
Fragment-Based Ab Initio Molecular Dynamics Simulation for Combustion

 ,
, 
Abstract
:1. Introduction
2. Results and Discussion
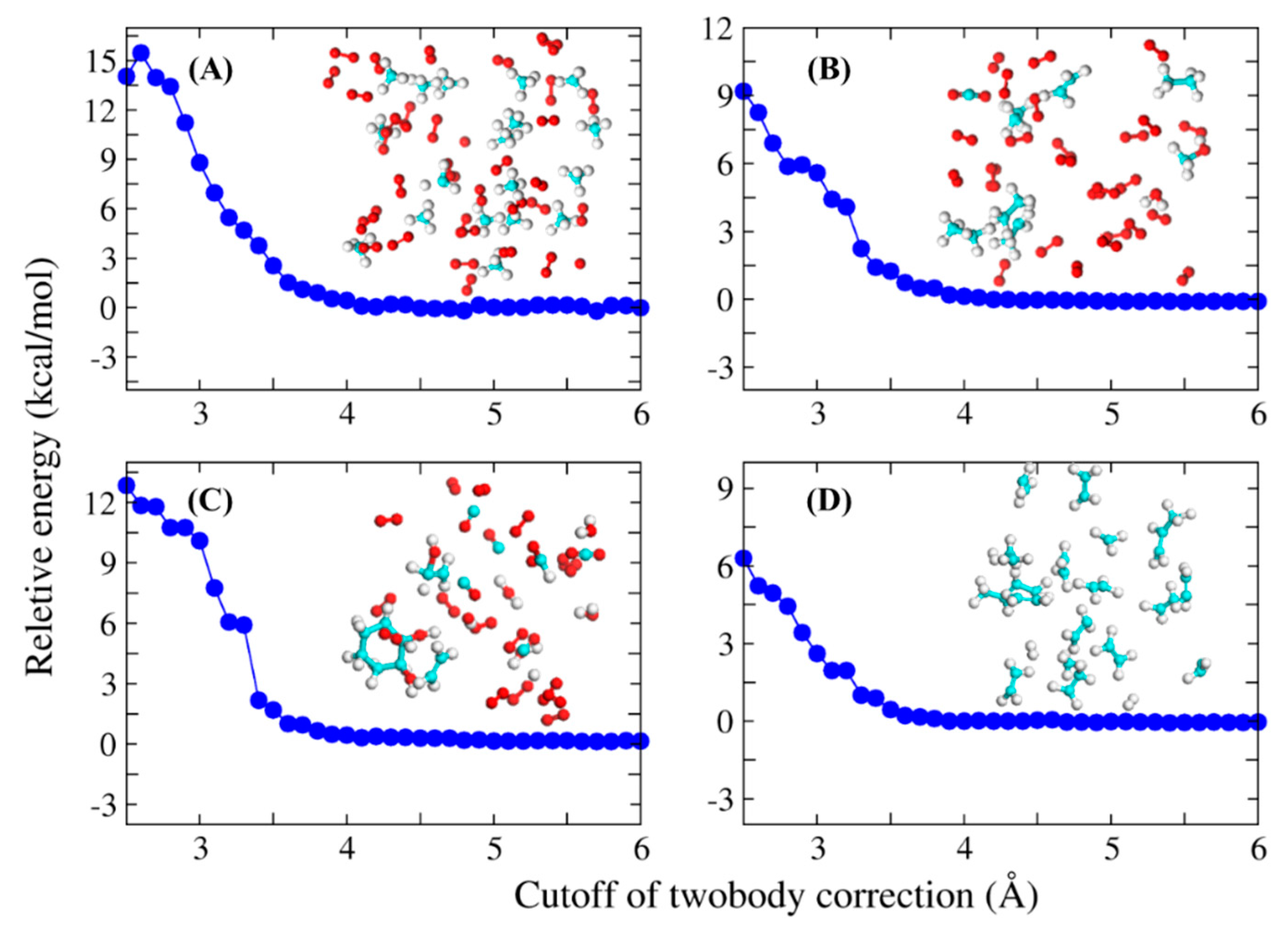
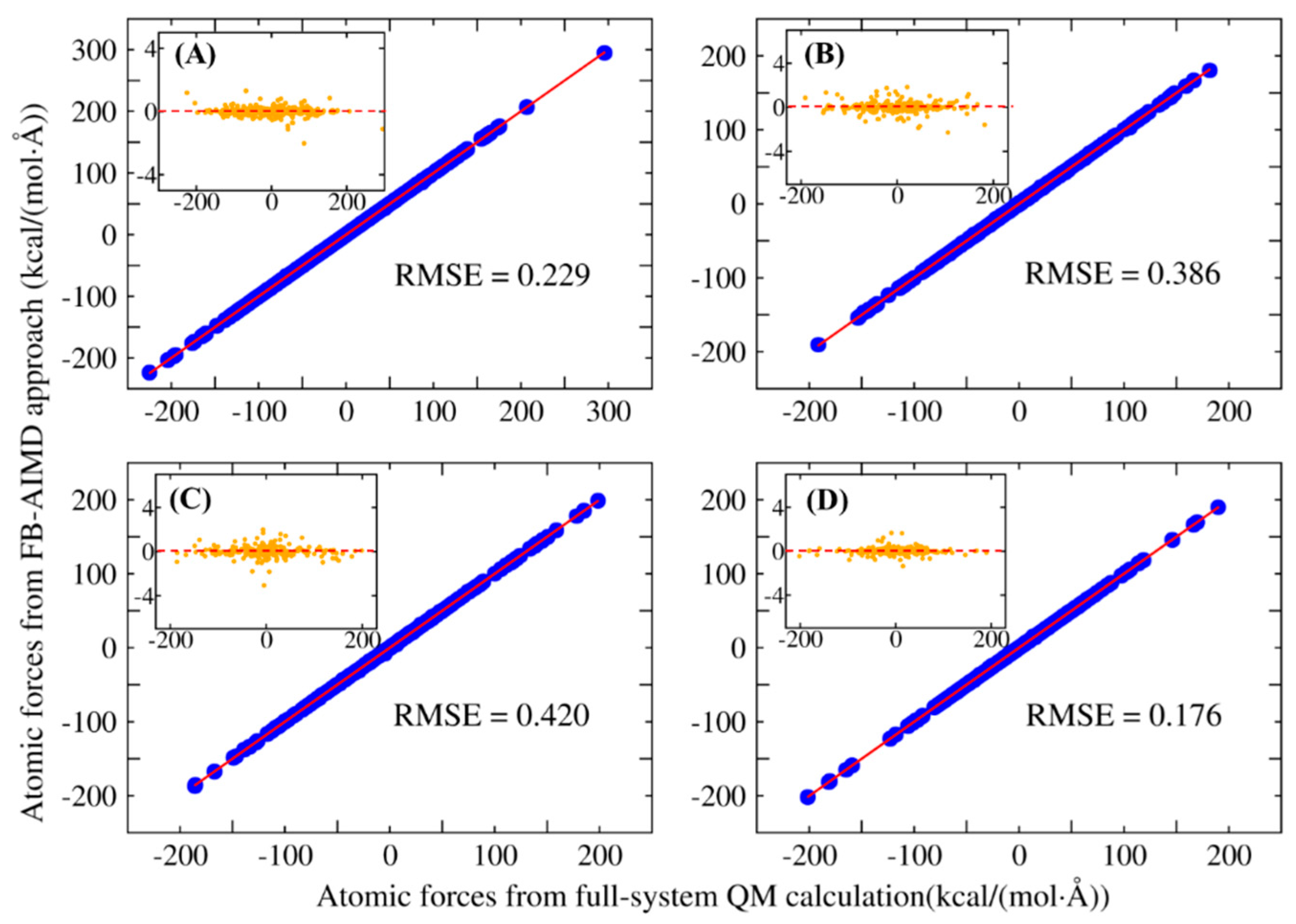
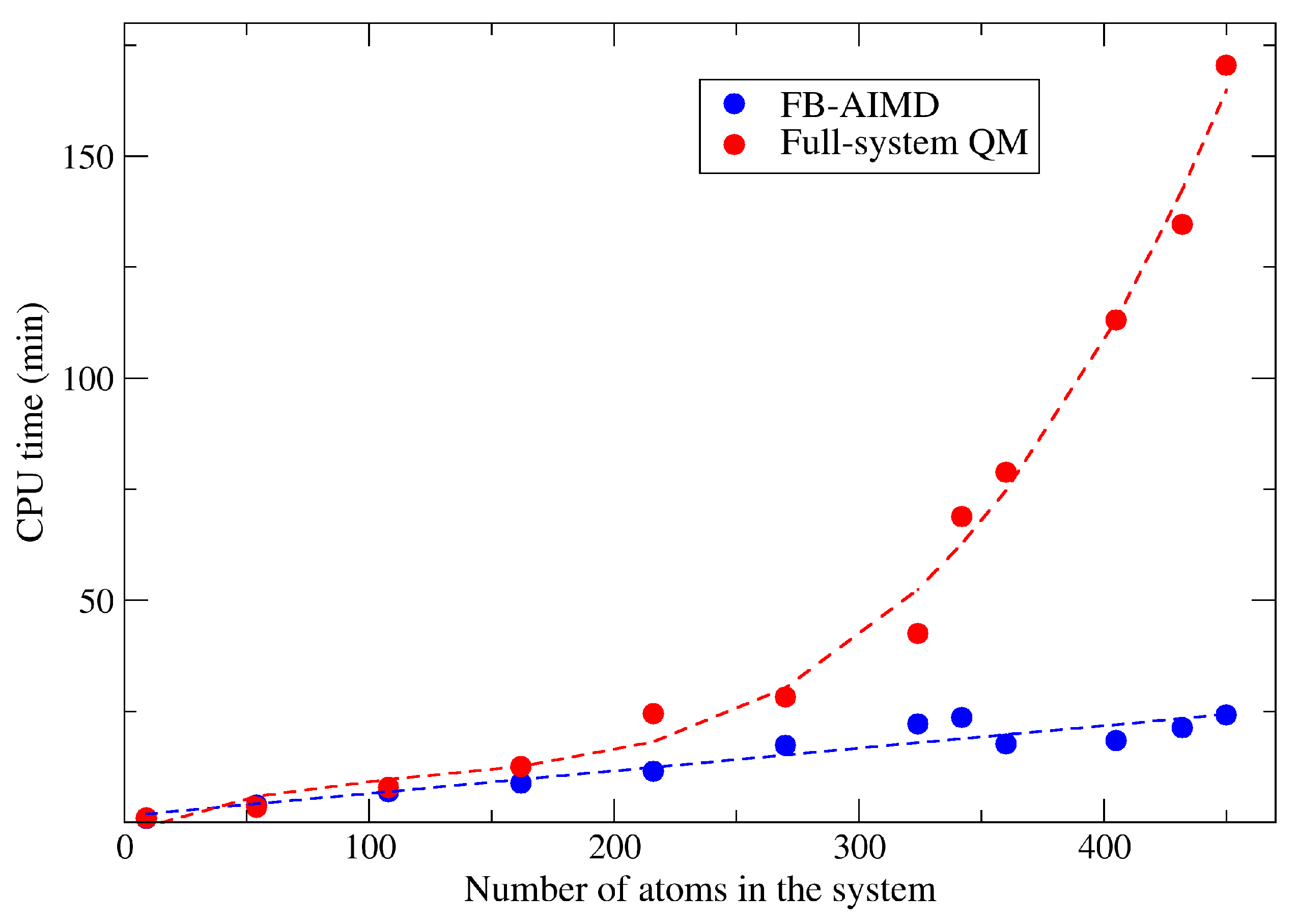
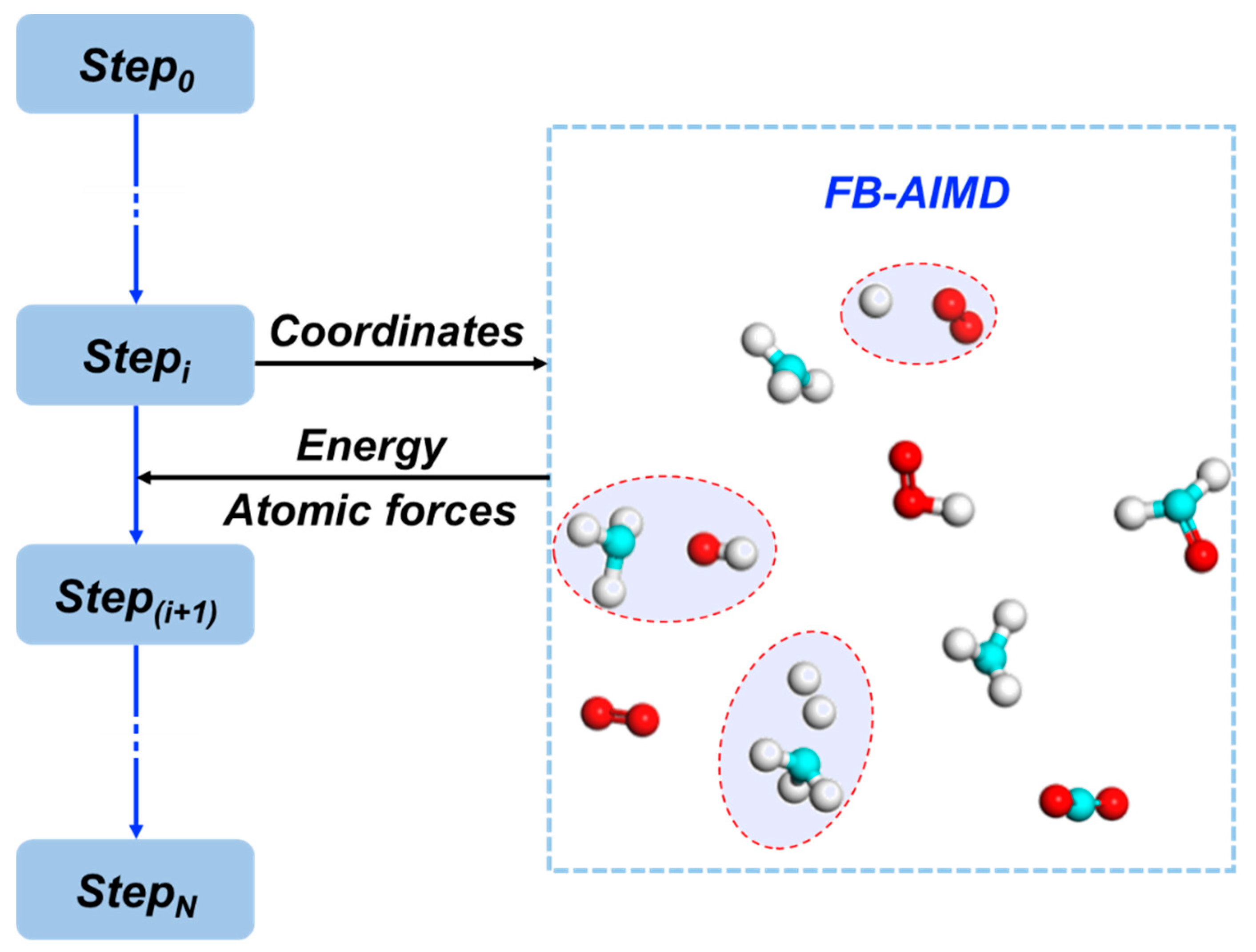
2.1. The Accuracy and Efficiency of the FB-AIMD Method
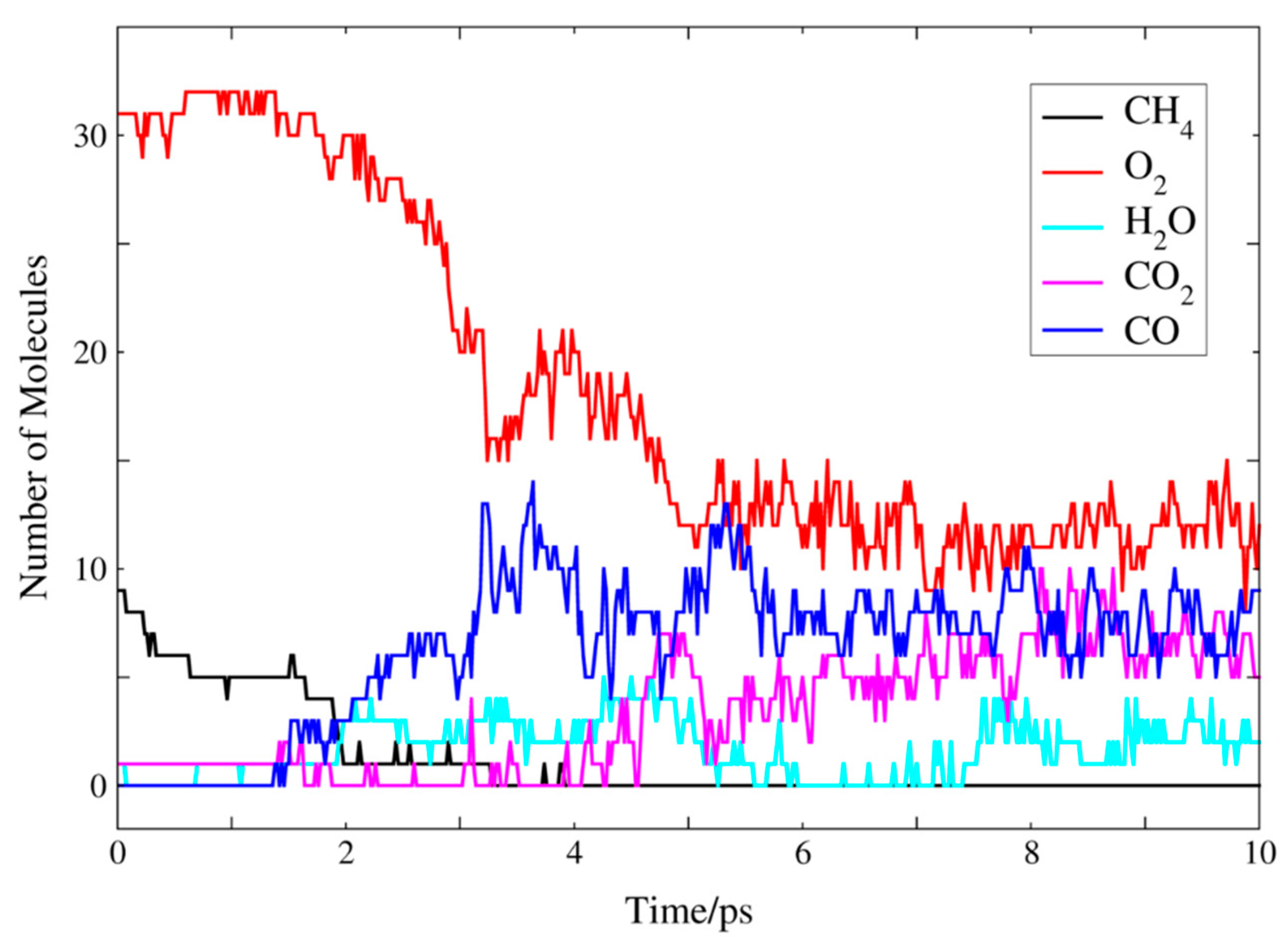
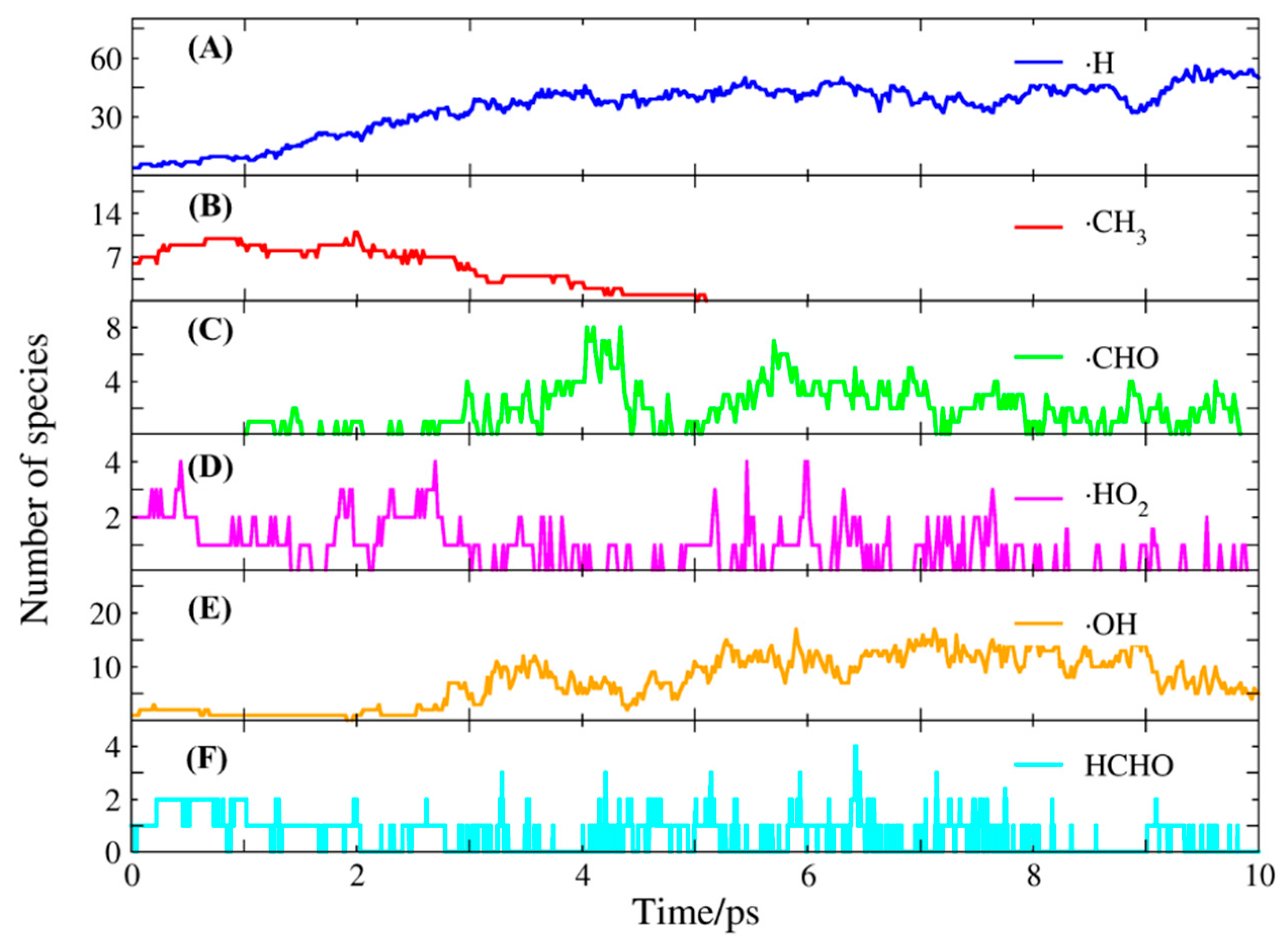
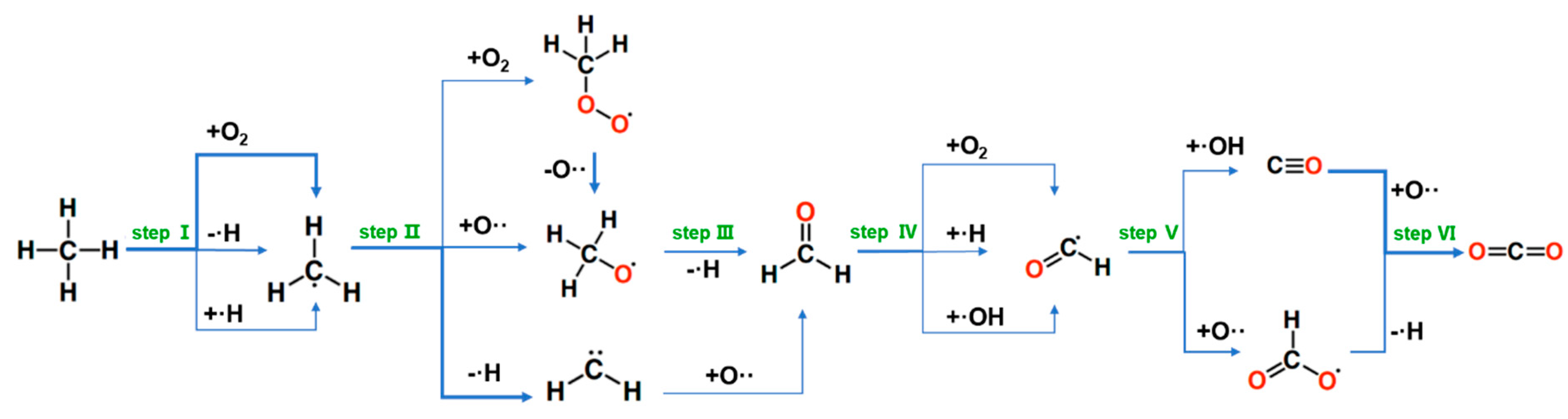
2.2. Molecular Dynamics Simulation of Methane Combustion
3. Theoretical Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Iii, W.A.G. ReaxFF: A Reactive Force Field for Hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.F., Jr.; van Duin, A.C. Atomistic-scale simulations of chemical reactions: Bridging from quantum chemistry to engineering. Nucl. Instrum. Methods Phys. Res. B 2011, 269, 1549–1554. [Google Scholar] [CrossRef]
- Jensen, B.D.; Wise, K.E.; Odegard, G.M. The effect of time step, thermostat, and strain rate on ReaxFF simulations of mechanical failure in diamond, graphene, and carbon nanotube. J. Comput. Chem. 2015, 36, 1587–1596. [Google Scholar] [CrossRef]
