
Antibody–Drug Conjugates—A Tutorial Review



Abstract
:
1. Introduction
2. Antibody–Drug Conjugate Components
2.1. Antibodies
2.1.1. Antibody Basics
2.1.2. Antibody Structure
2.1.3. Antibody Function
2.1.4. Targeting Cancer Cells with Antibodies
2.1.5. Types of Monoclonal Antibody
2.1.6. Factors Influencing the Choice of Monoclonal Antibodies for ADCs
2.2. Linkers
2.2.1. Cleavable Linkers
2.2.2. Non-Cleavable Linkers
2.2.3. Drug–Antibody Ratio and Homogeneity
2.2.4. Recent Advances in Linker Technologies
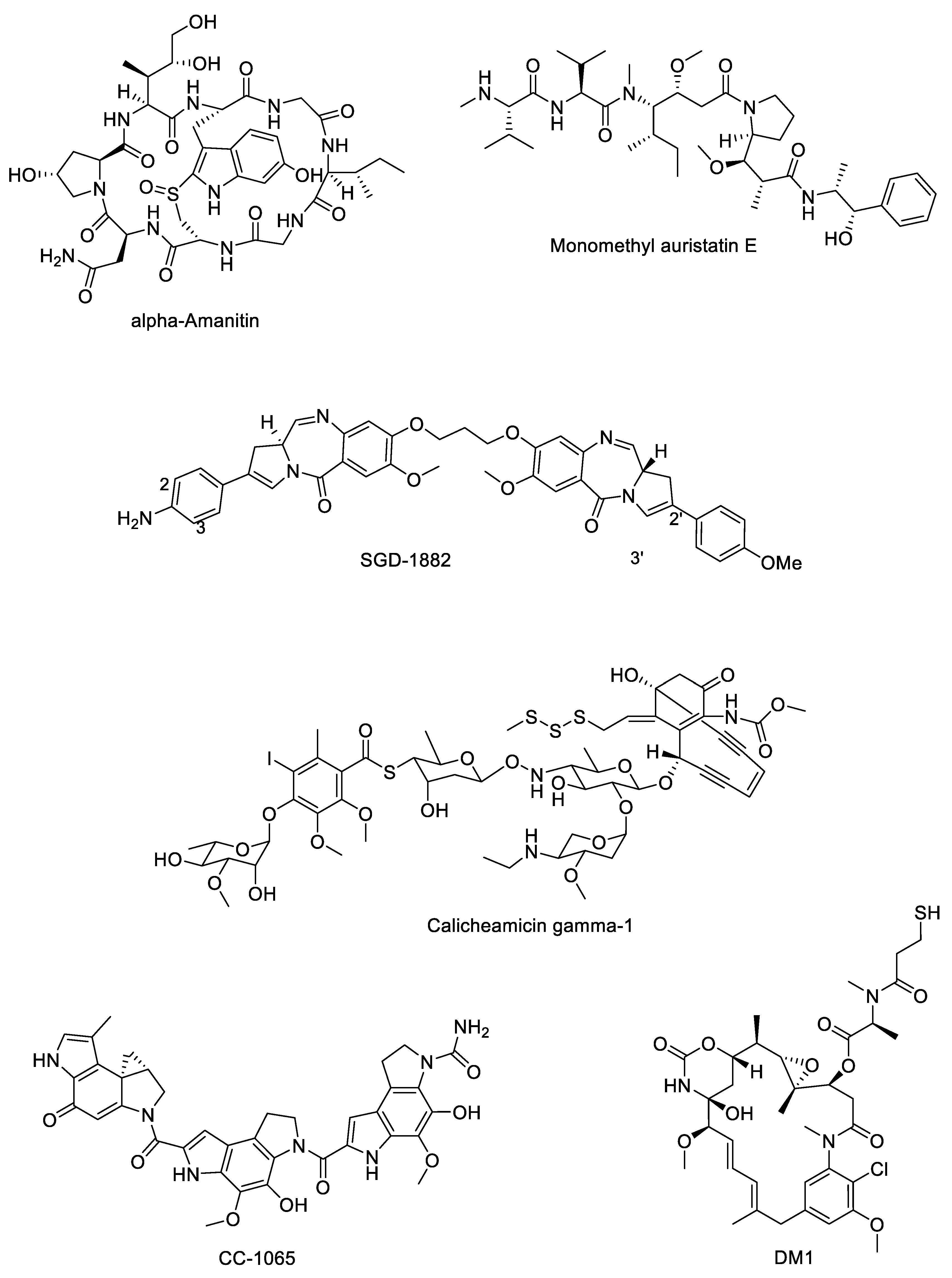
2.3. Payloads
Payload Classes
3. Antibody–Drug Conjugates in the Clinic
3.1. Currently Marketed Antibody–Drug Conjugates
3.2. General Mode of Action
3.3. Antibody–Drug Conjugate Case Studies
3.3.1. Pfizer’s Mylotarg® (Gemtuzumab Ozogamicin)
3.3.2. Genentech’s Kadcyla® (Trastuzumab Emtansine)
4. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- How Chemotherapy Works. Available online: https://www.cancerresearchuk.org/about-cancer/cancer-in-general/treatment/chemotherapy/how-chemotherapy-works#:~:text=Examples%20of%20cancers%20where%20chemotherapy,with%20other%20types%20of%20treatment (accessed on 1 October 2020).
