
Theoretical Study on the Photo-Oxidation and Photoreduction of an Azetidine Derivative as a Model of DNA Repair

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
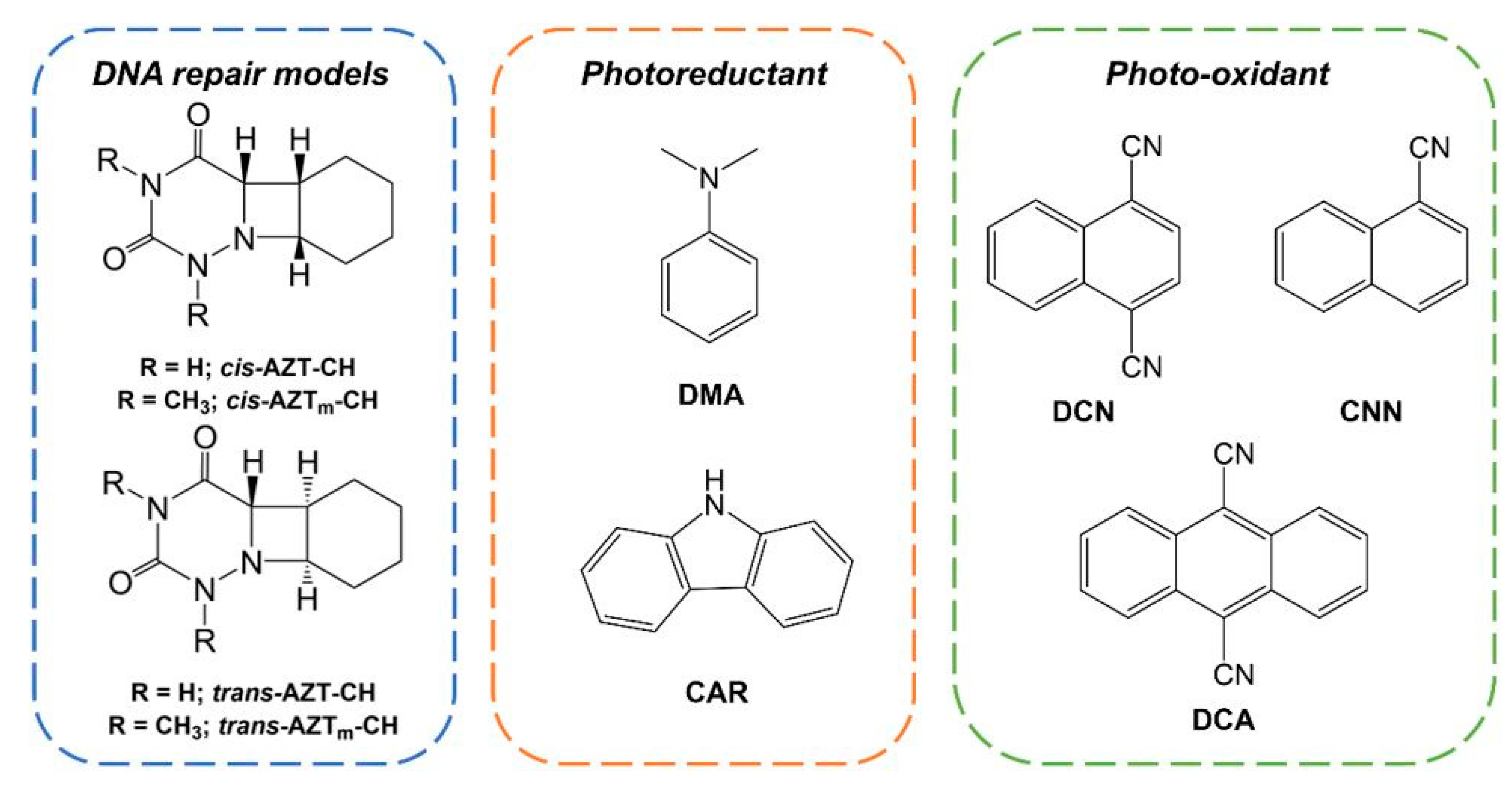
2. Results
2.1. Photoreductive and Photo-Oxidative Properties
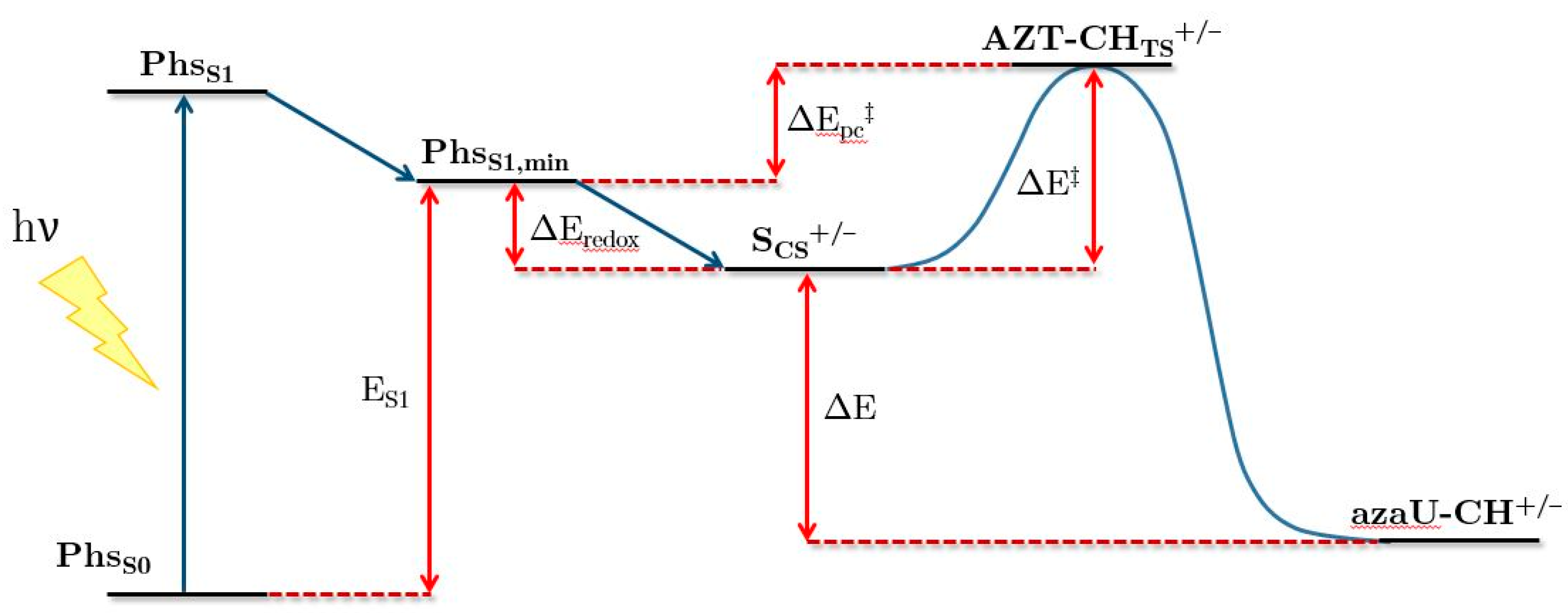
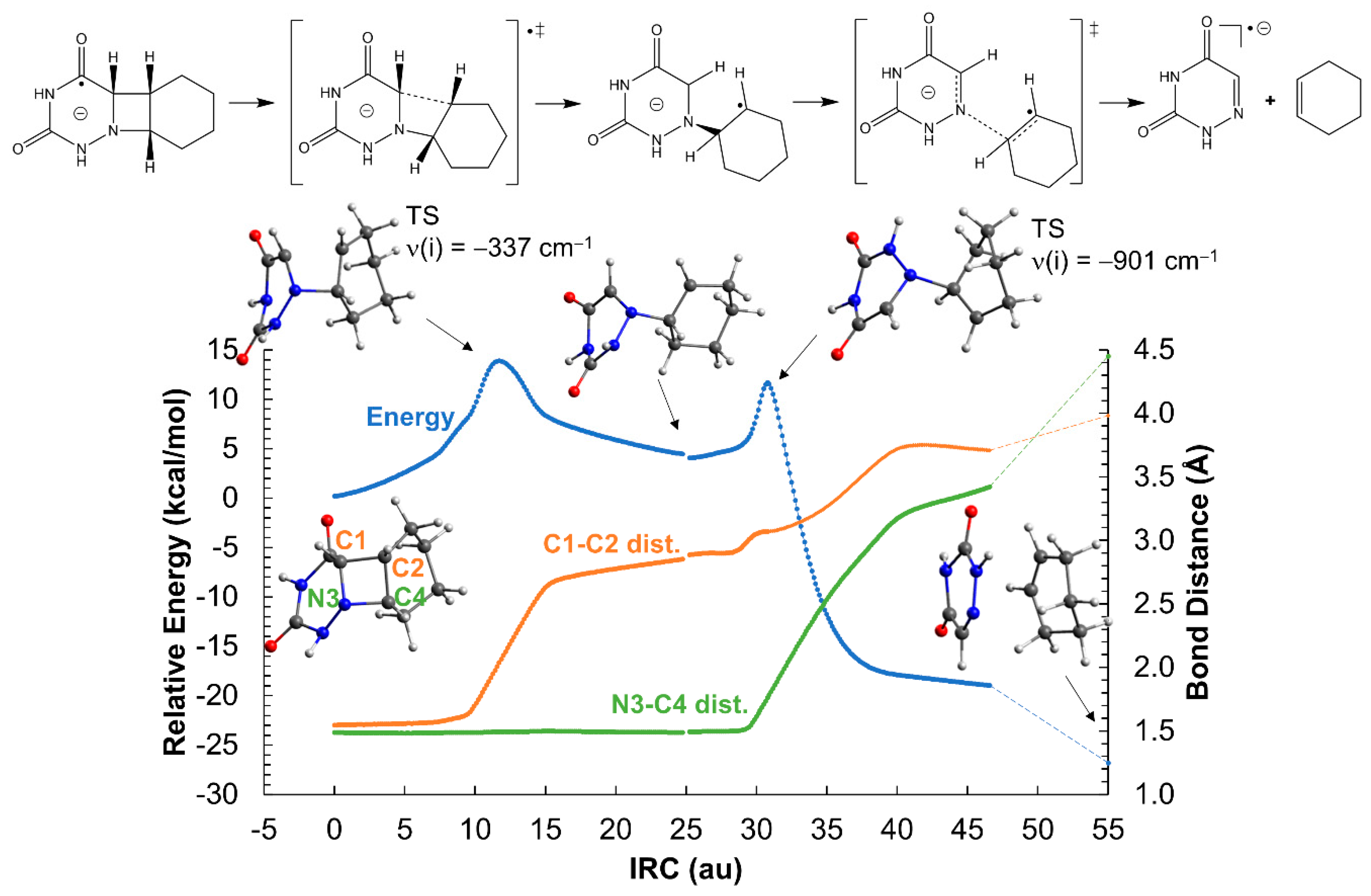
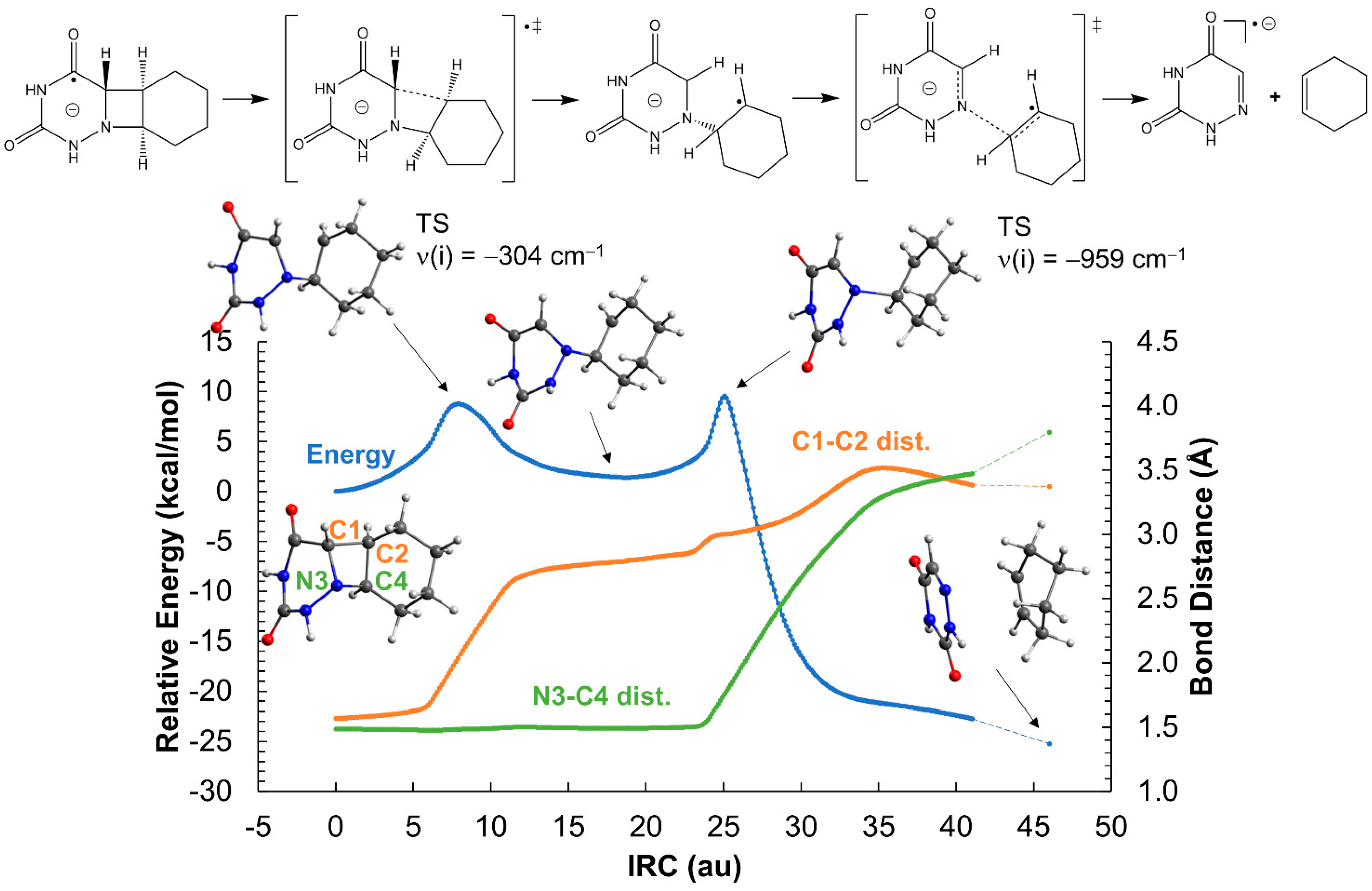
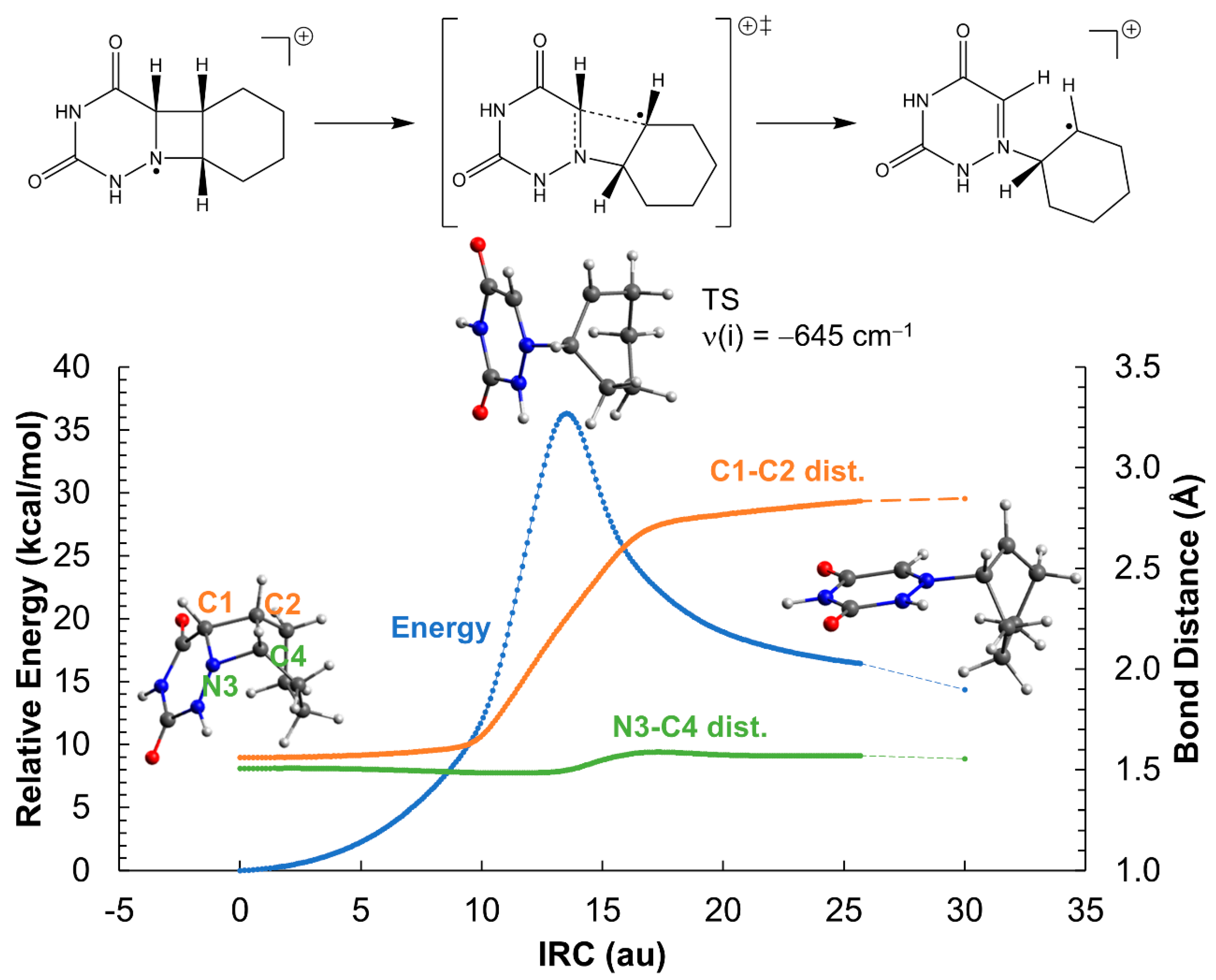
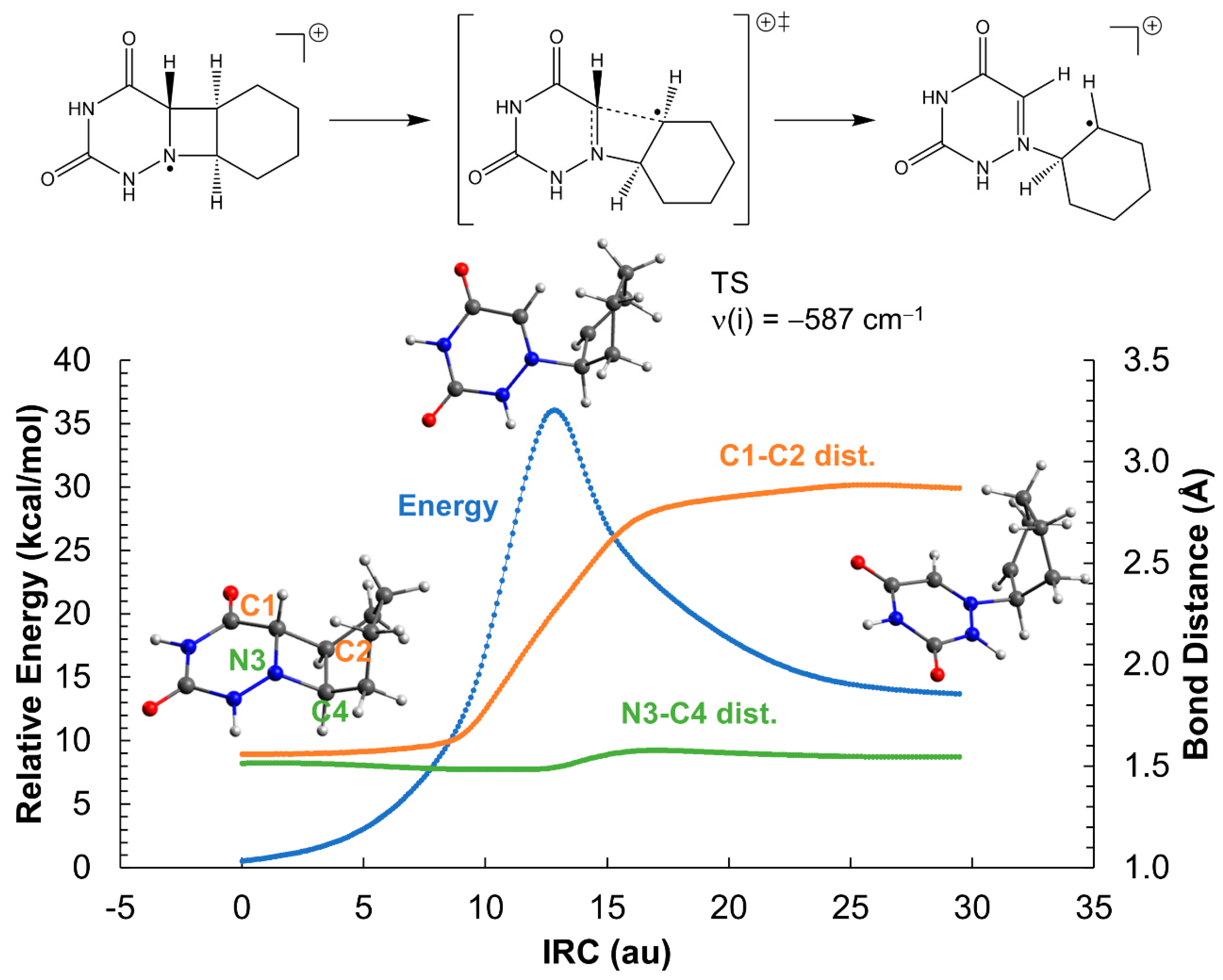
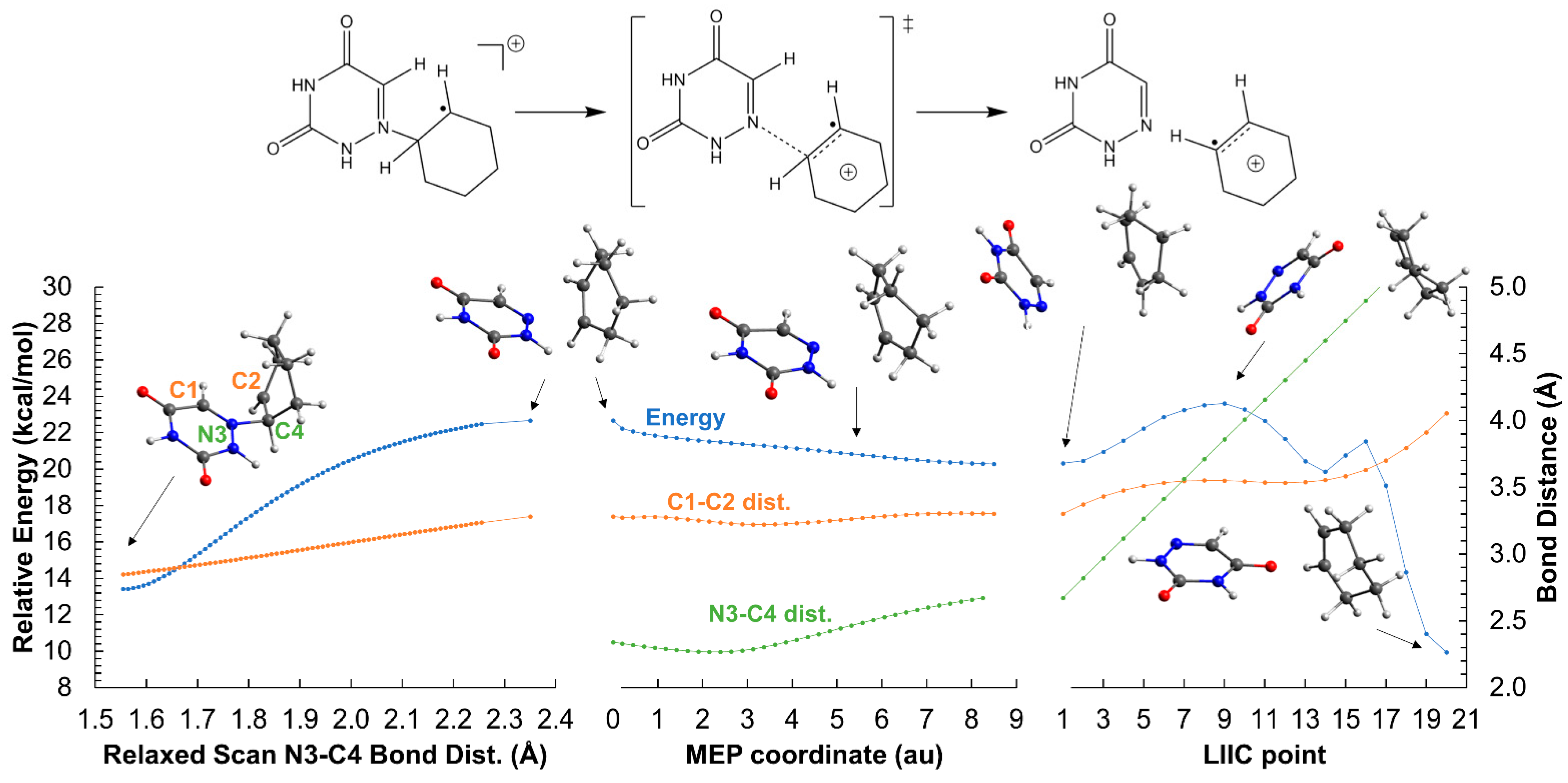
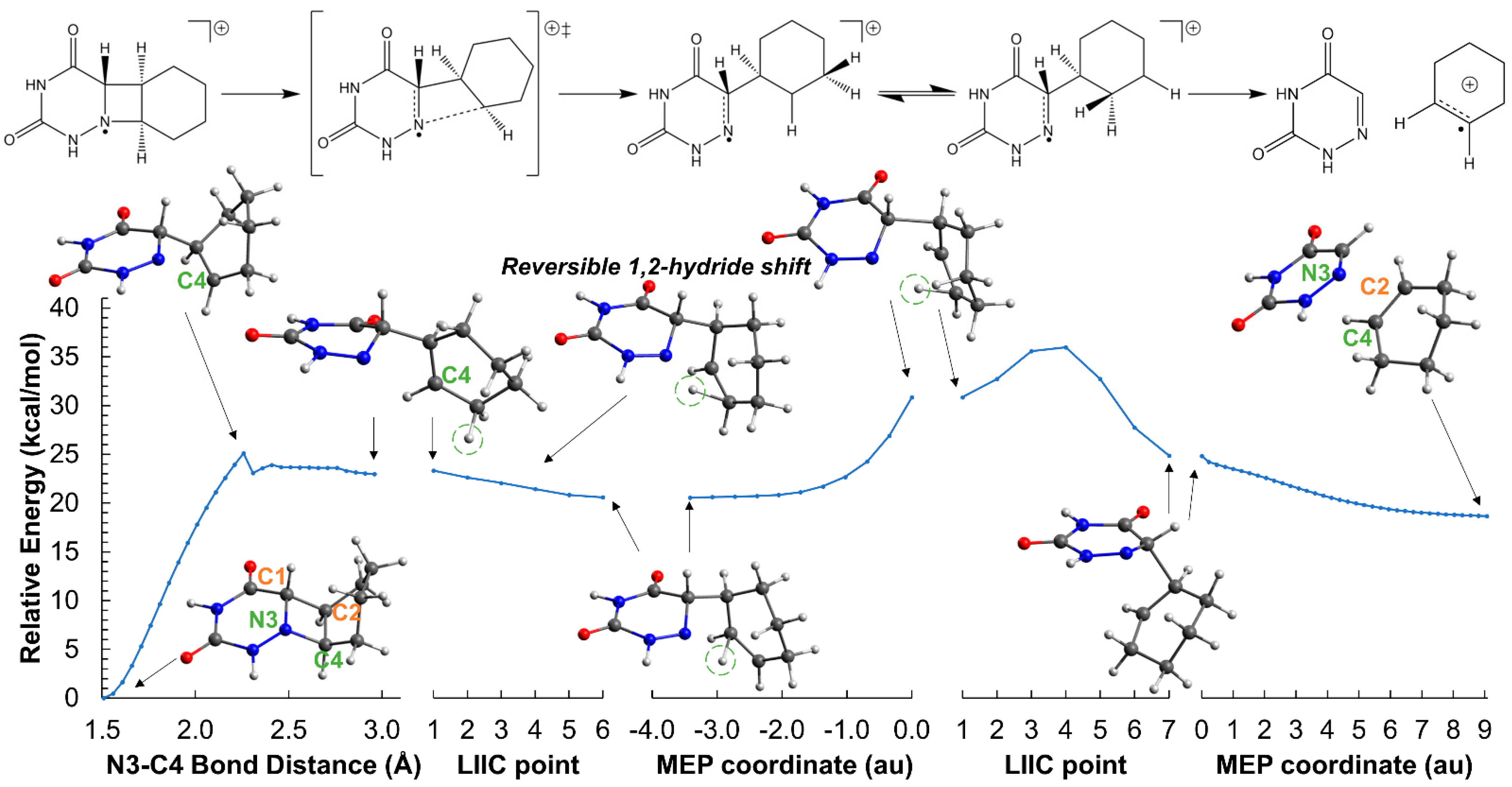
2.2. Ring-Opening Mechanisms of cis- and trans-AZT-CH Radical Anion and Cation
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Improta, R.; Santoro, F.; Blancafort, L. Quantum mechanical studies on the photophysics and the photochemistry of nucleic acids and nucleobases. Chem. Rev. 2016, 116, 3540–3593. [Google Scholar] [CrossRef] [PubMed]
- Francés-Monerris, A.; Gillet, N.; Dumont, E.; Monari, A. DNA photodamage and repair: Computational photobiology in action. In QM/MM Studies of Light-Responsive Biological Systems; Springer: Cham, Switzerland, 2021; pp. 293–332. [Google Scholar]
- Fraga-Timiraos, A.B.; Lhiaubet-Vallet, V.; Miranda, M.A. Repair of a dimeric azetidine related to the thymine-cytosine (6-4) photoproduct by electron transfer photoreduction. Angew. Chem. Int. Ed. 2016, 55, 6037–6040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Luque, R.; Climent, T.; González-Ramírez, I. Singlet–triplet states interaction regions in DNA/RNA nucleobase hypersurfaces. J. Chem. Theory Comput. 