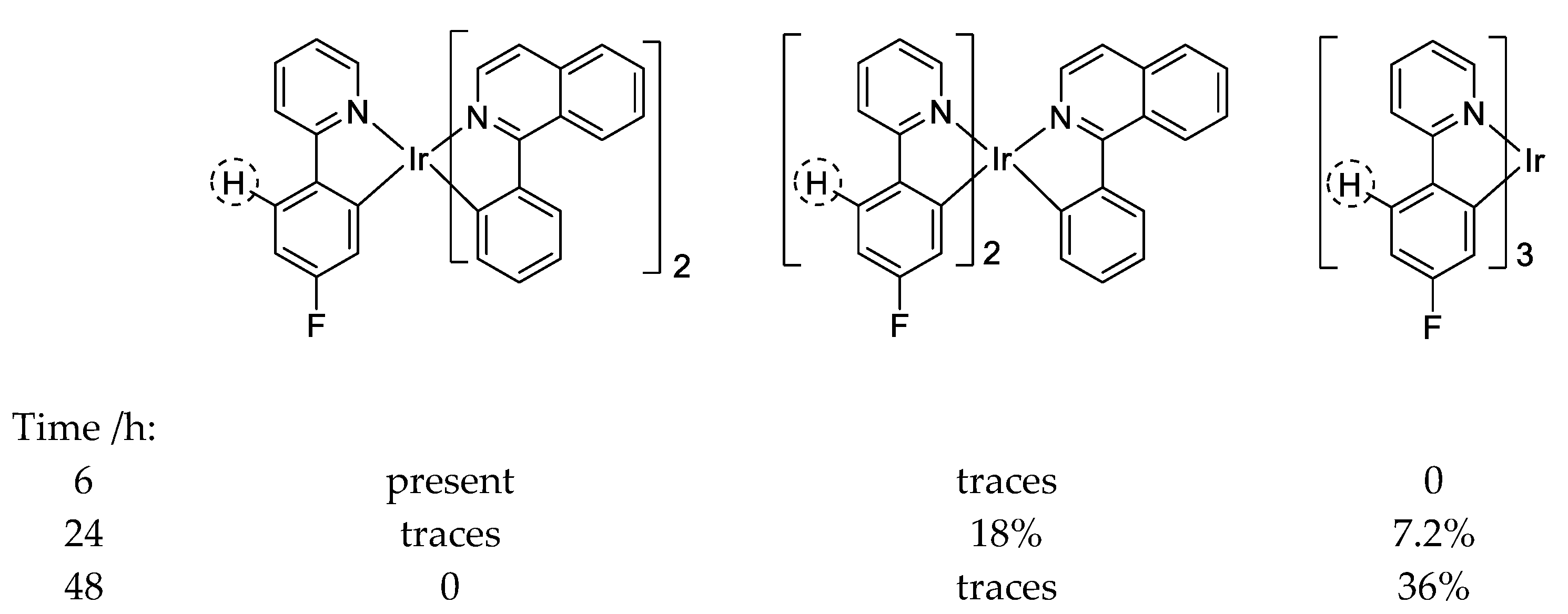
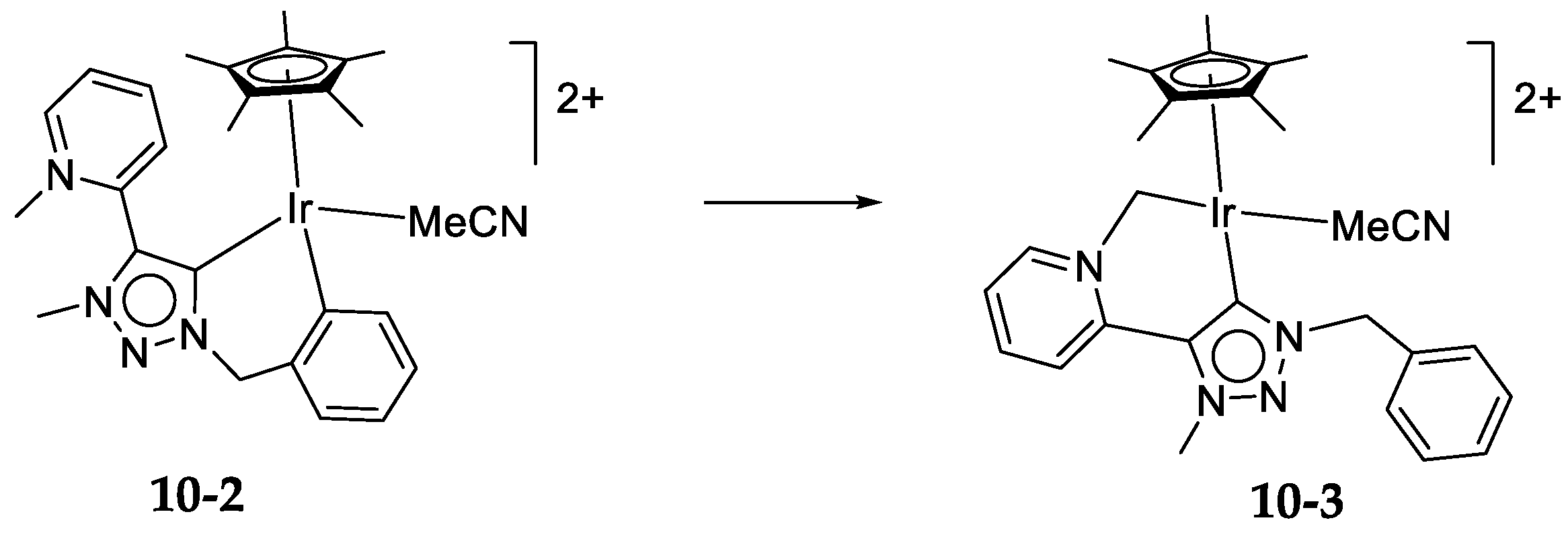
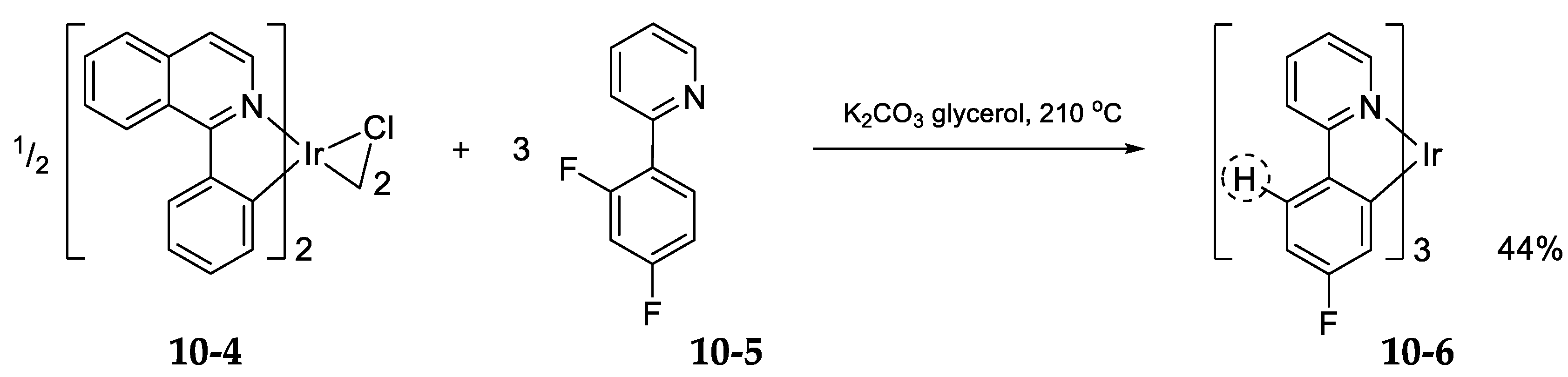
10.3. Iridium(III)
11. Summary and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Dehand, J.; Pfeffer, M. Cyclometallated compounds. Coord. Chem. Rev. 1976, 18, 327–352. [Google Scholar] [CrossRef]
- Ryabov, A.D. Mechanisms of intramolecular activation of carbon-hydrogen bonds in transition-metal complexes. Chem. Rev. 1990, 90, 403–424. [Google Scholar] [CrossRef]
- Yatsimirsky, A.K.; Ryabov, A.D.; Berezin, I.V. Kinetics and mechanism of oxidative olefin arylation by palladium(II). J. Mol. Catal. 1979, 5, 399–414. [Google Scholar] [CrossRef]
- Ryabov, A.D.; Yatsimirsky, A.K. Reaction of ortho-palladated dimethylbenzylamine with styrene: Unexpected salt effect. Tetrahedron Lett. 1980, 21, 2757–2760. [Google Scholar] [CrossRef]
- Ryabov, A.D. Cyclopalladated Complexes in Organic Synthesis. Synthesis 1985, 233–252. [Google Scholar] [CrossRef]
- Ryabov, A.D.; Sakodinskaya, I.K.; Yatsimirsky, A.K. Kinetics and mechanism of vinylation of ortho-palladated N,N-dialkylbenzylamines by para-substituted styrenes. J. Chem. Soc. Perkin Trans. 1983, 1511–1518. [Google Scholar] [CrossRef]
- Ryabov, A.D.; Polyakov, V.A.; Yatsimirsky, A.K. Reaction paths in the cyclopalladated N,N-dialkylbenzylamine-substituted styrene system in acetic acid as solvent. The structure of palladated 2-dialkylaminomethylstilbenes. J. Chem. Soc. Perkin Trans. 1983, 1503–1509. [Google Scholar] [CrossRef]
- Ryabov, A.D.; Kazankov, G.M. The exchange of cyclometalated ligands. II. Attempts to prepare six-membered palladocycles via the ligand exchange reaction. J. Organomet. Chem. 1984, 268, 85–90. [Google Scholar] [CrossRef]
- Ryabov, D.A.; Kazankov, G.M. Exchange of cyclopalladated ligands in palladium acetato dimeric complexes. Koord. Khim. 1986, 12, 540–542. [Google Scholar]
- Ryabov, D.A.; Kazankov, G.M. Exchange of cyclopalladated ligands involving optically active complexes. Vestn. Mosk. Univ. Ser. 2 Khim. 1988, 29, 220–221. [Google Scholar]
- Granell, J.; Sainz, D.; Sales, J.; Solans, X.; Font-Altaba, M. Ligand exchange reactions in cyclopalladated complexes of benzylideneanilines. Crystal structure of [Pd(C6H4CH[double bond, length half m-dash]NC6H5)Br(PPh3)2]. J. Chem. Soc. Dalton Trans. 1986, 1785–1790. [Google Scholar] [CrossRef]
- Dupont, J.; Beydoun, N.; Pfeffer, M. Reactions of Cyclopalladated Compounds. Part 21. Various Examples of Sulfur-Assisted Intramolecular Palladation of Aryl and Alkyl Groups. J. Chem. Soc. Dalton Trans. 1989, 1715–1720. [Google Scholar] [CrossRef]
- Ryabov, A.D.; Kazankov, G.M.; Yatsimirskii, A.K.; Kuz’Mina, L.G.; Burtseva, O.Y.; Dvortsova, N.V.; Polyakov, V.A. Synthesis by ligand exchange, structural characterization, and aqueous chemistry of ortho-palladated oximes. Inorg. Chem. 1992, 31, 3083–3090. [Google Scholar] [CrossRef]
- Selvakumar, K.; Vancheesan, S.; Varghese, B. Synthesis and characterization of cyclopalladated complexes of oximes by ligand-exchange method. Polyhedron 1997, 16, 2257–2262. [Google Scholar] [CrossRef]
- Smoliakova, I.P.; Keuseman, K.J.; Haagenson, D.C.; Wellmann, D.M.; Colligan, P.B.; Kataeva, N.A.; Churakov, A.V.; Kuz’Mina, L.G.; Dunina, V.V. Direct ortho-palladation of 2-phenyl-2-oxazoline. J. Organomet. Chem. 2000, 603, 86–97. [Google Scholar] [CrossRef]