- Castro-Marcano, F.; Kamat, A.M.; Russo, M.F., Jr.; van Duin, A.C.; Mathews, J.P. Combustion of an Illinois No. 6 coal char simulated using an atomistic char representation and the ReaxFF reactive force field. Combust. Flame 2012, 159, 1272–1285. [Google Scholar] [CrossRef]
- Zheng, M.; Li, X.; Liu, J.; Wang, Z.; Gong, X.; Guo, L.; Song, W. Pyrolysis of Liulin Coal Simulated by GPU-Based ReaxFF MD with Cheminformatics Analysis. Energy Fuels 2014, 28, 522–534. [Google Scholar] [CrossRef]
- Wang, E.; Ding, J.; Qu, Z.; Han, K. Development of a Reactive Force Field for Hydrocarbons and Application to Iso-octane Thermal Decomposition. Energy Fuels 2017, 32, 901–907. [Google Scholar] [CrossRef]
- Zádor, J.; Taatjes, C.A.; Fernandes, R.X. Kinetics of elementary reactions in low-temperature autoignition chemistry. Prog. Energy Combust. Sci. 2011, 37, 371–421. [Google Scholar] [CrossRef]
- Pilling, M.J. From elementary reactions to evaluated chemical mechanisms for combustion models. Proc. Combust. Inst. 2009, 32, 27–44. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [Green Version]
- Tuckerman, M.E. Ab initiomolecular dynamics: Basic concepts, current trends and novel applications. J. Phys. Condens. Matter 2002, 14, R1297–R1355. [Google Scholar] [CrossRef]