2010, 6, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, J.; Plaza, P.; Brettel, K. Repair of (6-4) lesions in DNA by (6-4) photolyase: 20 years of quest for the photoreaction mechanism. Photochem. Photobiol. 2017, 93, 51–66. [Google Scholar] [CrossRef] [Green Version]
- Faraji, S.; Dreuw, A. Insights into light-driven DNA repair by photolyases: Challenges and opportunities for electronic structure theory. Photochem. Photobiol. 2017, 93, 37–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maul, M.J.; Barends, T.R.M.; Glas, A.F.; Cryle, M.J.; Domratcheva, T.; Schneider, S.; Schlichting, I.; Carell, T. Crystal structure and mechanism of a DNA (6-4) photolyase. Angew. Chem. Int. Ed. 2008, 47, 10076–10080. [Google Scholar] [CrossRef]
- Glas, A.F.; Schneider, S.; Maul, M.J.; Hennecke, U.; Carell, T. Crystal structure of the T(6-4)C lesion in complex with a (6-4) DNA photolyase and repair of UV-induced (6-4) and dewar photolesions. Chem. Eur. J. 2009, 15, 10387–10396. [Google Scholar] [CrossRef]
- Sadeghian, K.; Bocola, M.; Merz, T.; Schütz, M. Theoretical study on the repair mechanism of the (6-4) photolesion by the (6-4) photolyase. J. Am. Chem. Soc. 2010, 132, 16285–16295. [Google Scholar] [CrossRef]
- Li, J.; Liu, Z.; Tan, C.; Guo, X.; Wang, L.; Sancar, A.; Zhong, D. Dynamics and mechanism of repair of ultraviolet-induced (6-4) photoproduct by photolyase. Nature 2010, 466, 887–890. [Google Scholar] [CrossRef]