- Bezsoudnova, E.Y.; Ryabov, A.D. Water-soluble cyclopalladated aryl oxime: A potent ‘green’ catalyst. J. Organomet. Chem. 2001, 622, 38–42. [Google Scholar] [CrossRef]
- Kazankov, M.G.; Polyakov, V.A.; Ryabov, A.D.; Yatsimirskii, A.K. Synthesis of o-palladated aryl oximes via the exchange of cyclopalladated ligands. Metalloorg. Khim. 1990, 3, 644–649. [Google Scholar]
- Nájera, C. Oxime-Derived Palladacycles: Applications in Catalysis. ChemCatChem 2016, 8, 1865–1881. [Google Scholar] [CrossRef] [Green Version]
- Yatsimirsky, A.K.; Kazankov, G.M.; Ryabov, A.D. Ester Hydrolysis Catalyzed by ortho-Palladated Aryl Oximes. J. Chem. Soc. Perkin Trans. 2010, 23, 1295–1300. [Google Scholar]
- Ceder, R.; Gómez, M.; Sales, J. Ligand exchange reactions of N-donor ligands in cyclopalladated complexes. J. Organomet. Chem. 1989, 361, 391–398. [Google Scholar] [CrossRef]
- Albert, J.; Ceder, R.M.; Gomez, M.; Granell, J.; Sales, J. Cyclopalladation of N-mesitylbenzylideneamines. Aromatic versus aliphatic carbon-hydrogen bond activation. Organometallics 1992, 11, 1536–1541. [Google Scholar] [CrossRef]
- Gomez, M.; Granell, J.; Martinez, M. Mechanisms of cyclopalladation reactions in acetic acid. Not so simple one-pot processes. Eur. J. Inorg. Chem. 2000, 217–224. [Google Scholar] [CrossRef]
- Albert, J.; Granell, J.; Durán-Mota, J.A.; Lozano, A.; Luque, A.; Mate, A.; Quirante, J.; Khosa, M.K.; Calvis, C.; Messeguer, R.; et al. Endo and exo cyclopalladated (E)-N-([1,1’-biphenyl]-2-yl)-1-mesitylmethanimines: Anticancer, antibacterial and antioxidant activities. J. Organomet. Chem. 2017, 839, 116–125. [Google Scholar] [CrossRef]
- Cocco, F.; Zucca, A.; Stoccoro, S.; Serratrice, M.; Guerri, A.; Cinellu, M.A. Synthesis and Characterization of Palladium(II) and Platinum(II) Adducts and Cyclometalated Complexes of 6,6′-Dimethoxy-2,2′-bipyridine: C(sp3)–H and C(sp2)–H Bond Activations. Organometallics 2014, 33, 3414–3424. [Google Scholar] [CrossRef]
- Lamb, J.R.; Stepanova, V.A.; Smoliakova, I.P. Transcyclopalladation on silica gel. Polyhedron 2013, 53, 202–207. [Google Scholar] [CrossRef]
- Ryabov, A.D.; Eliseev, A.V.; Sergeyenko, E.S.; Usatov, A.V.; Zakharkin, L.I.; Kalinin, V.N. Cyclopalladated o-carboranes. Palladium transfer from cyclopalladated N,N-dimethylbenzylamines, 1C-diphenylphosphino-o-carborane and η3-allyls in carboxylic acid solvents. Polyhedron 1989, 8, 1485–1496. [Google Scholar] [CrossRef]
- Hiraki, K.; Fuchita, Y.; Uchiyama, T. Cyclopalladation of benzyldiphenylphosphine by palladium(II) acetate. Inorganica Chim. Acta 1983, 69, 187–190. [Google Scholar] [CrossRef]
- Usyatinskii, Y.A.; Ryabov, A.D.; Bregadze, V.I.; Shcherbina, T.M.; Godovikov, N.N. Reaction of some carboranes and their carbon and boron thallated derivatives with palladium(II) acetate. Izv. Akad. Nauk SSSR Ser. Khim. 1982, 1598–1603. [Google Scholar]
- Sokolov, V.I. Introduction to Theoretical Stereochemistry; Nauka: Moscow, Russia, 1979. [Google Scholar]
- Sokolov, V.I. Asymmetric cyclopalladation as a tool of enantioselective synthesis. Pure Appl. Chem. 1983, 55, 1837–1844. [Google Scholar] [CrossRef]
- Sokolov, V. Optically active organometallic compounds (a personal account from the inside). J. Organomet. Chem. 1995, 500, 299–306. [Google Scholar] [CrossRef]
- Dunina, V.V. Chiral Cyclopalladated Compounds: New Structures, Methodologies and Applications. A Personal Account. Curr. Org. Chem. 2011, 15, 3415–3440. [Google Scholar] [CrossRef]