- Schlegel, H.B.; Millam, J.M.; Iyengar, S.S.; Voth, G.A.; Daniels, A.D.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. J. Chem. Phys. 2001, 114, 9758–9763. [Google Scholar] [CrossRef]
- Schlegel, H.B.; Iyengar, S.S.; Li, X.; Millam, J.M.; Voth, G.A.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. III. Comparison with Born–Oppenheimer dynamics. J. Chem. Phys. 2002, 117, 8694–8704. [Google Scholar] [CrossRef] [Green Version]
- Schlegel, H.B. Ab Initio Molecular Dynamics with Born-Oppenheimer and Extended Lagrangian Methods Using Atom Centered Basis Functions. Bull. Korean Chem. Soc. 2003, 24, 837–842. [Google Scholar]
- Iyengar, S.S.; Schlegel, H.B.; Voth, G.A.; Millam, J.M.; Scuseria, G.E.; Frisch, M.J. Ab initio molecular dynamics: Propagating the density matrix with gaussian orbitals. IV. Formal analysis of the deviations from born-oppenheimer dynamics. Isr. J. Chem. 2002, 42, 191–202. [Google Scholar] [CrossRef]
- Rega, N.; Iyengar, S.S.; Voth, G.A.; Schlegel, H.B.; Vreven, T.; Frisch, M.J. Hybrid Ab-Initio/Empirical Molecular Dynamics: Combining the ONIOM Scheme with the Atom-Centered Density Matrix Propagation (ADMP) Approach. J. Phys. Chem. B 2004, 108, 4210–4220. [Google Scholar] [CrossRef]