- Yamamoto, J.; Martin, R.; Iwai, S.; Plaza, P.; Brettel, K. Repair of the (6-4) photoproduct by DNA photolyase requires two photons. Angew. Chem. Int. Ed. 2013, 52, 7432–7436. [Google Scholar] [CrossRef]
- Faraji, S.; Zhong, D.; Dreuw, A. Characterization of the intermediate in and identification of the repair mechanism of (6-4) photolesions by photolyases. Angew. Chem. Int. Ed. 2016, 55, 5175–5178. [Google Scholar] [CrossRef] [Green Version]
- Sancar, A. Structure and function of DNA photolyase and cryptochrome blue-light photoreceptors. Chem. Rev. 2003, 103, 2203–2237. [Google Scholar] [CrossRef]
- Pérez-Ruiz, R.; Jiménez, M.C.; Miranda, M.A. Hetero-cycloreversions mediated by photoinduced electron transfer. Acc. Chem. Res. 2014, 47, 1359–1368. [Google Scholar] [CrossRef] [Green Version]
- Fraga-Timiraos, A.B.; Francés-Monerris, A.; Rodríguez-Muñiz, G.M.; Navarrete-Miguel, M.; Miranda, M.A.; Roca-Sanjuán, D.; Lhiaubet-Vallet, V. Experimental and theoretical study on the cycloreversion of a nucleobase-derived azetidine by photoinduced electron transfer. Chem. Eur. J. 2018, 24, 15346–15354. [Google Scholar] [CrossRef]
- Arnold, A.R.; Grodick, M.A.; Barton, J.K. DNA charge transport: From chemical principles to the cell. Cell Chem. Biol. 2016, 23, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Dandliker, P.J.; Erik Holmlin, R.; Barton, J.K. Oxidative thymine dimer repair in the DNA helix. Science 1997, 275, 1465–1468. [Google Scholar] [CrossRef]
