
Pharmacophore-Based Virtual Screening of Novel Competitive Inhibitors of the Neurodegenerative Disease Target Kynurenine-3-Monooxygenase


Abstract
:
1. Introduction
2. Results
2.1. Homology Modeling and Structure Validation
- Model 1: Competitive inhibitor model (Type II), which represents the Ro 61-8084 binding mode in ScKMO. As mentioned previously, Ro 61-8084 inhibits KMO without producing H2O2 in ScKMO and hKMO.
- Model 2: Non-substrate effector model (Type I) created based on a high-resolution crystal structure of PfKMO bound to the native substrate L-kyn.
- Model 3: Competitive inhibitor model (Type II) based on PfKMO, in which FAD is trapped in an unproductive, tilted conformation. This inhibitor binding mode is also reported not to cause H2O2 production.
2.2. Reference Inhibitor Docking Studies
2.3. Pharmacophore Generation
2.4. Virtual Screening
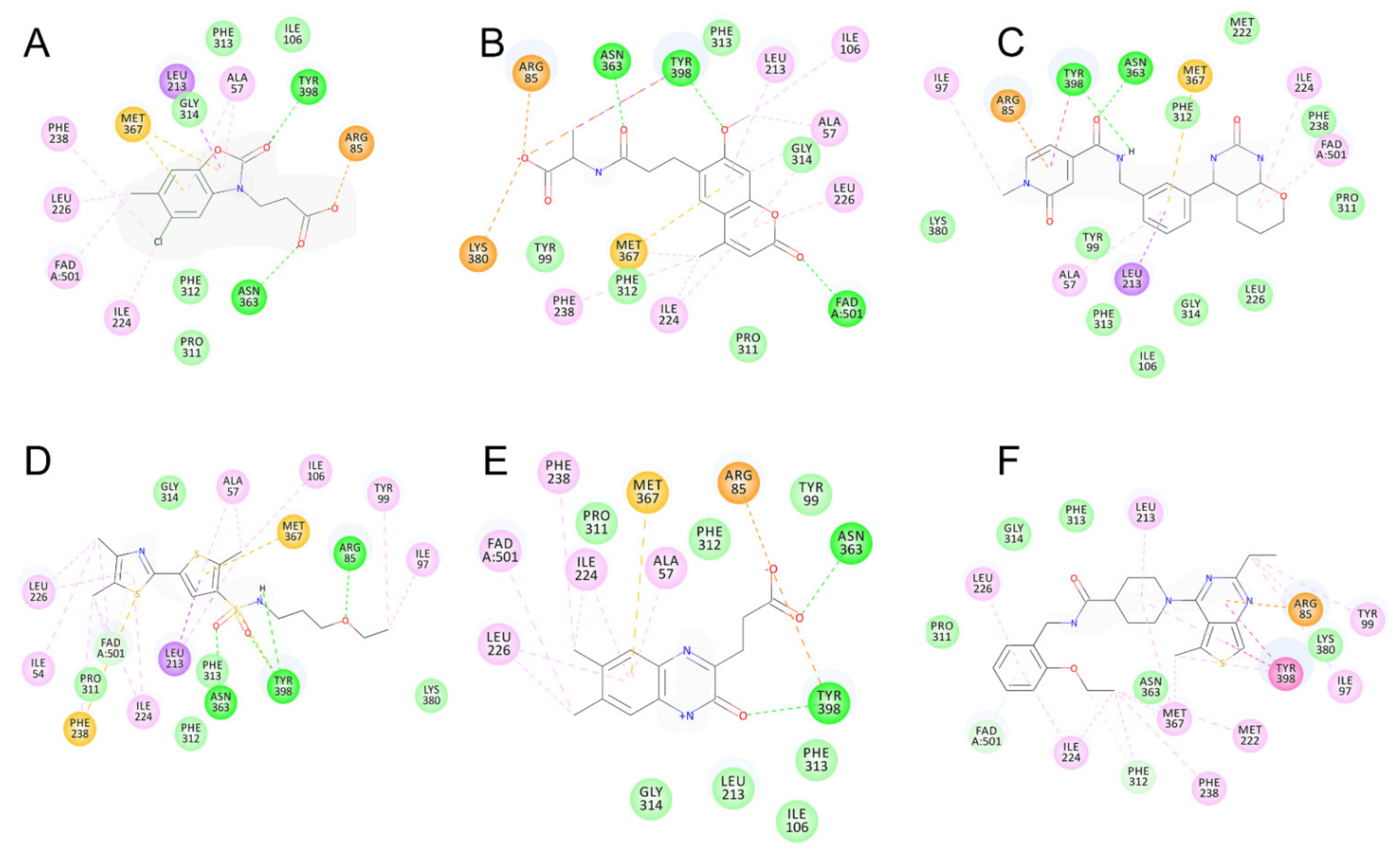
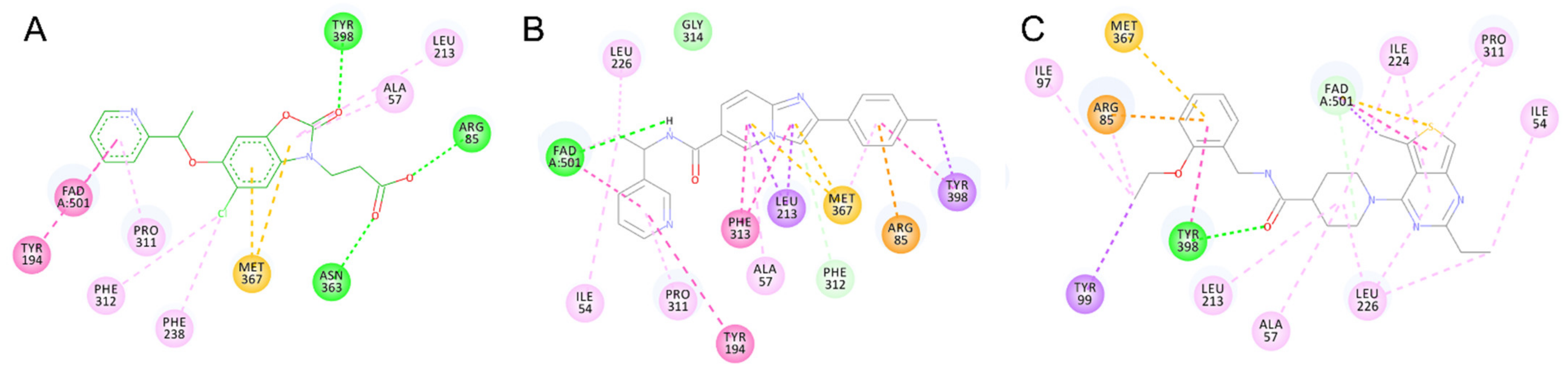
2.5. Identified Novel KMO Inhibitors
3. Discussion
4. Materials and Methods
4.1. hKMO Homology Modeling
4.2. Docking Protocol
4.3. Pharmacophore Generation and Validation
4.4. Compound Library Generation
4.5. Virtual Screening
4.6. Structure Similarity and BBB Permeability Calculations
4.7. KMO Inhibitor Screening Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maddison, D.C.; Giorgini, F. The kynurenine pathway and neurodegenerative disease. Semin. Cell Dev. Biol. 2015, 40, 134–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, R.S.; Iradukunda, E.C.; Hughes, T.; Bowen, J.P. Modulation of Enzyme Activity in the Kynurenine Pathway by Kynurenine Monooxygenase Inhibition. Front. Mol. Biosci. 2019, 6, 3. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.-Y.; Li, Z.; Wang, H.-M.; Liu, J.; Chen, L.; Zhang, H.-W.; Wang, X.; Yang, J. Targeting the kynurenine pathway as a potential strategy to prevent and treat Alzheimer’s disease. Med. Hypotheses 2011, 77, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Thevandavakkam, M.A.; Schwarcz, R.; Muchowski, P.J.; Giorgini, F. Targeting Kynurenine 3-Monooxygenase (KMO): Implications for Therapy in Huntingtons Disease. CNS Neurol. Disord. Drug Targets 2010, 9, 791–800. [Google Scholar] [CrossRef]
- Campesan, S.; Green, E.W.; Breda, C.; Sathyasaikumar, K.V.; Muchowski, P.J.; Schwarcz, R.; Kyriacou, C.P.; Giorgini, F. The Kynurenine Pathway Modulates Neurodegeneration in a Drosophila Model of Huntington’s Disease. Curr. Biol. 2011, 21, 961–966. [Google Scholar] [CrossRef] [Green Version]