- Sokolov, V.; Troitskaya, L.; Reutov, O. Asymmetric cyclopalladation of (dimethylaminomethyl)ferrocene. J. Organomet. Chem. 1979, 182, 537–546. [Google Scholar] [CrossRef]
- Dunina, V.V.; Razmyslova, E.D.; Gorunova, O.N.; Livantsov, M.V.; Grishin, Y.K. Asymmetric exchange of cyclopalladated ligands with a high level of asymmetric induction: A new route to optically active phosphapalladacycles. Tetrahedron: Asymmetry 2003, 14, 2331–2333. [Google Scholar] [CrossRef]
- Dunina, V.V.; Turubanova, E.I.; Livantsov, M.V.; Lyssenko, K.A.; Grishin, Y.K. New approach to chiral phosphapalladacycles: First optically active phosphinite PC-palladacycle with non-metallocenic planar chirality. Tetrahedron: Asymmetry 2008, 19, 1519–1522. [Google Scholar] [CrossRef]
- Ryabov, A.D. Cyclopalladium ligand exchange. Equilibria, kinetics, and mechanism. Zh. Obshch. Khim. 1987, 57, 249–265. [Google Scholar]
- Ryabov, D.A.; Sakodinskaya, I.K.; Yatsimirsky, A.K. Kinetics and mechanism of ortho-palladation of ring-substituted N,N-dimethylbenzylamines. J. Chem. Soc. Dalton Trans. 1985, 2629–2638. [Google Scholar] [CrossRef]
- Ryabov, A.D. Thermodynamics, kinetics, and mechanism of exchange of cyclopalladated ligands. Inorg. Chem. 1987, 26, 1252–1260. [Google Scholar] [CrossRef]
- Ryabov, D.A.; Kuz’mina, L.; Polyakov, V.A.; Kazankov, G.M.; Ryabova, E.S.; Pfeffer, M.; van Eldik, R. Kinetics and mechanism of halogen-bridge cleavage in dimethylaminomethylphenyl-C1,N pallada- and platinacycles by pyridines. Pressure effects, and crystal structures of the N,N-cis reaction product, its N,N-trans orthometallated analog and a dimer of similar reactivity. J. Chem. Soc. Dalton Trans. 1995, 999–1006. [Google Scholar] [CrossRef]
- Ryabov, A.D. Lithium chloride-induced dissociation of cyclopalladated ligands from chloro(ligand-C,N)triphenylphosphinepalladium(II) complexes in acetic acid solvent. J. Organomet. Chem. 1984, 268, 91–96. [Google Scholar] [CrossRef]
- Ryabov, A.; Yatsimirsky, A.K.; Abicht, H.-P. Protolytic stability of palladocycles. Polyhedron 1987, 6, 1619–1620. [Google Scholar] [CrossRef]
- Yagyu, T.; Aizawa, S.-I.; Funahashi, S. Kinetics and Mechanism of Cyclopalladation for [Pd(S)(Bn2Medptn)](BF4)2(S = Solvent, Bn2Medptn = N,N″-Dibenzyl-N′-methyl-3,3′-diaminodipropylamine) and Its Derivatives. Chem. Lett. 1996, 1107–1108. [Google Scholar] [CrossRef]
- Gomez, M.J.; Granell, J.; Martinez, M. Solution behavior, kinetics and mechanism of the acid-catalyzed cyclopalladation of imines. J. Chem. Soc. Dalton Trans. 1998, 37–44. [Google Scholar] [CrossRef]
- Yagyu, T.; Aizawa, S.-I.; Funahashi, S. Mechanistic Studies on Cyclopalladation of the Solvated Palladium(II) Complexes withN-Benzyl Triamine Ligands in Various Solvents. Crystal Structures of [Pd(Sol)(Bn2Medptn)](BF4)2(Sol = Acetonitrile and N,N-Dimethylformamide; Bn2Medptn = N,N′-Dibenzyl-4-methyl-4-azaheptane-1,7-diamine) and [Pd(H–1Bn2Medptn-C,N,N′,N″)]CF3SO3. Bull. Chem. Soc. Jpn. 1998, 71, 619–629. [Google Scholar] [CrossRef]
- Campora, J.; Lopez, J.A.; Palma, P.; Valerga, P.; Spillner, E.; Carmona, E. Cleavage of palladium metallacycles by acids: A probe for the study of the cyclometalation reaction. Angew. Chem. Int. Ed. 1999, 38, 147–151. [Google Scholar] [CrossRef]
- O’Keefe, B.J.; Steel, P.J. Cyclometallated compounds XIV. Kinetic versus thermodynamic control in the reversible cyclopalladation of 2-(2-naphthyloxy)pyridine. Inorg. Chem. Commun. 1999, 2, 10–13. [Google Scholar] [CrossRef]