- Vicic, D.A.; Odom, D.T.; Nunez, M.E.; Gianolio, D.A.; McLaughlin, L.W.; Barton, J.K. Oxidative repair of a thymine dimer in DNA from a distance by a covalently linked organic intercalator. J. Am. Chem. Soc. 2000, 122, 8603–8611. [Google Scholar] [CrossRef]
- Fraga-Timiraos, A.B.; Rodríguez-Muñiz, G.M.; Peiro-Penalba, V.; Miranda, M.A.; Lhiaubet-Vallet, V. Stereoselective fluorescence quenching in the electron transfer photooxidation of nucleobase-related azetidines by cyanoaromatics. Molecules 2016, 21, 1683. [Google Scholar] [CrossRef] [Green Version]
- Roca-Sanjuán, D.; Rubio, M.; Merchán, M.; Serrano-Andrés, L. Ab initio determination of the ionization potentials of DNA and RNA nucleobases. J. Chem. Phys. 2006, 125, 084302. [Google Scholar] [CrossRef] [Green Version]
- Roca-Sanjuán, D.; Merchán, M.; Serrano-Andrés, L.; Rubio, M. Ab initio determination of the electron affinities of DNA and RNA nucleobases. J. Chem. Phys. 2008, 129, 095104. [Google Scholar] [CrossRef] [Green Version]
- Tóth, Z.; Kubečka, J.; Muchová, E.; Slavíček, P. Ionization energies in solution with the QM:QM approach. Phys. Chem. Chem. Phys. 2020, 22, 10550–10560. [Google Scholar] [CrossRef]
- Tomaník, L.; Muchová, E.; Slavíček, P. Solvation energies of ions with ensemble cluster-continuum approach. Phys. Chem. Chem. Phys. 2020, 22, 22357–22368. [Google Scholar] [CrossRef]