- Venkatesan, D.; Iyer, M.; Narayanasamy, A.; Siva, K.; Vellingiri, B. Kynurenine pathway in Parkinson’s disease—An update. eNeurologicalSci 2020, 21, 100270. [Google Scholar] [CrossRef]
- Kindler, J.; Lim, C.K.; Weickert, C.S.; Boerrigter, D.; Galletly, C.; Liu, D.; Jacobs, K.R.; Balzan, R.; Bruggemann, J.; O’Donnell, M.; et al. Dysregulation of kynurenine metabolism is related to proinflammatory cytokines, attention, and prefrontal cortex volume in schizophrenia. Mol. Psychiatry 2020, 25, 2860–2872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wonodi, I.; Stine, O.C.; Sathyasaikumar, K.V.; Roberts, R.C.; Mitchell, B.D.; Hong, L.E.; Kajii, Y.; Thaker, G.K.; Schwarcz, R. Downregulated Kynurenine 3-Monooxygenase Gene Expression and Enzyme Activity in Schizophrenia and Genetic Association with Schizophrenia Endophenotypes. Arch. Gen. Psychiatry 2011, 68, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.L.; Llenos, I.C.; Cwik, M.; Walkup, J.; Weis, S. Alterations in kynurenine precursor and product levels in schizophrenia and bipolar disorder. Neurochem. Int. 2008, 52, 1297–1303. [Google Scholar] [CrossRef]
- Parrott, J.M.; Redus, L.; Coelho, D.S.; Morales, J.; Gao, X.; O’Connor, J.C. Neurotoxic kynurenine metabolism is increased in the dorsal hippocampus and drives distinct depressive behaviors during inflammation. Transl. Psychiatry 2016, 6, e918. [Google Scholar] [CrossRef]
- Laumet, G.; Zhou, W.; Dantzer, R.; Edralin, J.D.; Huo, X.; Budac, D.P.; Lee, A.W.; Heijnen, C.J.; Audie, L.; Antonio, S.; et al. Upregulation of neuronal kynurenine 3-monooxygenase mediates depression-like behavior in a mouse model of neuropathic pain Geoffroy. Brain Behav. Immun. 2018, 66, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Rojewska, E.; Ciapała, K.; Piotrowska, A.; Makuch, W.; Mika, J. Pharmacological Inhibition of Indoleamine 2,3-Dioxygenase-2 and Kynurenine 3-Monooxygenase, Enzymes of the Kynurenine Pathway, Significantly Diminishes Neuropathic Pain in a Rat Model. Front. Pharmacol. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojewska, E.; Piotrowska, A.; Makuch, W.; Przewlocka, B.; Mika, J. Pharmacological kynurenine 3-monooxygenase enzyme inhibition significantly reduces neuropathic pain in a rat model. Neuropharmacology 2016, 102, 80–91. [Google Scholar] [CrossRef]