- Rogge, T.; Oliveira, J.C.A.; Kuniyil, R.; Hu, L.; Ackermann, L. Reactivity-Controlling Factors in Carboxylate-Assisted C–H Activation under 4d and 3d Transition Metal Catalysis. ACS Catal. 2020, 10, 10551–10558. [Google Scholar] [CrossRef]
- Lapointe, D.; Fagnou, K. Overview of the Mechanistic Work on the Concerted Metallation–Deprotonation Pathway. Chem. Lett. 2010, 39, 1118–1126. [Google Scholar] [CrossRef]
- Alharis, R.A.; McMullin, C.L.; Davies, D.L.; Singh, K.; MacGregor, S.A. The Importance of Kinetic and Thermodynamic Control when Assessing Mechanisms of Carboxylate-Assisted C–H Activation. J. Am. Chem. Soc. 2019, 141, 8896–8906. [Google Scholar] [CrossRef]
- Davies, D.L.; Donald, A.S.M.A.; MacGregor, S.A. Computational Study of the Mechanism of Cyclometalation by Palladium Acetate. J. Am. Chem. Soc. 2005, 127, 13754–13755. [Google Scholar] [CrossRef]
- Nonoyama, M.; Nakajima, K.; Kita, M. Cyclometallation of N,N-dimethyl-2-bromothiobenzamide and some related thioamides with palladium(0) and palladium(II). Polyhedron 1995, 14, 1035–1043. [Google Scholar] [CrossRef]
- Zucca, A.; Cinellu, M.A.; Pinna, M.V.; Stoccoro, S.; Minghetti, G.; Manassero, M.; Sansoni, M. Cyclopalladation of 6-Substituted-2,2‘-bipyridines. Metalation of Unactivated Methyl Groups vs Aromatic C–H Activation. Organometallics 2000, 19, 4295–4304. [Google Scholar] [CrossRef]
- Cope, A.C.; Friedrich, E.C. Electrophilic aromatic substitution reactions by platinum(II) and palladium(II) chlorides on N,N-dimethylbenzylamines. J. Am. Chem. Soc. 2005, 90, 909–913. [Google Scholar] [CrossRef]
- Fuchita, Y.; Tsuchiya, H. First ortho-palladation of unsubstituted benzylamine by palladium(II) acetate. Polyhedron 1993, 12, 2079–2080. [Google Scholar] [CrossRef]
- Fuchita, Y.; Tsuchiya, H.; Miyafuji, A. Cyclopalladation of secondary and primary benzylamines. Inorganica Chim. Acta 1995, 233, 91–96. [Google Scholar] [CrossRef]
- Vincente, J.; Saura-Llamas, I.; Jones, P.G. Orthometallated primary amines. Part 1. Facile preparation of the first optically active cyclopalladated primary amines. J. Chem. Soc. Dalton Trans. 1993, 3619–3624. [Google Scholar] [CrossRef]
- Kurzeev, S.A.; Kazankov, G.M.; Ryabov, A.D. Second- and inverse order pathways in the mechanism of orthopalladation of primary amines. Inorganica Chim. Acta 2002, 340, 192–196. [Google Scholar] [CrossRef]
- Ryabov, A.D.; Eliseev, A.V.; Yatsimirsky, A.K. Palladium(II)-catalysed Hydrogen/Deuterium allylic exchange in alkenes: An intermediacy of palladium(IV)? Appl. Organomet. Chem. 1988, 2, 101–107. [Google Scholar] [CrossRef]
- Canty, A.J. The +IV Oxidation State in Organopalladium Chemistry. Recent Advances and Potential Intermediates in Organic Synthesis and Catalysis. Platinum Met. Rev. 1993, 37, 2–7. [Google Scholar] [CrossRef]
- Canty, A.J.; Van Koten, G. Mechanisms of d8 organometallic reactions involving electrophiles and intramolecular assistance by nucleophiles. Accounts Chem. Res. 1995, 28, 406–413. [Google Scholar] [CrossRef]
- Anklin, C.; Pregosin, P.S.; Wombacher, F.J.; Ruegg, H.J. Mechanistic aspects of the cyclometalation of quinoline-8-carboxaldehyde with platinum(II). Organometallics 1990, 9, 1953–1958. [Google Scholar] [CrossRef]