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T., Jr.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, R.S. Paul Ehrlich’s magic bullets. N. Engl. J. Med. 2004, 350, 1079–1080. [Google Scholar] [CrossRef] [PubMed]
- Ford, C.H.; Newman, C.E.; Johnson, J.R.; Woodhouse, C.S.; Reeder, T.A.; Rowland, G.F.; Simmonds, R.G. Localisation and toxicity study of a vindesine-anti-CEA conjugate in patients with advanced cancer. Br. J. Cancer 1983, 47, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, H.L.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody-drug conjugates: Current status and future directions. Drug Discov. Today 2014, 19, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.L.; Goulet, D.R.; Teplyakov, A.; Gilliland, G.L. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies 2019, 8, 55. [Google Scholar] [CrossRef] [Green Version]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. The Adaptive Immune System. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002; Chapter 24. [Google Scholar]
- Lipman, N.S.; Jackson, L.R.; Trudel, L.J.; Weis-Garcia, F. Monoclonal versus polyclonal antibodies: Distinguishing characteristics, applications, and information resources. Ilar J. 2005, 46, 258–268. [Google Scholar] [CrossRef] [Green Version]
- Peters, C.; Brown, S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [Green Version]
- Immunoglobulins: Classes and Subclasses. Available online: https://www.bio-rad-antibodies.com/immunoglobulins-classes-subclasses.html (accessed on 1 October 2020).
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [Green Version]
- Vu, T.; Claret, F.X. Trastuzumab: Updated mechanisms of action and resistance in breast cancer. Front. Oncol. 2012, 2, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, A.; Weiss, D.T. Structural and functional properties of human lambda-light-chain variable-region subgroups. Clin. Diagn Lab. Immunol. 1995, 2, 387–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seidel, U.J.; Schlegel, P.; Lang, P. Natural killer cell mediated antibody-dependent cellular cytotoxicity in tumor immunotherapy with therapeutic antibodies. Front. Immunol. 2013, 4, 76. [Google Scholar] [CrossRef] [Green Version]
- Gelderman, K.A.; Tomlinson, S.; Ross, G.D.; Gorter, A. Complement function in mAb-mediated cancer immunotherapy. Trends Immunol. 2004, 25, 158–164. [Google Scholar] [CrossRef]
- Attarwala, H. Role of antibodies in cancer targeting. J. Nat. Sci. Biol. Med. 2010, 1, 53–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jager, E.; Knuth, A. The discovery of cancer/testis antigens by autologous typing with T cell clones and the evolution of cancer vaccines. Cancer Immun. 2012, 12, 6. [Google Scholar]
- Jilani, I.; Estey, E.; Huh, Y.; Joe, Y.; Manshouri, T.; Yared, M.; Giles, F.; Kantarjian, H.; Cortes, J.; Thomas, D.; et al. Differences in CD33 intensity between various myeloid neoplasms. Am. J. Clin. Pathol. 2002, 118, 560–566. [Google Scholar] [CrossRef] [Green Version]
- Rudnick, S.I.; Lou, J.; Shaller, C.C.; Tang, Y.; Klein-Szanto, A.J.; Weiner, L.M.; Marks, J.D.; Adams, G.P. Influence of affinity and antigen internalization on the uptake and penetration of Anti-HER2 antibodies in solid tumors. Cancer Res. 2011, 71, 2250–2259. [Google Scholar] [CrossRef] [Green Version]
- Polson, A.G.; Ho, W.Y.; Ramakrishnan, V. Investigational antibody-drug conjugates for hematological malignancies. Expert Opin. Investig. Drugs 2011, 20, 75–85. [Google Scholar] [CrossRef]