- Jovanovic, F.; Candido, K.D.; Knezevic, N.N. The Role of the Kynurenine Signaling Pathway in Different Chronic Pain Conditions and Potential Use of Therapeutic Agents. Int. J. Mol. Sci. 2020, 21, 6045. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Zhang, Y.; You, H.; Tao, X.; Wang, C.; Jin, G.; Wang, N.; Ruan, H.; Gu, D.; Huo, X.; et al. Prognostic significance of kynurenine 3-monooxygenase and effects on proliferation, migration and invasion of human hepatocellular carcinoma. Sci. Rep. 2015, 5, srep10466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mole, D.J.; Webster, S.P.; Uings, I.; Zheng, X.; Binnie, M.; Wilson, K.; Hutchinson, J.P.; Mirguet, O.; Walker, A.P.; Beaufils, B.; et al. Kynurenine-3-monooxygenase inhibition prevents multiple organ failure in rodent models of acute pancreatitis. Nat. Med. 2016, 22, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Platten, M.; Nollen, E.A.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef]
- Röver, S.; Cesura, A.M.; Huguenin, P.; Kettler, R.; Szente, A. Synthesis and Biochemical Evaluation ofN-(4-Phenylthiazol-2-yl)benzenesulfonamides as High-Affinity Inhibitors of Kynurenine 3-Hydroxylase. J. Med. Chem. 1997, 40, 4378–4385. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Suda, H.; Kassai, M.; Endo, M.; Deai, Y.; Yahata, M.; Miyajima, M.; Isobe, Y. N-(6-phenylpyridazin-3-yl)benzenesulfonamides as highly potent, brain-permeable, and orally active kynurenine monooxygenase inhibitors. Bioorg. Med. Chem. Lett. 2021, 33, 127753. [Google Scholar] [CrossRef]
- Toledo-Sherman, L.M.; Prime, M.E.; Mrzljak, L.; Beconi, M.G.; Beresford, A.; Brookfield, F.A.; Brown, C.J.; Cardaun, I.; Courtney, S.M.; Dijkman, U.; et al. Development of a Series of Aryl Pyrimidine Kynurenine Monooxygenase Inhibitors as Potential Therapeutic Agents for the Treatment of Huntington’s Disease. J. Med. Chem. 2015, 58, 1159–1183. [Google Scholar] [CrossRef]
- Giordani, A.; Corti, L.; Cini, M.; Bormetti, R.; Marconi, M.; Veneroni, O.; Speciale, C.; Varasi, M. Enantiospecific Synthesis and in vitro Activity of Selective Inhibitors of Rat Brain Kynureninase and Kynurenine-3-Hydroxylase. In Recent Advances in Tryptophan Research: Tryptophan and Serotonin Pathways; Filippini, G.A., Costa, C.V.L., Bertazzo, A., Eds.; Springer: Boston, MA, USA, 1996; pp. 531–534. ISBN 978-1-4613-0381-7. [Google Scholar]
- Giordani, A.; Pevarello, P.; Cini, M.; Bormetti, R.; Greco, F.; Toma, S.; Speciale, C.; Varasi, M. 4-Phenyl-4-oxo-butanoic acid derivatives inhibitors of kynurenine 3-hydroxylase. Bioorg. Med. Chem. Lett. 1998, 8, 2907–2912. [Google Scholar] [CrossRef]