- Gordon, M.S.; Fedorov, D.G.; Pruitt, S.R.; Slipchenko, L.V. Fragmentation Methods: A Route to Accurate Calculations on Large Systems. Chem. Rev. 2012, 112, 632–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collins, M.A.; Bettens, R.P.A. Energy-Based Molecular Fragmentation Methods. Chem. Rev. 2015, 115, 5607–5642. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, W.; Ma, J. Generalized Energy-Based Fragmentation Approach and Its Applications to Macromolecules and Molecular Aggregates. Accounts Chem. Res. 2014, 47, 2712–2720. [Google Scholar] [CrossRef]
- He, X.; Zhang, J.Z.H. The generalized molecular fractionation with conjugate caps/molecular mechanics method for direct calculation of protein energy. J. Chem. Phys. 2006, 124, 184703. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, T.; Wang, X.; He, X.; Zhang, J.Z.H. Quantum Fragment Based ab Initio Molecular Dynamics for Proteins. J. Chem. Theory Comput. 2015, 11, 5897–5905. [Google Scholar] [CrossRef]
- Wang, X.; Liu, J.; Zhang, J.Z.H.; He, X. Electrostatically Embedded Generalized Molecular Fractionation with Conjugate Caps Method for Full Quantum Mechanical Calculation of Protein Energy. J. Phys. Chem. A 2013, 117, 7149–7161. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; He, X.; Zhang, J.Z. Fragment density functional theory calculation of NMR chemical shifts for proteins with implicit solvation. Phys. Chem. Chem. Phys. 2012, 14, 7837–7845. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Zhang, J.Z.H.; He, X. Automated Fragmentation QM/MM Calculation of Amide Proton Chemical Shifts in Proteins with Explicit Solvent Model. J. Chem. Theory Comput. 2013, 9, 2104–2114. [Google Scholar] [CrossRef]
- Fedorov, D.G.; Kitaura, K. Second order Møller-Plesset perturbation theory based upon the fragment molecular orbital method. J. Chem. Phys. 2004, 121, 2483–2490. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, D.G.; Kitaura, K. Extending the Power of Quantum Chemistry to Large Systems with the Fragment Molecular Orbital Method. J. Phys. Chem. A 2007, 111, 6904–6914. [Google Scholar] [CrossRef]
- Collins, M.A.; Deev, V.A. Accuracy and efficiency of electronic energies from systematic molecular fragmentation. J. Chem. Phys. 2006, 125, 104104. [Google Scholar] [CrossRef]
- Mullin, J.M.; Roskop, L.B.; Pruitt, S.R.; Collins, M.A.; Gordon, M.S. Systematic Fragmentation Method and the Effective Fragment Potential: An Efficient Method for Capturing Molecular Energies. J. Phys. Chem. A 2009, 113, 10040–10049. [Google Scholar] [CrossRef] [Green Version]