- Dudley, A.C. Tumor endothelial cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006536. [Google Scholar] [CrossRef]
- Brekke, O.H.; Sandlie, I. Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat. Rev. Drug Discov. 2003, 2, 52–62. [Google Scholar] [CrossRef]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. MAbs 2010, 2, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Kohler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Milstein, C. The hybridoma revolution: An offshoot of basic research. Bioessays 1999, 21, 966–973. [Google Scholar] [CrossRef]
- Skerra, A.; Pluckthun, A. Assembly of a functional immunoglobulin Fv fragment in Escherichia coli. Science 1988, 240, 1038–1041. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef]
- Van de Donk, N.W.; Dhimolea, E. Brentuximab vedotin. MAbs 2012, 4, 458–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Wang, E.Q.; Balthasar, J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2008, 84, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Panowski, S.; Bhakta, S.; Raab, H.; Polakis, P.; Junutula, J.R. Site-specific antibody drug conjugates for cancer therapy. MAbs 2014, 6, 34–45. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, R.M.; Coumbe, B.G.T.; Josephs, D.H.; Mele, S.; Ilieva, K.M.; Cheung, A.; Tutt, A.N.; Spicer, J.F.; Thurston, D.E.; Crescioli, S. Antibody structure and engineering considerations for the design and function of Antibody Drug Conjugates (ADCs). Oncoimmunology 2018, 7, e1395127. [Google Scholar] [CrossRef] [PubMed]
- Yoo, E.M.; Wims, L.A.; Chan, L.A.; Morrison, S.L. Human IgG2 Can Form Covalent Dimers. J. Immunol. 2003, 170, 3134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jefferis, R. Antibody therapeutics: Isotype and glycoform selection. Expert Opin. Biol. Ther. 2007, 7, 1401–1413. [Google Scholar] [CrossRef]
- Van der Neut Kolfschoten, M.; Schuurman, J.; Losen, M.; Bleeker, W.K.; Martinez-Martinez, P.; Vermeulen, E.; den Bleker, T.H.; Wiegman, L.; Vink, T.; Aarden, L.A.; et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science 2007, 317, 1554–1557. [Google Scholar] [CrossRef] [Green Version]
- Drachman, J.G.; Senter, P.D. Antibody-drug conjugates: The chemistry behind empowering antibodies to fight cancer. Hematol. Am. Soc. Hematol Educ Program. 2013, 2013, 306–310. [Google Scholar] [CrossRef]
- Goldmacher, V.S.; Kovtun, Y.V. Antibody-drug conjugates: Using monoclonal antibodies for delivery of cytotoxic payloads to cancer cells. Ther. Deliv. 2011, 2, 397–416. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Linkers Having a Crucial Role in Antibody-Drug Conjugates. Int. J. Mol. Sci. 2016, 17, 561. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, Y.V.; Audette, C.A.; Ye, Y.; Xie, H.; Ruberti, M.F.; Phinney, S.J.; Leece, B.A.; Chittenden, T.; Blattler, W.A.; Goldmacher, V.S. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 2006, 66, 3214–3221. [Google Scholar] [CrossRef] [Green Version]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC Linker Chemistry. Pharm Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, B.J.; Lang, C.A. Differential distribution of free and bound glutathione and cyst(e)ine in human blood. Biochem. Pharmacol. 1996, 52, 401–406. [Google Scholar] [CrossRef]