- Liddle, J.; Beaufils, B.; Binnie, M.; Bouillot, A.; Denis, A.A.; Hann, M.M.; Haslam, C.P.; Holmes, D.S.; Hutchinson, J.P.; Kranz, M.; et al. The discovery of potent and selective kynurenine 3-monooxygenase inhibitors for the treatment of acute pancreatitis. Bioorg. Med. Chem. Lett. 2017, 27, 2023–2028. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.L.; Ancellin, N.; Beaufils, B.; Bergeal, M.; Binnie, M.; Bouillot, A.; Clapham, D.; Denis, A.; Haslam, C.P.; Holmes, D.S.; et al. Development of a Series of Kynurenine 3-Monooxygenase Inhibitors Leading to a Clinical Candidate for the Treatment of Acute Pancreatitis. J. Med. Chem. 2017, 60, 3383–3404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amori, L.; Guidetti, P.; Pellicciari, R.; Kajii, Y.; Schwarcz, R. On the relationship between the two branches of the kynurenine pathway in the rat brainin vivo. J. Neurochem. 2009, 109, 316–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwilling, D.; Huang, S.-Y.; Sathyasaikumar, K.V.; Francesca, V.; Notarangelo, F.M.; Guidetti, P.; Wu, H.-Q.; Lee, J.; Truong, J.; Andrews-Zwilling, Y.; et al. Kynurenine 3-Monooxygenase Inhibition in Blood Ameliorates Neurodegeneration. Cell 2011, 145, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Speciale, C.; Wu, H.-Q.; Cini, M.; Marconi, M.; Varasi, M.; Schwarcz, R. (R,S)-3,4-dichlorobenzoylalanine (FCE 28833A) causes a large and persistent increase in brain kynurenic acid levels in rats. Eur. J. Pharmacol. 1996, 315, 263–267. [Google Scholar] [CrossRef]
- Lovejoy, D.B.; Jacobs, K.R. Inhibiting the kynurenine pathway in spinal cord injury: Multiple therapeutic potentials? Neural Regen. Res. 2018, 13, 2073–2076. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Collier, M.E.W.; Heyes, D.J.; Giorgini, F.; Scrutton, N.S. Advantages of brain penetrating inhibitors of kynurenine-3-monooxygenase for treatment of neurodegenerative diseases. Arch. Biochem. Biophys. 2021, 697, 108702. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Sakuma, M.; Deora, G.S.; Levy, C.W.; Klausing, A.; Breda, C.; Read, K.D.; Edlin, C.D.; Ross, B.P.; Muelas, M.W.; et al. A brain-permeable inhibitor of the neurodegenerative disease target kynurenine 3-monooxygenase prevents accumulation of neurotoxic metabolites. Commun. Biol. 2019, 2, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimasu, S.; Yamagishi, H.; Kubo, S.; Kiyohara, M.; Matsuda, T.; Yahata, T.; Thomson, H.A.; Hupp, C.D.; Liu, J.; Okuda, T.; et al. Full-length in meso structure and mechanism of rat kynurenine 3-monooxygenase inhibition. Commun. Biol. 2021, 4, 1–10. [Google Scholar] [CrossRef]