- Francés-Monerris, A.; Segarra-Martí, J.; Merchán, M.; Roca-Sanjuán, D. Complete-active-space second-order perturbation theory (CASPT2//CASSCF) study of the dissociative electron attachment in canonical DNA nucleobases caused by low-energy electrons (0–3 eV). J. Chem. Phys. 2015, 143, 215101. [Google Scholar] [CrossRef] [PubMed]
- González-Ramírez, I.; Segarra-Martí, J.; Serrano-Andrés, L.; Merchán, M.; Rubio, M.; Roca-Sanjuán, D. On the N 1-H and N 3-H bond dissociation in uracil by low energy electrons: A CASSCF/CASPT2 study. J. Chem. Theory Comput. 2012, 8, 2769–2776. [Google Scholar] [CrossRef] [PubMed]
- Chyongjin, P.; Tomohito, O.; Yozo, S.; Shozo, Y.; Hiroshi, S. Photochemical reactions of aromatic compounds. XLII. Photosensitized reactions of some selected diarylcyclobutanes by aromatic nitriles and chloranil. Implications of charge-transfer contributions on exciplex reactivities. Bull. Chem. Soc. Jpn. 1986, 59, 1133–1139. [Google Scholar] [CrossRef] [Green Version]
- Ajò, D.; Casarin, M.; Granozzi, G.; Fragalà, I. UV photoelectron spectra of 5- and 6-azauracil. Chem. Phys. Lett. 1981, 80, 188–191. [Google Scholar] [CrossRef]
- Lambert, J.B.; Xue, L.; Bosch, R.J.; Taba, K.M.; Marko, D.E.; Urano, S.; LeBreton, P.R. Through space interactions of double bonds by photoelectron spectroscopy. J. Am. Chem. Soc. 1986, 108, 7575–7579. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01 2009; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Aranda, J.; Francés-Monerris, A.; Tunón, I.; Roca-Sanjuán, D. Regioselectivity of the OH radical addition to uracil in nucleic acids. A theoretical approach based on QM/MM simulations. J. Chem. Theory Comput. 2017, 13, 5089–5096. [Google Scholar] [CrossRef]