- Dahlke, E.E.; Truhlar, D.G. Electrostatically Embedded Many-Body Expansion for Simulations. J. Chem. Theory Comput. 2007, 4, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Exner, T.E.; Mezey, P.G. Evaluation of the field-adapted ADMA approach: Absolute and relative energies of crambin and derivatives. Phys. Chem. Chem. Phys. 2005, 7, 4061–4069. [Google Scholar] [CrossRef]
- Liu, J.; Herbert, J.M. Pair–Pair Approximation to the Generalized Many-Body Expansion: An Alternative to the Four-Body Expansion for ab Initio Prediction of Protein Energetics via Molecular Fragmentation. J. Chem. Theory Comput. 2016, 12, 572–584. [Google Scholar] [CrossRef]
- He, X.; Zhu, T.; Wang, X.W.; Liu, J.F.; Zhang, J.Z.H. Fragment Quantum Mechanical Calculation of Proteins and Its Applications. Acc. Chem. Res. 2014, 47, 2748–2757. [Google Scholar] [CrossRef]
- Barrientos, E.J.; Lapuerta, M.; Boehman, A.L. Group additivity in soot formation for the example of C-5 oxygenated hydrocarbon fuels. Combust. Flame 2013, 160, 1484–1498. [Google Scholar] [CrossRef]