- Crozier-Reabe, K.R.; Phillips, R.S.; Moran, G.R. Kynurenine 3-Monooxygenase fromPseudomonas fluorescens: Substrate-like Inhibitors both Stimulate Flavin Reduction and Stabilize the Flavin−Peroxo Intermediate yet Result in the Production of Hydrogen Peroxide. Biochemistry 2008, 47, 12420–12433. [Google Scholar] [CrossRef]
- Kim, H.T.; Na, B.K.; Chung, J.; Kim, S.; Kwon, S.K.; Cha, H.; Son, J.; Cho, J.M.; Hwang, K.Y. Structural Basis for Inhibitor-Induced Hydrogen Peroxide Production by Kynurenine 3-Monooxygenase. Cell Chem. Biol. 2018, 25, 426–438.e4. [Google Scholar] [CrossRef] [PubMed]
- Holtmann, D.; Hollmann, F. The Oxygen Dilemma: A Severe Challenge for the Application of Monooxygenases? ChemBioChem 2016, 17, 1391–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutchinson, J.P.; Rowland, P.; Taylor, M.R.D.; Christodoulou, E.M.; Haslam, C.; Hobbs, C.I.; Holmes, D.S.; Homes, P.; Liddle, J.; Mole, D.J.; et al. Structural and mechanistic basis of differentiated inhibitors of the acute pancreatitis target kynurenine-3-monooxygenase. Nat. Commun. 2017, 8, 15827. [Google Scholar] [CrossRef] [Green Version]
- Shave, S.; McGuire, K.; Pham, N.T.; Mole, D.J.; Webster, S.P.; Auer, M. Diclofenac Identified as a Kynurenine 3-Monooxygenase Binder and Inhibitor by Molecular Similarity Techniques. ACS Omega 2018, 3, 2564–2568. [Google Scholar] [CrossRef]
- Amin, S.A.; Adhikari, N.; Jha, T.; Gayen, S. First molecular modeling report on novel arylpyrimidine kynurenine monooxygenase inhibitors through multi-QSAR analysis against Huntington’s disease: A proposal to chemists! Bioorg. Med. Chem. Lett. 2016, 26, 5712–5718. [Google Scholar] [CrossRef]
- Phillips, R.S.; Anderson, A.D.; Gentry, H.G.; Güner, O.F.; Bowen, J.P. Substrate and inhibitor specificity of kynurenine monooxygenase from Cytophaga hutchinsonii. Bioorg. Med. Chem. Lett. 2017, 27, 1705–1708. [Google Scholar] [CrossRef]
- Özkılıç, Y.; Tüzün, N.Ş. In silico methods predict new blood-brain barrier permeable structure for the inhibition of kynurenine 3-monooxygenase. J. Mol. Graph. Model. 2020, 100, 1–15. [Google Scholar] [CrossRef]
- Winkler, D.; Beconi, M.; Toledo-Sherman, L.M.; Prime, M.; Ebneth, A.; Dominguez, C.; Muñoz-Sanjuan, I. Development of LC/MS/MS, High-Throughput Enzymatic and Cellular Assays for the Characterization of Compounds That Inhibit Kynurenine Monooxygenase (KMO). J. Biomol. Screen. 2013, 18, 879–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, K.R.; Guillemin, G.J.; Lovejoy, D.B. Development of a Rapid Fluorescence-Based High-Throughput Screening Assay to Identify Novel Kynurenine 3-Monooxygenase Inhibitor Scaffolds. SLAS Discov. Adv. Life Sci. R&D 2018, 23, 554–560. [Google Scholar] [CrossRef] [Green Version]