- Francés-Monerris, A.; Merchán, M.; Roca-Sanjuán, D. Theoretical study of the hydroxyl radical addition to uracil and photochemistry of the formed U6OH• adduct. J. Phys. Chem. B 2014, 118, 2932–2939. [Google Scholar] [CrossRef]
- Francés-Monerris, A.; Merchán, M.; Roca-Sanjuán, D. Mechanism of the OH radical addition to adenine from quantum-chemistry determinations of reaction paths and spectroscopic tracking of the intermediates. J. Org. Chem. 2017, 82, 276–288. [Google Scholar] [CrossRef]
- Borràs, V.J.; Francés-Monerris, A.; Roca-Sanjuán, D. Hydroxyl radical addition to thymine and cytosine and photochemistry of the adducts at the C6 position. ChemPhotoChem 2019, 3, 889–896. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R. Guanosine + OH radical reaction in aqueous solution: A reinterpretation of the UV-vis data based on thermodynamic and kinetic calculations. Org. Lett. 2009, 11, 5114–5117. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VIP | AIP | AEA | Ered,0,AZT-CH | |
|---|---|---|---|---|
| cis-AZT-CH | ||||
| gas-phase | 8.97 (206.8) | 8.11 (186.9) | −0.48 (−11.0) | - |
| Acetonitrile | 6.98 (160.9) | 6.12 (141.0) | 1.55 (35.8) | 1.26 |
| trans-AZT-CH | ||||
| gas phase | 9.28 (214.0) | 8.28 (190.8) | −0.61 (−14.0) | - |
| Acetonitrile | 7.28 (167.9) | 6.26 (144.4) | 1.58 (36.4) | 1.51 |