- Pregosin, P.S.; Wombacher, F.; Albinati, A.; Lianza, F. Facile cycloplatination of nitrogen compounds. Crystal structure of the cycloplatinated Schiff’s base tetralone derivative PtCl{(cyclohexyl)N:C(CH2)3C6H3 (CO). J. Organomet. Chem. 1991, 418, 249–267. [Google Scholar] [CrossRef]
- Navarro-Ranninger, C.; López-Solera, I.; Álvarez-Valdés, A.; Rodriguez-Ramos, J.H.; Masaguer, J.R.; Garcia-Ruano, J.L.; Solans, X. Cyclometalated complexes of palladium(II) and platinum(II) with N-benzyl- and N-(phenylethyl)-.alpha.-benzoylbenzylideneamine. Delocalization in the cyclometalated ring as a driving force for the orthometalation. Organometallics 1993, 12, 4104–4111. [Google Scholar] [CrossRef]
- Praefcke, K.; Bilgin, B.; Pickardt, J.; Borowski, M. Liquid-crystalline Compounds. 86. The First Disc-Shaped Dinuclear Platinum Mesogen. Chem. Ber. 1994, 127, 1543–1545. [Google Scholar] [CrossRef]
- Navarro-Ranninger, C.; López-Solera, I.; González, V.M.; Pérez, J.M.; Alvarez-Valdés, A.; Martín, A.; Raithby, P.R.; Masaguer, J.R.; Alonso, C. Cyclometalated Complexes of Platinum and Palladium withN-(4-Chlorophenyl)-α-benzoylbenzylideneamine. In Vitro Cytostatic Activity, DNA Modification, and Interstrand Cross-Link Studies. Inorg. Chem. 1996, 35, 5181–5187. [Google Scholar] [CrossRef]
- Ghedini, M.; Pucci, D.; Crispini, A.; Barberio, G. Oxidative Addition to Cyclometalated Azobenzene Platinum(II) Complexes: A Route to Octahedral Liquid Crystalline Materials. Organometallics 1999, 18, 2116–2124. [Google Scholar] [CrossRef]
- Praefcke, K.; Bilgin, B.; Pickardt, J.; Borowski, M. A novel platinum methylene complex. J. Organomet. Chem. 1999, 592, 155–161. [Google Scholar] [CrossRef]
- Gorla, F.; Venanzi, L.M.; Albinati, A. Synthesis of (1R,1’S)- and (1S,1’S),(1R,1’R)-bis[1-(diphenylphosphino)ethyl]benzene derivatives and their cyclometalation reactions with platinum(II) compounds. The x-ray crystal structures of [2,6-bis[(diphenylphosphino)methyl]phenyl]chloropalladium(II), [(1R,1’S)-2,6-bis[1-(diphenylphosphino)ethyl]phenyl]chloroplatinum(II), and [(1R,1’R),(1S,1’S)-2,6-bis[1-(diphenylphosphino)ethyl]phenyl]chloroplatinum(II). Organometallics 1994, 13, 43–54. [Google Scholar] [CrossRef]
- Helm, L.; Merbach, A.E. Inorganic and Bioinorganic Solvent Exchange Mechanisms. Chem. Rev. 2005, 105, 1923–1960. [Google Scholar] [CrossRef]
- Helm, L.; Merbach, A.E. The Periodic Table and Kinetics? Chim. Int. J. Chem. 2019, 73, 179–184. [Google Scholar] [CrossRef]
- Schmuelling, M.; Ryabov, A.D.; van Eldik, R. To what extent can the Pt-C bond of a metallacycle labilize the trans position? A temperature- and pressure-dependent mechanistic study. J. Chem. Soc. Dalton Trans. 1994, 1257–1263. [Google Scholar] [CrossRef]
- Ryabov, A.D.; van Eldik, R. A simple access to homo- and heteroleptic platinum complexes by cycloplatination without the use of organolithium compounds. Angew. Chem. Int. Ed. Engl. 1994, 33, 783–784. [Google Scholar] [CrossRef]
- Poveda, D.; Vivancos, Á.; Bautista, D.; González-Herrero, P. Visible light driven generation and alkyne insertion reactions of stable bis-cyclometalated Pt(iv) hydrides. Chem. Sci. 2020, 11, 12095–12102. [Google Scholar] [CrossRef]