- Gondi, C.S.; Rao, J.S. Cathepsin B as a cancer target. Expert Opin. Ther. Targets 2013, 17, 281–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zammarchi, F.; Corbett, S.; Adams, L.; Tyrer, P.C.; Kiakos, K.; Janghra, N.; Marafioti, T.; Britten, C.E.; Havenith, C.E.G.; Chivers, S.; et al. ADCT-402, a PBD dimer–containing antibody drug conjugate targeting CD19-expressing malignancies. Blood 2018, 131, 1094–1105. [Google Scholar] [CrossRef]
- Kovtun, Y.; Jones, G.E.; Adams, S.; Harvey, L.; Audette, C.A.; Wilhelm, A.; Bai, C.; Rui, L.; Laleau, R.; Liu, F.; et al. A CD123-targeting antibody-drug conjugate, IMGN632, designed to eradicate AML while sparing normal bone marrow cells. Blood Adv. 2018, 2, 848–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubowchik, G.M.; Firestone, R.A.; Padilla, L.; Willner, D.; Hofstead, S.J.; Mosure, K.; Knipe, J.O.; Lasch, S.J.; Trail, P.A. Cathepsin B-labile dipeptide linkers for lysosomal release of doxorubicin from internalizing immunoconjugates: Model studies of enzymatic drug release and antigen-specific in vitro anticancer activity. Bioconjug. Chem. 2002, 13, 855–869. [Google Scholar] [CrossRef]
- Chen, H.; Lin, Z.; Arnst, K.E.; Miller, D.D.; Li, W. Tubulin Inhibitor-Based Antibody-Drug Conjugates for Cancer Therapy. Molecules 2017, 22, 1281. [Google Scholar] [CrossRef] [Green Version]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [Green Version]
- Burke, P.J.; Hamilton, J.Z.; Jeffrey, S.C.; Hunter, J.H.; Doronina, S.O.; Okeley, N.M.; Miyamoto, J.B.; Anderson, M.E.; Stone, I.J.; Ulrich, M.L.; et al. Optimization of a PEGylated Glucuronide-Monomethylauristatin E Linker for Antibody-Drug Conjugates. Mol. Cancer Ther. 2017, 16, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Sochaj, A.M.; Swiderska, K.W.; Otlewski, J. Current methods for the synthesis of homogeneous antibody-drug conjugates. Biotechnol. Adv. 2015, 33 Pt 1, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Yoder, N.C.; Bai, C.; Tavares, D.; Widdison, W.C.; Whiteman, K.R.; Wilhelm, A.; Wilhelm, S.D.; McShea, M.A.; Maloney, E.K.; Ab, O.; et al. A Case Study Comparing Heterogeneous Lysine- and Site-Specific Cysteine-Conjugated Maytansinoid Antibody-Drug Conjugates (ADCs) Illustrates the Benefits of Lysine Conjugation. Mol. Pharm. 2019, 16, 3926–3937. [Google Scholar] [CrossRef]
- Jackson, D.Y. Processes for Constructing Homogeneous Antibody Drug Conjugates. Org. Process. Res. Dev. 2016, 20, 852–866. [Google Scholar] [CrossRef] [Green Version]
- Kern, J.C.; Cancilla, M.; Dooney, D.; Kwasnjuk, K.; Zhang, R.; Beaumont, M.; Figueroa, I.; Hsieh, S.; Liang, L.; Tomazela, D.; et al. Discovery of Pyrophosphate Diesters as Tunable, Soluble, and Bioorthogonal Linkers for Site-Specific Antibody-Drug Conjugates. J. Am. Chem. Soc. 2016, 138, 1430–1445. [Google Scholar] [CrossRef]
- Bargh, J.D.; Walsh, S.J.; Isidro-Llobet, A.; Omarjee, S.; Carroll, J.S.; Spring, D.R. Sulfatase-cleavable linkers for antibody-drug conjugates. Chem. Sci. 2020, 11, 2375–2380. [Google Scholar] [CrossRef] [Green Version]
- Faridoon; Shi, W.; Qin, K.; Tang, Y.; Li, M.; Guan, D.; Tian, X.; Jiang, B.; Dong, J.; Tang, F.; et al. New linker structures applied in glycosite-specific antibody drug conjugates. Org. Chem. Front. 2019, 6, 3144–3149. [Google Scholar] [CrossRef]
- Hess, C.; Venetz, D.; Neri, D. Emerging classes of armed antibody therapeutics against cancer. MedChemComm 2014, 5, 408–431. [Google Scholar] [CrossRef]
- Anami, Y.; Yamazaki, C.M.; Xiong, W.; Gui, X.; Zhang, N.; An, Z.; Tsuchikama, K. Glutamic acid-valine-citrulline linkers ensure stability and efficacy of antibody-drug conjugates in mice. Nat. Commun. 2018, 9, 2512. [Google Scholar] [CrossRef]