| ES1 | ES1,exp | AIP/AEA | Ered,0,Phs | cis-ΔEredox | trans-ΔEredox | cis-kq | trans-kq | |
|---|---|---|---|---|---|---|---|---|
| Photoreduction | ||||||||
| DMA | 4.39 (101.1) | 3.76 (86.7) | 5.41 (124.7) | 0.68 | −0.53 (−12.3) | −0.56 (−12.9) | N.D. | N.D. |
| CAR | 4.20 (96.8) | 3.50 (80.7) | 6.02 (138.9) | 0.96 | 0.27 (6.2) | 0.24 (5.6) | N.D. | N.D. |
| Photo-oxidation | ||||||||
| DCA | 2.74 (63.3) | 2.86 (66.0) | 3.57 (82.2) | −1.0 | −0.19 (−4.4) | −0.05 (−1.1) | 10 | 7.7 |
| DCN | 3.62 (83.5) | 3.75 (86.5) | 3.14 (72.5) | −0.93 | −0.65 (−15.0) | −0.51 (−11.6) | 6.4 | 4.6 |
| CNN | 3.95 (91.1) | 3.88 (89.5) | 2.43 (56.0) | −2.21 | −0.26 (−6.0) | −0.12 (−2.7) | 3.2 | 2.6 |
| Methodology | cis-AZT-CH•− | cis-AZT-CH•+ | ||
|---|---|---|---|---|
| ΔE | ΔE‡ | ΔE | ΔE‡ | |
| M06-2X | −26.79 | 13.90 | 10.89 | 36.32 |
| PCM-M06-2X | −24.74 | 16.21 | 14.67 | 35.03 |
| ΔE0 | ΔE0‡ | ΔE0 | ΔE0‡ | |
| M06-2X | −29.22 | 12.02 | 7.06 | 33.84 |
| PCM-M06-2X | −27.05 | 14.45 | 10.47 | 32.18 |
| ΔG | ΔG‡ | ΔG | ΔG‡ | |
| M06-2X | −32.21 | 11.38 | 2.63 | 33.40 |
| PCM-M06-2X | −30.16 | 13.69 | 6.40 | 32.11 |
| Methodology | trans-AZT-CH•− | trans-AZT-CH•+ | ||
|---|---|---|---|---|
| ΔE | ΔE‡ | ΔE | ΔE‡ | |
| M06-2X | −25.23 | 8.78 | 9.93 | 36.08 |
| PCM-M06-2X | −20.80 | 13.68 | 14.81 | 36.16 |
| ΔE0 | ΔE0‡ | ΔE0 | ΔE0‡ | |
| M06-2X | −27.65 | 6.85 | 6.36 | 33.85 |
| PCM-M06-2X | −23.19 | 11.77 | 11.02 | 33.71 |
| ΔG | ΔG‡ | ΔG | ΔG‡ | |