- Vivancos, Á.; Bautista, D.; González-Herrero, P. Luminescent Platinum(IV) Complexes Bearing Cyclometalated 1,2,3-Triazolylidene and Bi- or Terdentate 2,6-Diarylpyridine Ligands. Chem. Eur. J. 2019, 25, 6014–6025. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.A.; Phillips, J.M.; Clarkson, G.J.; Rourke, J.P. Trapping five-coordinate platinum(iv) intermediates. Dalton Trans. 2016, 45, 11397–11406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, P.A.; Phillips, J.M.; Newman, C.P.; Clarkson, G.J.; Rourke, J.P. Intramolecular transcyclometallation: The exchange of an aryl–Pt bond for an alkyl–Pt bond via an agostic intermediate. Chem. Commun. 2015, 51, 8365–8368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, P.A.; Clarkson, G.J.; Rourke, J.P. Oxidation of an o-tolyl phosphine complex of platinum: C-H activation and transcyclometallation. J. Organomet. Chem. 2017, 851, 115–121. [Google Scholar] [CrossRef] [Green Version]
- Shaw, P.A.; Clarkson, G.J.; Rourke, J.P. Reversible C–C bond formation at a triply cyclometallated platinum(iv) centre. Chem. Sci. 2017, 8, 5547–5558. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, M.; Dani, P.; Lutz, M.; Spek, A.L.; Van Koten, G. Transcyclometalation Processes with Late Transition Metals: Caryl–H Bond Activation via Noncovalent C–H·Interactions. J. Am. Chem. Soc. 2000, 122, 11822–11833. [Google Scholar] [CrossRef]
- Albrecht, M.; James, S.L.; Veldman, N.; Spek, A.L.; Van Koten, G. Transmetalation reactions with nitrogen-containing “pincer”-class ligands on platinum(II) centers. Can. J. Chem. 2001, 79, 709–718. [Google Scholar] [CrossRef]
- Dijkstra, H.P.; Albrecht, M.; Medici, S.; van Klink, G.P.M.; van Koten, G. Hexakis(PCP-platinum and -ruthenium) complexes by the transcyclometalation reaction and their use in catalysis. Adv. Synth. Catal. 2002, 344, 1135–1141. [Google Scholar] [CrossRef] [Green Version]
- Dijkstra, H.P.; Albrecht, M.; van Koten, G. Transcyclometalation, a versatile methodology for multiple metal-carbon bond formation with multisite ligands. Chem. Commun. 2002, 126–127. [Google Scholar] [CrossRef] [PubMed]
- Van Klink, G.P.M.; Dani, P.; van Koten, G. Bis(ortho-)chelated monoanionic bisphosphinoaryl ruthenium(II) complexes: Synthesis, characterization and reactivity. Chin. J. Chem. 2002, 20, 207–213. [Google Scholar] [CrossRef] [Green Version]
- Osakada, K. Transmetalation. Curr. Methods Inorg. Chem. 2003, 3, 233–291. [Google Scholar]
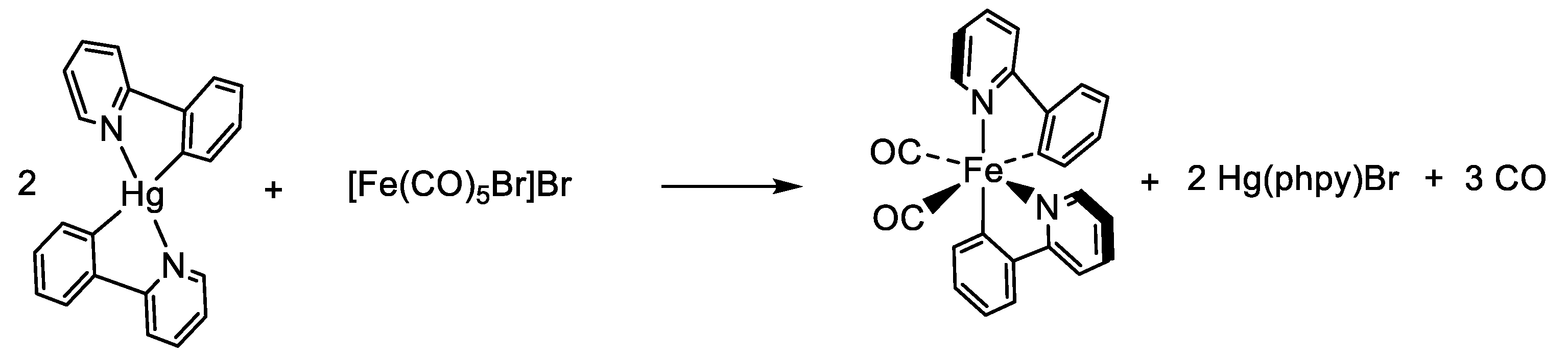
- Estrada-Montaño, A.S.; Gries, A.; Oviedo-Fortino, J.A.; Torres-Gutierrez, C.; Grain-Hayton, A.; Marcial-Hernández, R.; Shen, L.Q.; Ryabov, A.D.; Gaiddon, C.; Le Lagadec, R. Dibromine Promoted Transmetalation of an Organomercurial by Fe(CO)5: Synthesis, Properties, and Cytotoxicity of Bis(2-C6H4-2′-py-κC,N)dicarbonyliron(II). Organometallics 2020, 39, 1842–1854. [Google Scholar] [CrossRef]
- Cerón-Camacho, R.; Hernández, S.; Le Lagadec, R.; Ryabov, A.D. Cyclometalated [Os(C–N)x(N–N)3−x]m+ mimetics of tris(2,2′-bipyridine)osmium(ii): Covering a 2 V potential range by known (x = 0, 1) and new (x = 2, 3) species (C–N = o-2-phenylpyridinato). Chem. Commun. 2011, 47, 2823–2825. [Google Scholar] [CrossRef]
- Le Lagadec, R.; Alexandrova, L.; Estevez, H.; Pfeffer, M.; Laurinavičius, V.; Razumienė, J.; Ryabov, A.D. Bis-Ruthena(III)cycles [Ru(C∩N)2(N∩N)]PF6 as Low-Potential Mediators for PQQ Alcohol Dehydrogenase (C∩N = 2-phenylpyridinato or 4-(2-tolyl)pyridinato, N∩N = bpy or phen). Eur. J. Inorg. Chem. 2006, 2735–2738. [Google Scholar] [CrossRef]
- Ryabov, A.D.; Sukharev, V.S.; Alexandrova, L.; Le Lagadec, R.; Pfeffer, M. Low-Potential Cyclometalated Osmium(II) Mediators of Glucose Oxidase. Inorg. Chem. 2003, 42, 6598–6600. [Google Scholar] [CrossRef]