- Poudel, Y.B.; Chowdari, N.S.; Cheng, H.; Iwuagwu, C.I.; King, H.D.; Kotapati, S.; Passmore, D.; Rampulla, R.; Mathur, A.; Vite, G.; et al. Chemical Modification of Linkers Provides Stable Linker-Payloads for the Generation of Antibody-Drug Conjugates. ACS Med. Chem. Lett. 2020, 11, 2190–2194. [Google Scholar] [CrossRef]
- Finbloom, D.S.; Abeles, D.; Rifai, A.; Plotz, P.H. The specificity of uptake of model immune complexes and other protein aggregates by the murine reticuloendothelial system. J. Immunol. 1980, 125, 1060–1065. [Google Scholar]
- Zhao, R.Y.; Wilhelm, S.D.; Audette, C.; Jones, G.; Leece, B.A.; Lazar, A.C.; Goldmacher, V.S.; Singh, R.; Kovtun, Y.; Widdison, W.C.; et al. Synthesis and evaluation of hydrophilic linkers for antibody-maytansinoid conjugates. J. Med. Chem. 2011, 54, 3606–3623. [Google Scholar] [CrossRef]
- Stenton, B.J.; Oliveira, B.L.; Matos, M.J.; Sinatra, L.; Bernardes, G.J.L. A thioether-directed palladium-cleavable linker for targeted bioorthogonal drug decaging. Chem. Sci. 2018, 9, 4185–4189. [Google Scholar] [CrossRef] [Green Version]
- Pietersz, G.A.; Krauer, K. Antibody-targeted drugs for the therapy of cancer. J. Drug Target. 1994, 2, 183–215. [Google Scholar] [CrossRef]
- Sedlacek, H.H.; Seemann, G.; Hoffmann, D.; Czech, J.; Lorenz, P.; Kolar, C.; Bosslet, K. Antibodies as Carriers of Cytotoxicity; Karger Publishers: Basel, Switzerland, 1992; Volume 43. [Google Scholar]
- Chari, R.V.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. Engl. 2014, 53, 3796–3827. [Google Scholar] [CrossRef] [PubMed]
- Pahl, A.; Lutz, C.; Hechler, T. Amanitins and their development as a payload for antibody-drug conjugates. Drug Discov. Today Technol. 2018, 30, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Pahl, A.; Ko, J.; Breunig, C.; Figueroa, V.; Lehners, N.; Baumann, A.; Pálfi, A.; Mueller, C.; Lutz, C.; Hechler, T.; et al. HDP-101: Preclinical evaluation of a novel anti-BCMA antibody drug conjugates in multiple myeloma. J. Clin. Oncol. 2018, 36 (Suppl. S15), e14527. [Google Scholar] [CrossRef]
- Pettit, G.R.; Kamano, Y.; Fujii, Y.; Herald, C.L.; Inoue, M.; Brown, P.; Gust, D.; Kitahara, K.; Schmidt, J.M.; Doubek, D.L.; et al. Marine animal biosynthetic constituents for cancer chemotherapy. J. Nat. Prod. 1981, 44, 482–485. [Google Scholar] [CrossRef]
- Bai, R.; Pettit, G.R.; Hamel, E. Dolastatin 10, a powerful cytostatic peptide derived from a marine animal. Inhibition of tubulin polymerization mediated through the vinca alkaloid binding domain. Biochem Pharm. 1990, 39, 1941–1949. [Google Scholar] [CrossRef]
- Akaiwa, M.; Dugal-Tessier, J.; Mendelsohn, B.A. Antibody-Drug Conjugate Payloads; Study of Auristatin Derivatives. Chem. Pharm Bull. 2020, 68, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Park, M.H.; Lee, B.I.; Byeon, J.J.; Shin, S.H.; Choi, J.; Park, Y.; Shin, Y.G. Pharmacokinetic and Metabolism Studies of Monomethyl Auristatin F via Liquid Chromatography-Quadrupole-Time-of-Flight Mass Spectrometry. Molecules 2019, 24, 2754. [Google Scholar] [CrossRef] [Green Version]
- Zein, N.; Sinha, A.M.; McGahren, W.J.; Ellestad, G.A. Calicheamicin gamma 1I: An antitumor antibiotic that cleaves double-stranded DNA site specifically. Science 1988, 240, 1198–1201. [Google Scholar] [CrossRef]
- Nicolaou, K.C. Chemistry and biology of the calicheamicins. Chem. Biol. 1994, 1, xxvi–xxvii. [Google Scholar] [CrossRef]