- Fujigaki, H.; Yamamoto, Y.; Saito, K. L-Tryptophan-kynurenine pathway enzymes are therapeutic target for neuropsychiatric diseases: Focus on cell type differences. Neuropharmacology 2017, 112, 264–274. [Google Scholar] [CrossRef]
- Gao, J.; Yao, L.; Xia, T.; Liao, X.; Zhu, D.; Xiang, Y. Biochemistry and structural studies of kynurenine 3-monooxygenase reveal allosteric inhibition by Ro 61-8048. FASEB J. 2018, 32, 2036–2045. [Google Scholar] [CrossRef] [Green Version]
- Hirai, K.; Kuroyanagi, H.; Tatebayashi, Y.; Hayashi, Y.; Hirabayashi-Takahashi, K.; Saito, K.; Haga, S.; Uemura, T.; Izumi, S. Dual role of the carboxyl-terminal region of pig liver L-kynurenine 3-monooxygenase: Mitochondrial-targeting signal and enzymatic activity. J. Biochem. 2010, 148, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.; Levy, C.; Heyes, D.J.; Lafite, P.; Outeiro, T.F.; Giorgini, F.; Leys, D.; Scrutton, N.S. Structural basis of kynurenine 3-monooxygenase inhibition. Nature 2013, 496, 382–385. [Google Scholar] [CrossRef]
- Wilson, K.; Mole, D.J.; Binnie, M.; Homer, N.Z.M.; Zheng, X.; Yard, B.A.; Iredale, J.P.; Auer, M.; Webster, S.P. Bacterial expression of human kynurenine 3-monooxygenase: Solubility, activity, purification. Protein Expr. Purif. 2014, 95, 96–103. [Google Scholar] [CrossRef] [Green Version]
- Breton, J.; Avanzi, N.; Magagnin, S.; Covini, N.; Magistrelli, G.; Cozzi, L.; Isacchi, A. Functional characterization and mechanism of action of recombinant human kynurenine 3-hydroxylase. JBIC J. Biol. Inorg. Chem. 2000, 267, 1092–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özkılıç, Y.; Tüzün, N.Ş. Mechanism of Kynurenine 3-Monooxygenase-Catalyzed Hydroxylation Reaction: A Quantum Cluster Approach. J. Phys. Chem. A 2019, 123, 3149–3159. [Google Scholar] [CrossRef]
- Beaupre, B.A.; Reabe, K.R.; Roman, J.V.; Moran, G.R. Hydrogen movements in the oxidative half-reaction of kynurenine 3-monooxygenase from Pseudomonas fluorescens reveal the mechanism of hydroxylation. Arch. Biochem. Biophys. 2020, 690, 108474. [Google Scholar] [CrossRef]
- Fischer, A.; Smieško, M.; Sellner, M.; Lill, M.A. Decision Making in Structure-Based Drug Discovery: Visual Inspection of Docking Results. J. Med. Chem. 2021, 64, 2489–2500. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA, Dassault Systèmes. Discovery Studio Modeling Environment, Release 2019; Dassault Systèmes: San Diego, CA, USA, 2021. [Google Scholar]