- Saavedra-Díaz, O.; Cerón-Camacho, R.; Hernández, S.; Ryabov, A.D.; Le Lagadec, R. Denial of Tris(C,N-cyclometalated) Ruthenacycle: Nine-Membered η6-N,N-transor η2-N,N-cisRuIIChelates of 2,2′-Bis(2-pyridinyl)-1,1′-biphenyl. Eur. J. Inorg. Chem. 2008, 4866–4869. [Google Scholar] [CrossRef]
- Jolliet, P.; Gianini, M.; Von Zelewsky, A.; Bernardinelli, G.; Stoeckli-Evans, H. Cyclometalated Complexes of Palladium(II) and Platinum(II): Cis-Configured Homoleptic and Heteroleptic Compounds with Aromatic C⌒N Ligands. Inorg. Chem. 1996, 35, 4883–4888. [Google Scholar] [CrossRef]
- Sanna, G.; Pilo, M.I.; Spano, N.; Minghetti, G.; Cinellu, M.A.; Zucca, A.; Seeber, R. Electrochemical behaviour of cyclometallated gold(III) complexes. Evidence of transcyclometallation in the fate of electroreduced species. J. Organomet. Chem. 2001, 622, 47–53. [Google Scholar] [CrossRef]
- Aydin, J.; Selander, N.; Szabó, K.J. Strategies for fine-tuning the catalytic activity of pincer-complexes. Tetrahedron Lett. 2006, 47, 8999–9001. [Google Scholar] [CrossRef]
- Dani, P.; Albrecht, M.; Van Klink, G.P.M.; Van Koten, G. Transcyclometalation: A Novel Route to (Chiral) Bis-Ortho-Chelated Bisphosphinoaryl Ruthenium(II) Complexes. Organometallics 2000, 19, 4468–4476. [Google Scholar] [CrossRef] [Green Version]
- Gagliardo, M.; Chase, P.A.; Lutz, M.; Spek, A.L.; Hartl, F.; Havenith, R.W.A.; Van Klink, G.P.M.; Van Koten, G. A Ruthenium(II) Complex Stabilized by a Highly Fluorinated PCP Pincer Ligand. Organometallics 2005, 24, 4553–4557. [Google Scholar] [CrossRef] [Green Version]
- Gagliardo, M.; Dijkstra, H.P.; Coppo, P.; De Cola, L.; Lutz, M.; Spek, A.L.; Van Klink, G.P.M.; Van Koten, G. Synthesis, Crystal Structure, and Redox and Photophysical Properties of Novel Bisphosphinoaryl RuII-Terpyridine Complexes. Organometallics 2004, 23, 5833–5840. [Google Scholar] [CrossRef] [Green Version]
- Medici, S.; Gagliardo, M.; Williams, S.B.; Chase, P.A.; Gladiali, S.; Lutz, M.; Spek, A.L.; Van Klink, G.P.M.; Van Koten, G. Novel P-Stereogenic PCP Pincer-Aryl Ruthenium(II) Complexes and Their Use in the Asymmetric Hydrogen Transfer Reaction of Acetophenone. Helv. Chim. Acta 2005, 88, 694–705. [Google Scholar] [CrossRef] [Green Version]
- Gagliardo, M.; Perelaer, J.; Hartl, F.; Van Klink, G.P.M.; Van Koten, G. Mono- and Heterodimetallic FeII and RuII Complexes Based on a Novel Heteroditopic 4′-{Bis(phosphanyl)aryl}-2,2′:6′,2″-terpyridine Ligand. Eur. J. Inorg. Chem. 2007, 2111–2120. [Google Scholar] [CrossRef]
- Dani, P.; Toorneman, M.A.M.; Van Klink, G.P.M.; Van Koten, G. Complexes of Bis-ortho-cyclometalated Bisphosphinoaryl Ruthenium(II) Cations with Neutral Meta-bisphosphinoarene Ligands Containing an Agostic C–H·Ru Interaction. Organometallics 2000, 19, 5287–5296. [Google Scholar] [CrossRef]
- Djukic, J.-P.; Maisse, A.; Pfeffer, M. Cyclomanganated (η6-arene)tricarbonylchromium complexes: Synthesis and reactivity. J. Organomet. Chem. 1998, 567, 65–74. [Google Scholar] [CrossRef]
- Djukic, J.-P.; Maisse, A.; Pfeffer, M.; De Cian, A.; Fischer, J. Synthesis and Reactivity of New Cyclomanganated (η6-Arene)tricarbonylchromium Complexes. Organometallics 1997, 16, 657–667. [Google Scholar] [CrossRef]
- Bruce, M.; Liddell, M.; Snow, M.R.; Tiekink, E.R. Cyclometallation Reactions. XXI. Some Chemistry of Binuclear Manganese Complexes Derived From Azobenzene: X-Ray Crystal-Structures of {Mn (CO)4}2(μ-C6H4N=NC6H4) and {Mn(CO)4}{Mn(CO)3[P(OPh)3]}(μ-C6H4N=NC6H4). Aust. J. Chem. 1988, 41, 1407–1415. [Google Scholar] [CrossRef]