- Boger, D.L.; Johnson, D.S. CC-1065 and the Duocarmycins: Understanding their Biological Function through Mechanistic Studies. Angew. Chem. Int. Ed. Engl. 1996, 35, 1438–1474. [Google Scholar] [CrossRef]
- Cacciari, B.; Romagnoli, R.; Baraldi, P.G.; Ros, T.D.; Spalluto, G. CC-1065 and the duocarmycins: Recent developments. Expert Opin. Ther. Pat. 2000, 10, 1853–1871. [Google Scholar] [CrossRef]
- McGovren, J.P.; Clarke, G.L.; Pratt, E.A.; DeKoning, T.F. Preliminary toxicity studies with the DNA-binding antibiotic, CC-1065. J. Antibiot. 1984, 37, 63–70. [Google Scholar] [CrossRef]
- Rinnerthaler, G.; Gampenrieder, S.P.; Greil, R. HER2 Directed Antibody-Drug-Conjugates beyond T-DM1 in Breast Cancer. Int. J. Mol. Sci. 2019, 20, 1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupchan, S.M.; Komoda, Y.; Court, W.A.; Thomas, G.J.; Smith, R.M.; Karim, A.; Gilmore, C.J.; Haltiwanger, R.C.; Bryan, R.F. Maytansine, a novel antileukemic ansa macrolide from Maytenus ovatus. J. Am. Chem. Soc. 1972, 94, 1354–1356. [Google Scholar] [CrossRef]
- Bhattacharyya, B.; Wolff, J. Maytansine binding to the vinblastine sites of tubulin. FEBS Lett. 1977, 75, 159–162. [Google Scholar] [CrossRef] [Green Version]
- Cabanillas, F.; Bodey, G.P.; Burgess, M.A.; Freireich, E.J. Results of a phase II study of maytansine in patients with breast carcinoma and melanoma. Cancer Treat. Rep. 1979, 63, 507–509. [Google Scholar]
- Tang, W.; Deng, X.; Ou, Z.; Gan, J.; Dong, Q.; Tan, B.; Lu, L.; Chen, B.; Bao, C.; Li, S.; et al. Abstract P6-17-39: BAT8001, a potent anti-HER2 antibody-drug conjugate with a novel stable linker for the treatment of HER2-positive breast cancer. Cancer Res. 2019, 79 (Suppl. S4), P6-17-39. [Google Scholar]
- Zaro, J.L.; Wang, J.; Shen, W. Summary and Future Directions of ADCs. In Antibody-Drug Conjugates: The 21st Century Magic Bullets for Cancer; Wang, J., Shen, W., Zaro, J.L., Eds.; Springer Publishing: New York, NY, USA, 2015; Volume 17. [Google Scholar]
- Leimgruber, W.; Stefanovic, V.; Schenker, F.; Karr, A.; Berger, J. Isolation and characterization of anthramycin, a new antitumor antibiotic. J. Am. Chem. Soc. 1965, 87, 5791–5793. [Google Scholar] [CrossRef]
- Rahman, K.M.; James, C.H.; Thurston, D.E. Observation of the reversibility of a covalent pyrrolobenzodiazepine (PBD) DNA adduct by HPLC/MS and CD spectroscopy. Org. Biomol. Chem. 2011, 9, 1632–1641. [Google Scholar] [CrossRef]
- Corcoran, D.B.; Lewis, T.; Nahar, K.S.; Jamshidi, S.; Fegan, C.; Pepper, C.; Thurston, D.E.; Rahman, K.M. Effects of Systematic Shortening of Noncovalent C8 Side Chain on the Cytotoxicity and NF-κB Inhibitory Capacity of Pyrrolobenzodiazepines (PBDs). J. Med. Chem. 2019, 62, 2127–2139. [Google Scholar] [CrossRef]
- Rahman, K.M.; Jackson, P.J.; James, C.H.; Basu, B.P.; Hartley, J.A.; de la Fuente, M.; Schatzlein, A.; Robson, M.; Pedley, R.B.; Pepper, C. GC-targeted C8-linked pyrrolobenzodiazepine–biaryl conjugates with femtomolar in vitro cytotoxicity and in vivo antitumor activity in mouse models. J. Med. Chem. 2013, 56, 2911–2935. [Google Scholar] [CrossRef]
- Rahman, K.M.; Thompson, A.S.; James, C.H.; Narayanaswamy, M.; Thurston, D.E. The pyrrolobenzodiazepine dimer SJG-136 forms sequence-dependent intrastrand DNA cross-links and monoalkylated adducts in addition to interstrand cross-links. J. Am. Chem. Soc. 2009, 131, 13756–13766. [Google Scholar] [CrossRef]
- Our Pipeline. Available online: https://adctherapeutics.com/our-pipeline/#adct602 (accessed on 15 April 2021).