| M06-2X | −30.28 | 6.75 | 2.39 | 33.64 |
| PCM-M06-2X | −25.84 | 11.65 | 7.28 | 33.72 |
| ΔEpc‡ cis-AZT-CH | ΔEpc‡ trans-AZT-CH | |
|---|---|---|
| CAR | 22.4 | 19.3 |
| DMA | 4.0 | 0.8 |
| DCA | 30.6 | 35.1 |
| DCN | 20.1 | 24.5 |
| CNN | 29.0 | 33.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navarrete-Miguel, M.; Francés-Monerris, A.; Miranda, M.A.; Lhiaubet-Vallet, V.; Roca-Sanjuán, D. Theoretical Study on the Photo-Oxidation and Photoreduction of an Azetidine Derivative as a Model of DNA Repair. Molecules 2021, 26, 2911. https://doi.org/10.3390/molecules26102911
Navarrete-Miguel M, Francés-Monerris A, Miranda MA, Lhiaubet-Vallet V, Roca-Sanjuán D. Theoretical Study on the Photo-Oxidation and Photoreduction of an Azetidine Derivative as a Model of DNA Repair. Molecules. 2021; 26(10):2911. https://doi.org/10.3390/molecules26102911
Chicago/Turabian StyleNavarrete-Miguel, Miriam, Antonio Francés-Monerris, Miguel A. Miranda, Virginie Lhiaubet-Vallet, and Daniel Roca-Sanjuán. 2021. "Theoretical Study on the Photo-Oxidation and Photoreduction of an Azetidine Derivative as a Model of DNA Repair" Molecules 26, no. 10: 2911. https://doi.org/10.3390/molecules26102911
APA StyleNavarrete-Miguel, M., Francés-Monerris, A., Miranda, M. A., Lhiaubet-Vallet, V., & Roca-Sanjuán, D. (2021). Theoretical Study on the Photo-Oxidation and Photoreduction of an Azetidine Derivative as a Model of DNA Repair. Molecules, 26(10), 2911. https://doi.org/10.3390/molecules26102911