- Shen, M.-Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, R.; Šali, A. Evaluation of comparative protein structure modeling by MODELLER-3. Proteins Struct. Funct. Genet. 1997, 29, 50–58. [Google Scholar] [CrossRef]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles Roland. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017-3; Maestro Schrödinger LLC: New York, NY, USA, 2021.
- Wu, G.; Robertson, D.H.; Brooks, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER -A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Lauri, G. Prediction of intestinal permeability. Adv. Drug Deliv. Rev. 2002, 54, 273–289. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Validation Parameter | Model 1 | Model 2 | Model 3 |
|---|---|---|---|
| Ramachandran plot residues in allowed region, % | 95.5 | 97 | 96.7 |
| PDF total energy | −8089.32 | 2595 | |
| RMSD to template, Å | 0.939 | 0.503 | |
| 3D-Profile score, verify score/expected high score | 165.55/170.026 | 169.78/202.672 | 171.19/202.672 |
| Pharmacophore Characteristic | Model 1 | Model 2 | Model 3 |
|---|---|---|---|
| Pharmacophore Ro 61-8084 Based | Pharmacophore GSK428 Based | Pharmacophore GSK775 Based | |
| Total Features | 5 | 5 | 5 |
| Feature Set * | AAADH | AAHNS | AHHNS |
| Sensitivity | 0.846 | 0.956 | 1.00 |
| Specificity | 0.816 | 0.843 | 0.981 |
| AUC ** | 0.858 | 0.951 | 0.999 |
| № | Screening Step | Model 1 | Model 2 | Model3 | Total |
|---|---|---|---|---|---|
| 1 | Pharmacophore filter | 143,205 | 1043 | 1109 | 145,357 |
| 2 | Libdock filter | 120,006 | 1039 | 1058 | 122,103 |
| 3 | Toxicity filter | 61,067 | 654 | 672 | 62,393 |
| 4 | Selected for docking | 6925 | 654 | 672 | 8251 |
| 5 | Final Selection | 163 | 50 | 42 | 255 |
| Vendor Code | Vendor | Structure | MW | % Inhibition, µM | -CDOCKER Score, kcal/mol | ||||
|---|---|---|---|---|---|---|---|---|---|
| 10 | 1 | 0.1 | Model 1 | Model 2 | Model 3 | ||||
| P323-0389 | Chemdiv |  VS1 | 356.43 | 97.8 | 21.3 | 6.3 | 27.49 | 28.01 | 27.56 |
| D715-2857 | Chemdiv |  VS2 | 332.33 | 109.4 | 23.9 | 3.1 | * ND | 40.48 | ND |
| Z354-0210 | Chemdiv |  VS3 | 396.45 | 107.5 | 15.1 | 3.6 | 37.99 | 37.53 | ND |
| L921-0479 | Chemdiv |  VS4 | 374.53 | 109.7 | 21.2 | 8.4 | 42.11 | 42.32 | ND |
| BDE 33672567 | Asinex |  VS5 | 246.27 | 66.3 | 17.8 | 16.8 | ND | 45.25 | ND |
| F6548-0495 | Life Chemical |  VS6 | 438.59 | 60.4 | 27.1 | 4.3 | 41.52 | 25.82 | 35.53 |
 Ro 61-8084 | 421.449 | 97.3 | 60.5 | 7.5 | 23.43 | ND | ND | ||