- Wu, J.; Ning, H.; Ma, L.; Zhang, P.; Ren, W. Cascaded group-additivity ONIOM: A new method to approach CCSD(T)/CBS energies of large aliphatic hydrocarbons. Combust. Flame 2019, 201, 31–43. [Google Scholar] [CrossRef]
- Jin, X.; Zhang, J.Z.H.; He, X. Full QM Calculation of RNA Energy Using Electrostatically Embedded Generalized Molecular Fractionation with Conjugate Caps Method. J. Phys. Chem. A 2017, 121, 2503–2514. [Google Scholar] [CrossRef]
- Xu, M.; He, X.; Zhu, T.; Zhang, J.Z.H. A Fragment Quantum Mechanical Method for Metalloproteins. J. Chem. Theory Comput. 2018, 15, 1430–1439. [Google Scholar] [CrossRef]
- Simmie, J.M. Detailed chemical kinetic models for the combustion of hydrocarbon fuels. Prog. Energy Combust. Sci. 2003, 29, 599–634. [Google Scholar] [CrossRef]
- Hughes, K.J.; Turanyi, T.; Clague, A.R.; Pilling, M.J. Development and testing of a comprehensive chemical mechanism for the oxidation of methane. Int. J. Chem. Kinet. 2001, 33, 513–538. [Google Scholar] [CrossRef]
- Dagaut, P.; Boettner, J.C.; Cathonnet, M. Methane Oxidation: Experimental and Kinetic Modeling Study. Combust. Sci. Technol. 1991, 77, 127–148. [Google Scholar] [CrossRef]
- Smith, G.P. GRI-Mech 3.0. 1999. Available online: http://combustion.berkeley.edu/gri-mech/ (accessed on 30 April 2021).