- Robinson, N.P.; Main, L.; Nicholson, B.K. Orthomanganated arenes in synthesis VIII. Mono- and dicyclomanganation of diacetyl benzenes. J. Organomet. Chem. 1992, 430, 79–86. [Google Scholar] [CrossRef]
- Vanderweide, A.I.; Brennessel, W.W.; Jones, W.D. Reversible Concerted Metalation–Deprotonation C–H Bond Activation by [Cp*RhCl2]2. J. Org. Chem. 2019, 84, 12960–12965. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.F.; Lalrempuia, R.; Müller-Bunz, H.; Clot, E.; Albrecht, M. Controlling the Selectivity of C–H Activation in Pyridinium Triazolylidene Iridium Complexes: Mechanistic Details and Influence of Remote Substituents. Organometallics 2015, 34, 858–869. [Google Scholar] [CrossRef]
- Lepeltier, M.; Dumur, F.; Marrot, J.; Contal, E.; Bertin, D.; Gigmes, D.; Mayer, C.R. Unprecedented combination of regioselective hydrodefluorination and ligand exchange reaction during the syntheses of tris-cyclometalated iridium(iii) complexes. Dalton Trans. 2013, 42, 4479–4486. [Google Scholar] [CrossRef] [PubMed]
- Perera, S.D.; Shaw, B.L.; Thornton-Pett, M. Cyclometallation of a pentafluorobenzaldehyde azine phosphine via a C-F bond fission by irridium(I): Crystal structure of [IrCl2(CO){PPh2CH2C(but):NN:CH(C6F4)}]. Inorganica Chim. Acta 1995, 233, 103–107. [Google Scholar] [CrossRef]























































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
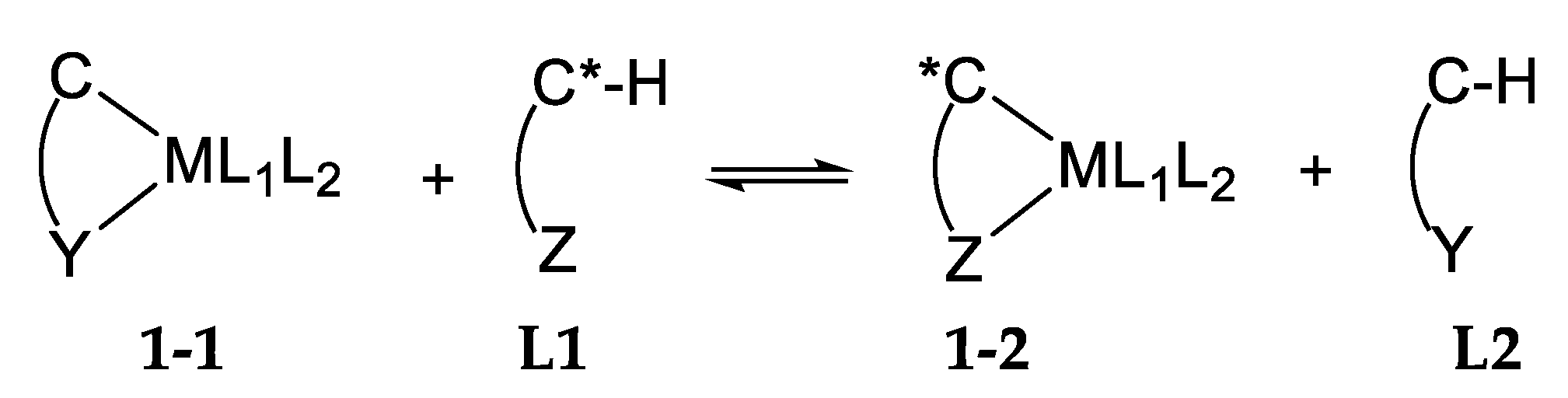{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
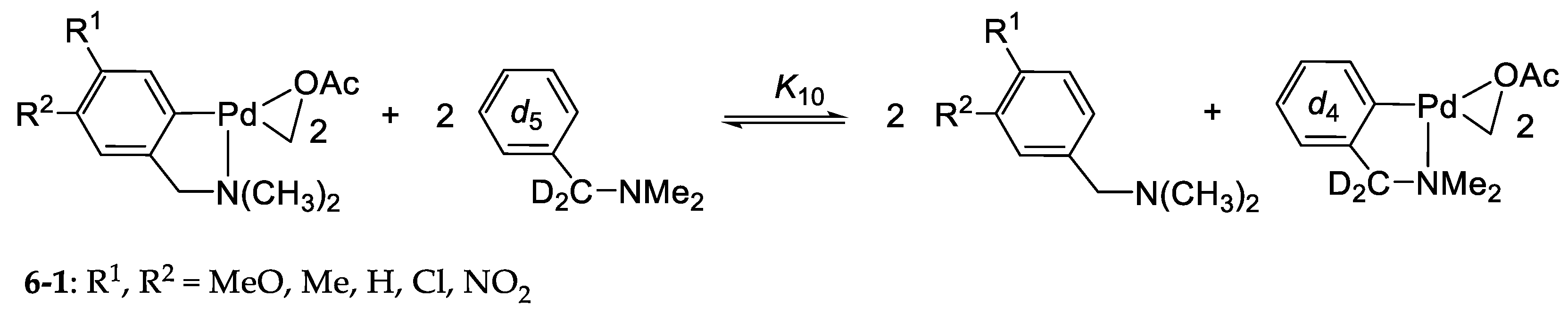{kind=link}
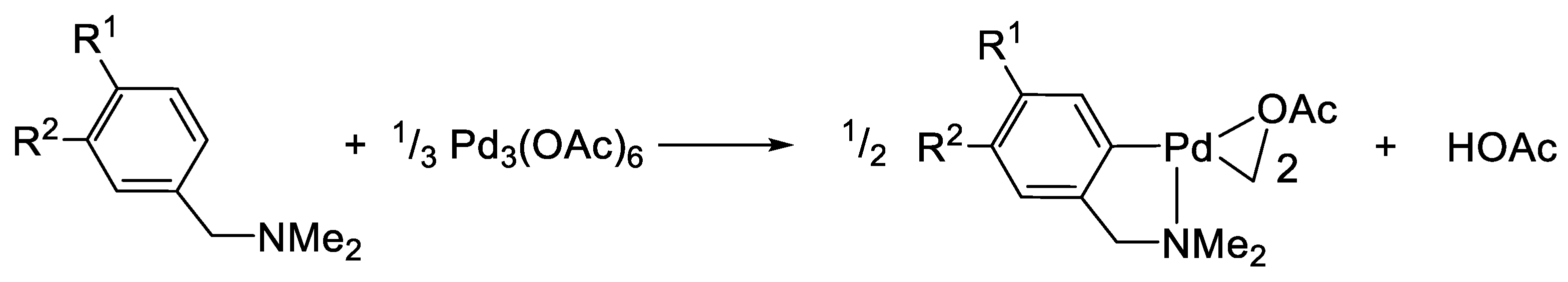{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
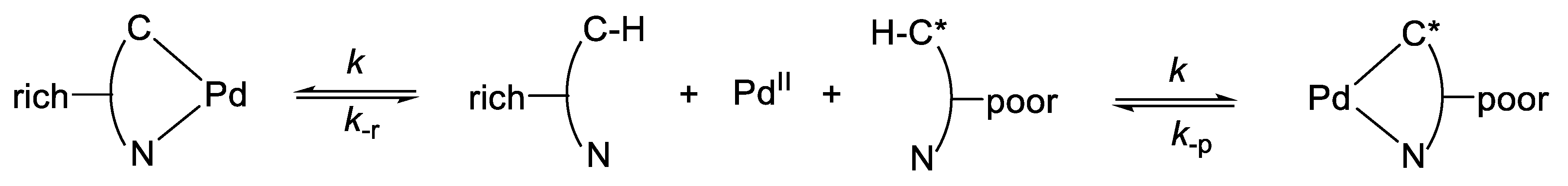{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
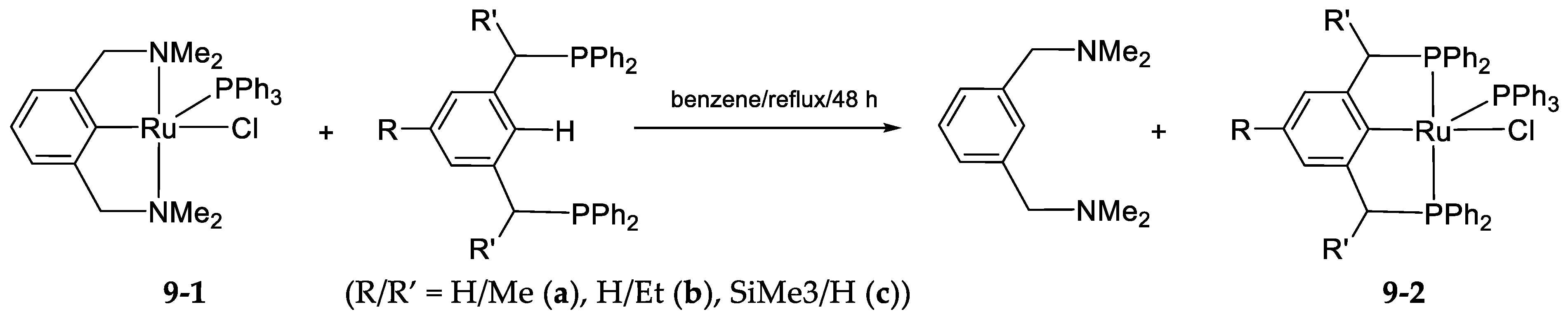{kind=link}
{kind=link}
{kind=link}
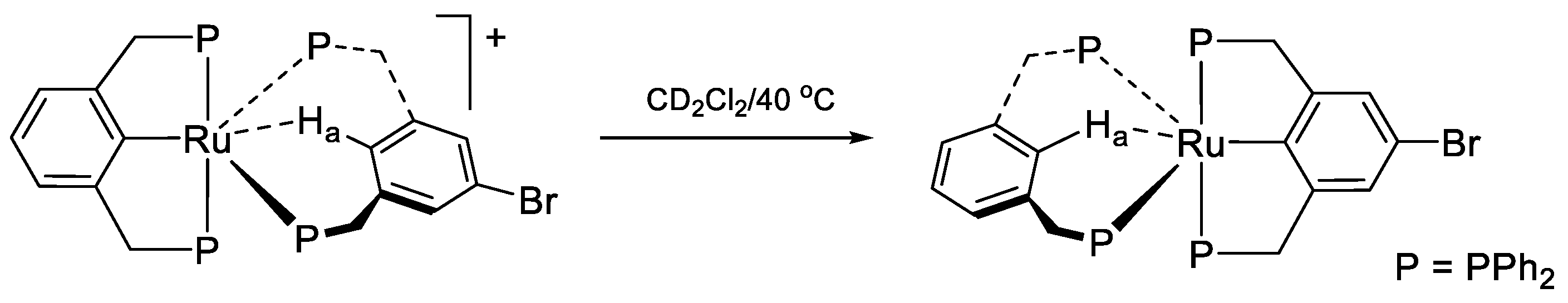{kind=link}
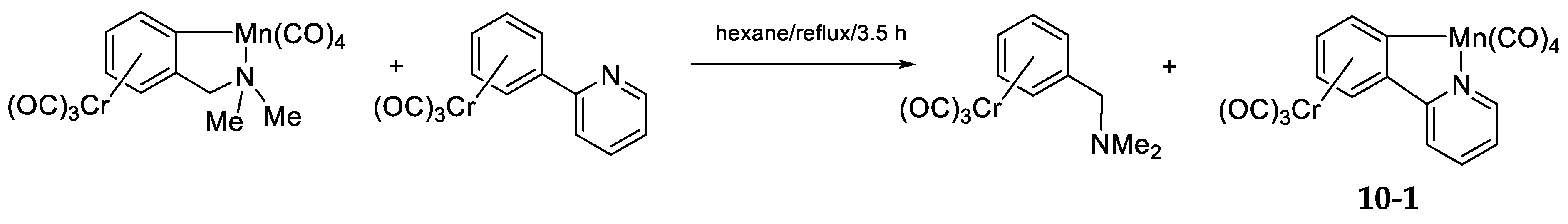{kind=link}
{kind=link}
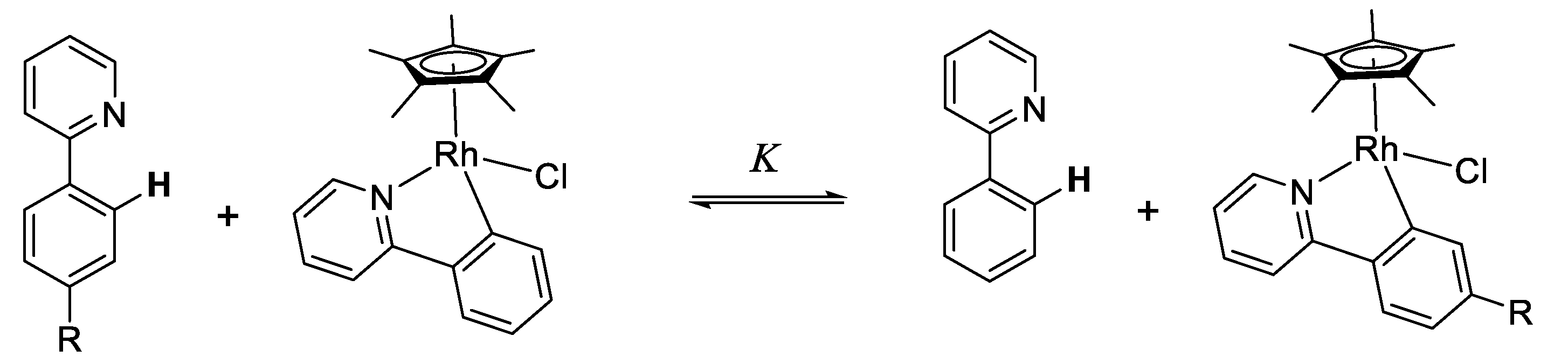{kind=link}
{kind=link}
{kind=link}
| Entry | Complex | Ligand | Product | Yield | Reference |
|---|---|---|---|---|---|
| 1 |  3-1 |  |  | 67% | [8] |
| 2 |  |  |  | 46% | [8] |
| 3 |  |  |  | 64% | [8] |
| 4 |  3-2 |  |  | 83 | [9] |
| 5 |  3-3 |  |  | 68% | [8] |
| 6 |  3-4 |  |  | 79% (R = H) 55% (R = MeR) 52% (R = MeS) | [10] |
| 7 |  |  |  | 95 | [9] |
| 8 |  |  |  | 38 | [9] |
| 9 |  |  |  | 80 | [9] |
| 10 |  |  |  | 93 | [9] |
| 11 |  |  |  3-5 | 60% (R = Cl) 70% (R = NO2) | [11] |
| 12 |  |  |  | 92% | [12] |
| 13 |  |  |  | 97% | [12] |
| 14 |  | N,N,N’,N’-tetramethyl-thiourea |  | 82% | [12] |
| 15 |  |  |  3-6 | R = H (81%), Me (92%), nPr (95%), Ph (86%) | [13,14] |
| 16 |  |  |  | 90% | [15] |
| 17 |  |  |  3-7 | 72% | [16] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryabov, A.D. The Exchange of Cyclometalated Ligands. Molecules 2021, 26, 210. https://doi.org/10.3390/molecules26010210
Ryabov AD. The Exchange of Cyclometalated Ligands. Molecules. 2021; 26(1):210. https://doi.org/10.3390/molecules26010210
Chicago/Turabian StyleRyabov, Alexander D. 2021. "The Exchange of Cyclometalated Ligands" Molecules 26, no. 1: 210. https://doi.org/10.3390/molecules26010210
APA StyleRyabov, A. D. (2021). The Exchange of Cyclometalated Ligands. Molecules, 26(1), 210. https://doi.org/10.3390/molecules26010210