 L-kyn | 208.22 | ND | 46.09 | ND | |||||
 GSK065 | 376.77 | ND | ND | 43.07 | |||||
| Comp | Presumed Binding Mode | Tanimoto Similarity | logBB | Penetration Ability |
|---|---|---|---|---|
| VS1 | Competitive inhibitor | 0.709 | −0.057 | Medium |
| VS2 | Non-substrate effector | 0.532 | −1.574 | Low |
| VS3 | Undetermined, both possible | 0.395 | −1.677 | Low |
| VS4 | Undetermined, both possible | 0.482 | −0.641 | Low |
| VS5 | Non-substrate effector | 0.605 | −1.42 | Low |
| VS6 | Competitive inhibitor | 0.596 | 0.02 | High |
| Vendor | Library Name | Total Compounds | Available at |
|---|---|---|---|
| ChemDiv | CNS Library | 22,306 | https://www.chemdiv.com/Accessed Accessed 17 May 2019 |
| Representative Diversity Library | 150,000 | ||
| 3D-Biodiversity Library | 30,073 | ||
| New Chemistry (NC) Library | 325,760 | ||
| Smart Library | 52,504 | ||
| Life Chemical | Stock HTS compounds | 45,589 | https://lifechemicals.com/Accessed Accessed 17 May 2019 |
| CNS screening Library | 7024 | ||
| Low MW Fragment Library | 36,080 | ||
| Asinex | Signature Library | 7815 | http://www.asinex.com/Accessed Accessed 6 May 2019 |
| BioDesign Library | 195,039 | ||
| Specs | Screening Compounds (10mg) | 210,419 | https://www.specs.net/Accessed Accessed 7 May 2019 |
| Building blocks Library | 8985 | ||
| Total | 1,091,594 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gotina, L.; Seo, S.H.; Kim, C.W.; Lim, S.M.; Pae, A.N. Pharmacophore-Based Virtual Screening of Novel Competitive Inhibitors of the Neurodegenerative Disease Target Kynurenine-3-Monooxygenase. Molecules 2021, 26, 3314. https://doi.org/10.3390/molecules26113314
Gotina L, Seo SH, Kim CW, Lim SM, Pae AN. Pharmacophore-Based Virtual Screening of Novel Competitive Inhibitors of the Neurodegenerative Disease Target Kynurenine-3-Monooxygenase. Molecules. 2021; 26(11):3314. https://doi.org/10.3390/molecules26113314
Chicago/Turabian StyleGotina, Lizaveta, Seon Hee Seo, Chae Won Kim, Sang Min Lim, and Ae Nim Pae. 2021. "Pharmacophore-Based Virtual Screening of Novel Competitive Inhibitors of the Neurodegenerative Disease Target Kynurenine-3-Monooxygenase" Molecules 26, no. 11: 3314. https://doi.org/10.3390/molecules26113314
APA StyleGotina, L., Seo, S. H., Kim, C. W., Lim, S. M., & Pae, A. N. (2021). Pharmacophore-Based Virtual Screening of Novel Competitive Inhibitors of the Neurodegenerative Disease Target Kynurenine-3-Monooxygenase. Molecules, 26(11), 3314. https://doi.org/10.3390/molecules26113314