- Mantaj, J.; Jackson, P.J.M.; Rahman, K.M.; Thurston, D.E. From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody-Drug Conjugates (ADCs). Angew. Chem. Int. Ed. 2017, 56, 462–488. [Google Scholar] [CrossRef] [Green Version]
- Joubert, N.; Beck, A.; Dumontet, C.; Denevault-Sabourin, C. Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals 2020, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- Smets, L.A. Programmed cell death (apoptosis) and response to anti-cancer drugs. Anticancer Drugs 1994, 5, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Tridente, G. Gemtuzumab. In Adverse Events with Biomedicines; Springer-Verlag Italia Srl.: Milan, Italy, 2014; pp. 211–217. [Google Scholar]
- DeLeve, L.D. Chapter 30—Cancer Chemotherapy. In Drug-Induced Liver Disease, 3rd ed.; Kaplowitz, N., DeLeve, L.D., Eds.; Academic Press: Boston, MA, USA, 2013; pp. 541–567. [Google Scholar]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef] [Green Version]
- Giles, F.J.; Kantarjian, H.M.; Kornblau, S.M.; Thomas, D.A.; Garcia-Manero, G.; Waddelow, T.A.; David, C.L.; Phan, A.T.; Colburn, D.E.; Rashid, A.; et al. Mylotarg (gemtuzumab ozogamicin) therapy is associated with hepatic venoocclusive disease in patients who have not received stem cell transplantation. Cancer 2001, 92, 406–413. [Google Scholar] [CrossRef]
- Selby, C.; Yacko, L.R.; Glode, A.E. Gemtuzumab Ozogamicin: Back Again. J. Adv. Pr. Oncol. 2019, 10, 68–82. [Google Scholar]
- Gbadamosi, M.; Meshinchi, S.; Lamba, J.K. Gemtuzumab ozogamicin for treatment of newly diagnosed CD33-positive acute myeloid leukemia. Future Oncol. 2018, 14, 3199–3213. [Google Scholar] [CrossRef] [PubMed]
- Albanell, J.; Codony, J.; Rovira, A.; Mellado, B.; Gascon, P. Mechanism of action of anti-HER2 monoclonal antibodies: Scientific update on trastuzumab and 2C4. Adv. Exp. Med. Biol. 2003, 532, 253–268. [Google Scholar]
- Kovtun, Y.V.; Goldmacher, V.S. Cell killing by antibody-drug conjugates. Cancer Lett. 2007, 255, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Erickson, H.K.; Park, P.U.; Widdison, W.C.; Kovtun, Y.V.; Garrett, L.M.; Hoffman, K.; Lutz, R.J.; Goldmacher, V.S.; Blattler, W.A. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006, 66, 4426–4433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhillon, S. Trastuzumab emtansine: A review of its use in patients with HER2-positive advanced breast cancer previously treated with trastuzumab-based therapy. Drugs 2014, 74, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Wedam, S.; Fashoyin-Aje, L.; Gao, X.; Bloomquist, E.; Tang, S.; Sridhara, R.; Goldberg, K.B.; King-Kallimanis, B.L.; Theoret, M.R.; Ibrahim, A.; et al. FDA Approval Summary: Ado-trastuzumab emtansine for the adjuvant treatment of HER2-positive early breast cancer. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef] [Green Version]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Morisaki, J.H.; et al. Novel antibody-antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Suffix | Origin | Therapeutic Potential | Example | Structure |
|---|---|---|---|---|---|