- He, Z.; Li, X.-B.; Liu, L.-M.; Zhu, W. The intrinsic mechanism of methane oxidation under explosion condition: A combined ReaxFF and DFT study. Fuel 2014, 124, 85–90. [Google Scholar] [CrossRef]
- Lee, J.H.; Trimm, D.L. Catalytic combustion of methane. Fuel Process. Technol. 1995, 42, 339–359. [Google Scholar] [CrossRef]
- Su, S.; Beath, A.; Guo, H.; Mallett, C. An assessment of mine methane mitigation and utilisation technologies. Prog. Energy Combust. Sci. 2005, 31, 123–170. [Google Scholar] [CrossRef]
- Zeng, J.; Cao, L.; Chin, C.-H.; Ren, H.; Zhang, J.Z.H.; Zhu, T. ReacNetGenerator: An automatic reaction network generator for reactive molecular dynamics simulations. Phys. Chem. Chem. Phys. 2020, 22, 683–691. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.A.; James, C.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Tarjan, R. Depth-First Search and Linear Graph Algorithms. In Proceedings of the 12th Annual Symposium on Switching and Automata Theory (swat 1971), East Lansing, MI, USA, 13–15 October 1971; pp. 114–121. [Google Scholar] [CrossRef] [Green Version]
- Lao, K.U.; Liu, K.-Y.; Richard, R.M.; Herbert, J.M. Understanding the many-body expansion for large systems. II. Accuracy considerations. J. Chem. Phys. 2016, 144, 164105. [Google Scholar] [CrossRef] [PubMed]
- Dahlke, E.E.; Truhlar, D.G. Electrostatically Embedded Many-Body Correlation Energy, with Applications to the Calculation of Accurate Second-Order Møller-Plesset Perturbation Theory Energies for Large Water Clusters. J. Chem. Theory Comput. 2007, 3, 1342–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Petersson, G.; Nakatsuji, H. Gaussian 16; Revision A; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Haoyu, S.Y.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn-Sham global-hybrid exchange-correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. [Google Scholar]
- LAMMPS. Available online: http://lammps.sandia.gov (accessed on 31 January 2021).
- Wisdom, J.; Holman, M. Symplectic maps for the n-body problem. Astron. J. 1991, 102, 1528–1538. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Number of Atoms | Full-System (au) | FB-AIMD (au) | Energy Deviation (kcal/mol) |
|---|---|---|---|---|
| System Ⅰ | 162 | −6132.09287 | −6132.08737 | 3.452 |
| System Ⅱ | 112 | −4762.42315 | −4762.41884 | 2.703 |
| System Ⅲ | 96 | −4232.35592 | −4232.35346 | 1.541 |
| System Ⅳ | 114 | −1415.00577 | −1415.00222 | 2.229 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, L.; Zeng, J.; Xu, M.; Chin, C.-H.; Zhu, T.; Zhang, J.Z.H. Fragment-Based Ab Initio Molecular Dynamics Simulation for Combustion. Molecules 2021, 26, 3120. https://doi.org/10.3390/molecules26113120
Cao L, Zeng J, Xu M, Chin C-H, Zhu T, Zhang JZH. Fragment-Based Ab Initio Molecular Dynamics Simulation for Combustion. Molecules. 2021; 26(11):3120. https://doi.org/10.3390/molecules26113120
Chicago/Turabian StyleCao, Liqun, Jinzhe Zeng, Mingyuan Xu, Chih-Hao Chin, Tong Zhu, and John Z. H. Zhang. 2021. "Fragment-Based Ab Initio Molecular Dynamics Simulation for Combustion" Molecules 26, no. 11: 3120. https://doi.org/10.3390/molecules26113120
APA StyleCao, L., Zeng, J., Xu, M., Chin, C. -H., Zhu, T., & Zhang, J. Z. H. (2021). Fragment-Based Ab Initio Molecular Dynamics Simulation for Combustion. Molecules, 26(11), 3120. https://doi.org/10.3390/molecules26113120