| Murine | -onab | 100% derived from mouse genes (both light and heavy chains) | Problems with immunogenicity, short half-lives and limited tumour site penetration | Muromonab |  |
| Chimeric | -ximab | 35% mouse, 65% human (murine variable regions, human constant region on each chain) | Reduced immunogenicity, improved half-lives versus murine mAbs | Rituximab |  |
| Humanised | -zumab | 95% human (part of the variable domain in each chain is either synthetic or animal-derived) | Reduced immunogenicity versus chimeric mAbs | Alemtuzumab |  |
| Human | -mumab | 100% human (both chain types are of human origin) | Broadly reduced immunogenicity versus humanised mAbs | Adalimumab |  |
| ADC Name | Indication | Target Antigen | mAb | Linker | Payload | Approval Date |
|---|---|---|---|---|---|---|
| Brentuximab vedotin (Adcetris®) | Relapsed/refractory Hodgkin lymphoma, systemic anaplastic large cell lymphoma | CD30 | Chimeric IgG1 | Val–Cit | MMAE | 25 October 2012 (EMA) 19 August 2011 (FDA) |
| Enfortumab vedotin (Padcev®) | Locally advanced/metastatic urothelial cancer | Nectin-4 | Human IgG1κ | Val–Cit | MMAE | 18 December 2019 (FDA) |
| Gemtuzumab ozogamicin (Mylotarg®) | Newly diagnosed, relapsed or refractory CD33-positive acute myeloid leukaemia | CD33 | Humanised IgG4κ | Cleavable acid-labile hydrazone | Calicheamicin | 19 April 2018 (EMA) 2 September 2017 (FDA) |
| Inotuzumab ozogamicin (Besponsa®) | Acute lymphoblastic leukaemia | CD22 | Humanised IgG4 | Cleavable acid-labile hydrazone | Calicheamicin | 29 June 2017 (EMA) 17 August 2017 (FDA) |
| Polatuzumab vedotin (Polivy®) | Diffuse large B cell lymphoma | CD79b | Humanised IgG1 | Val–Cit | MMAE | 16 January 2020 (EMA) 10 June 2019 (FDA) |
| Sacituzumab govitecan (Trodelvy®) | Metastatic triple-negative breast cancer | TROP2 | Humanised IgG1κ | CL2A | SN-38 | 22 April 2020 (FDA) |
| Trastuzumab deruxtecan (Enhertu®) | Unresectable/metastatic HER2-positive breast cancer | HER2 | Humanised IgG1 | Maleimide–GGFG | DXd | 18 January 2021 (EMA) 20 December 2019 (FDA) |
| Trastuzumab emtansine (Kadcyla®) | Metastatic HER2-positive breast cancer | HER2 | Humanised IgG1 | MCC | DM1 | 15 November 2013 (EMA) 22 February 2013 (FDA) |
| Belantamab mafodotin (Blenrep®) | Relapsed or refractory multiple myeloma | BCMA | Humanised IgG1 | MC | MMAF | 25 August 2020 (EMA) 5 August 2020 (FDA) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baah, S.; Laws, M.; Rahman, K.M. Antibody–Drug Conjugates—A Tutorial Review. Molecules 2021, 26, 2943. https://doi.org/10.3390/molecules26102943
Baah S, Laws M, Rahman KM. Antibody–Drug Conjugates—A Tutorial Review. Molecules. 2021; 26(10):2943. https://doi.org/10.3390/molecules26102943
Chicago/Turabian StyleBaah, Stephanie, Mark Laws, and Khondaker Miraz Rahman. 2021. "Antibody–Drug Conjugates—A Tutorial Review" Molecules 26, no. 10: 2943. https://doi.org/10.3390/molecules26102943
APA StyleBaah, S., Laws, M., & Rahman, K. M. (2021). Antibody–Drug Conjugates—A Tutorial Review. Molecules, 26(10), 2943. https://doi.org/10.3390/molecules26102943