
Synthesis and Antinociceptive Effect of Some Thiazole-Piperazine Derivatives: Involvement of Opioidergic System in the Activity

 , , ,
, , ,
Abstract
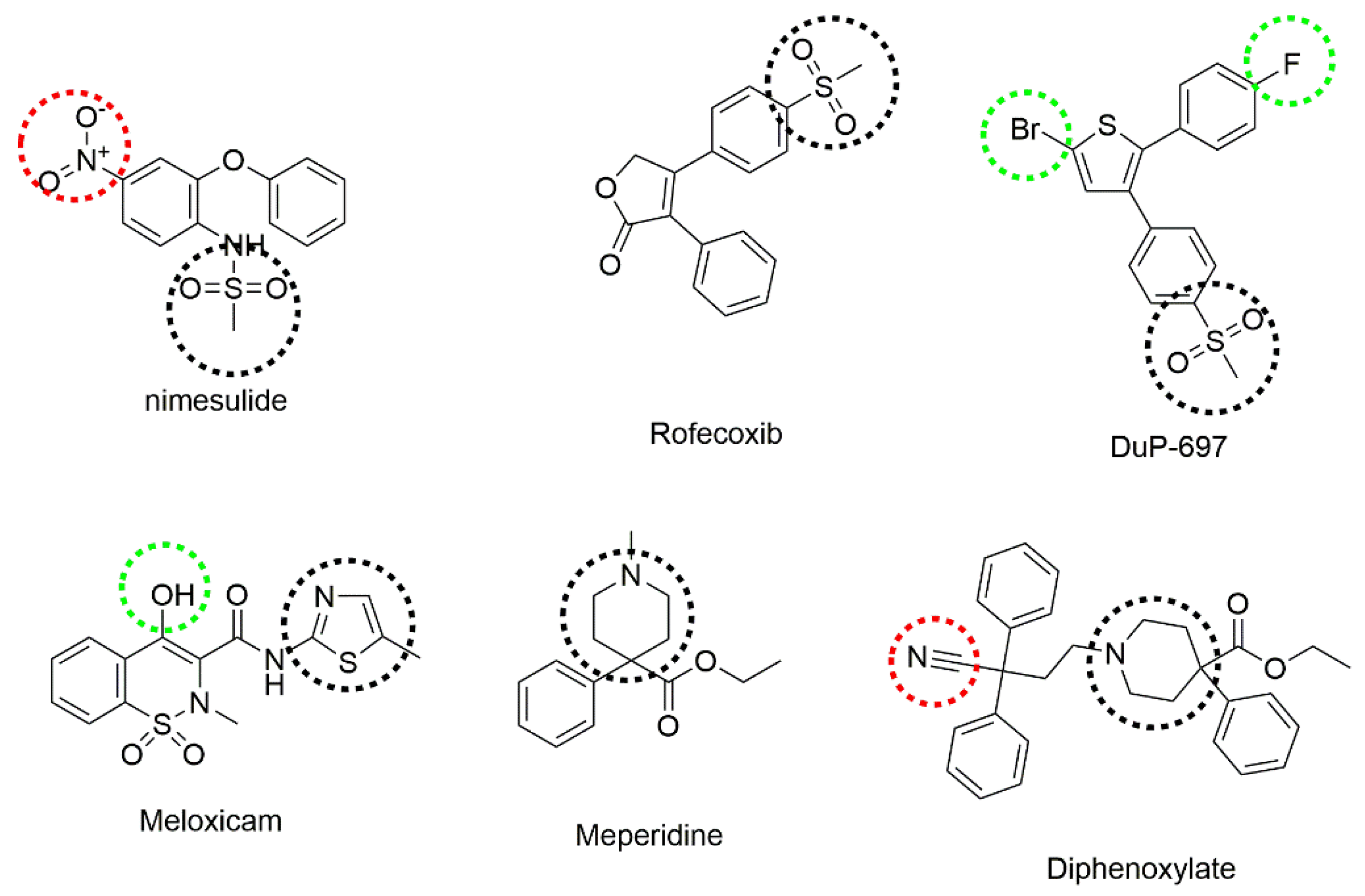
:1. Introduction
2. Results
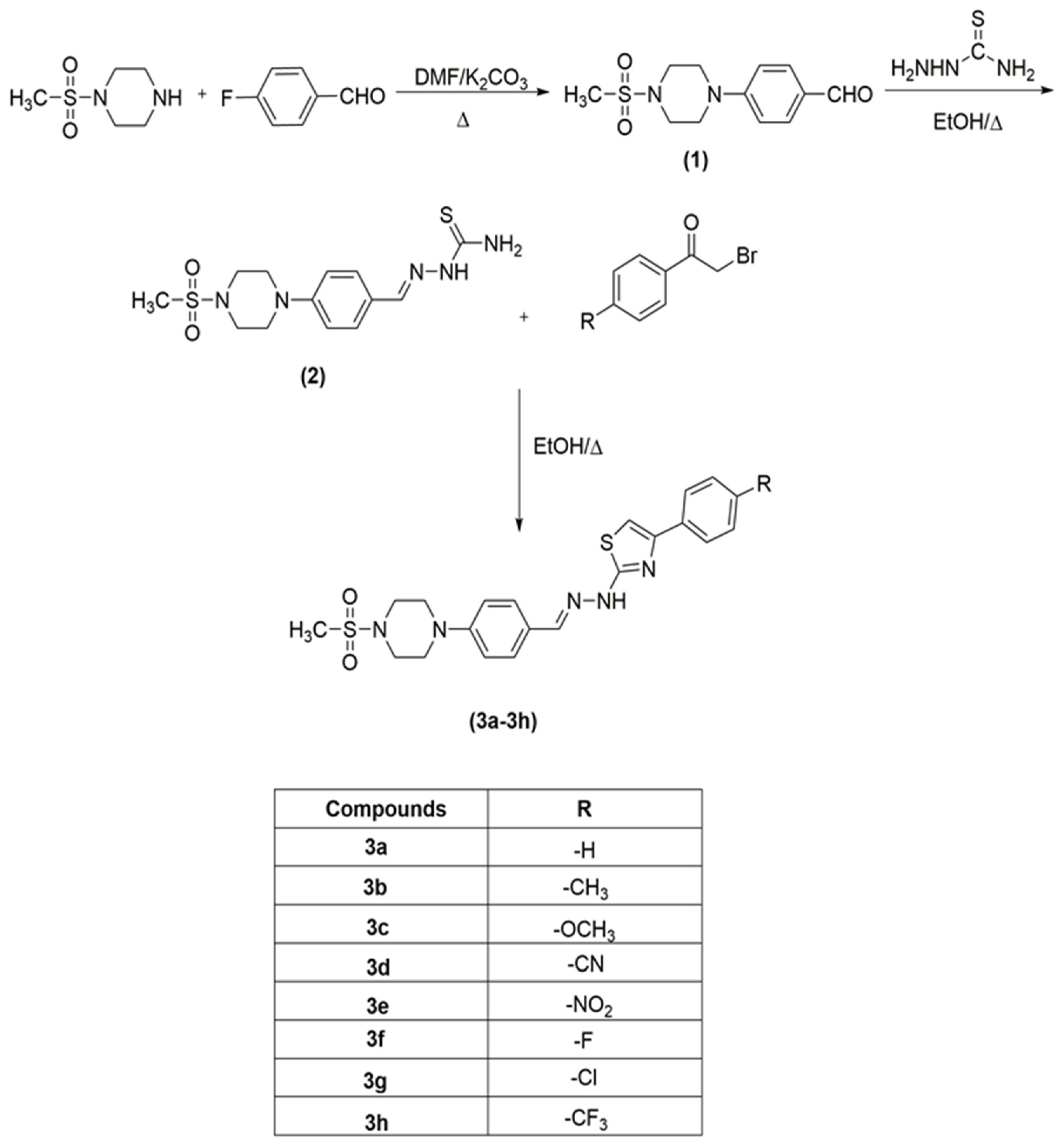
2.1. Chemistry
2.2. Prediction of ADME Parameters
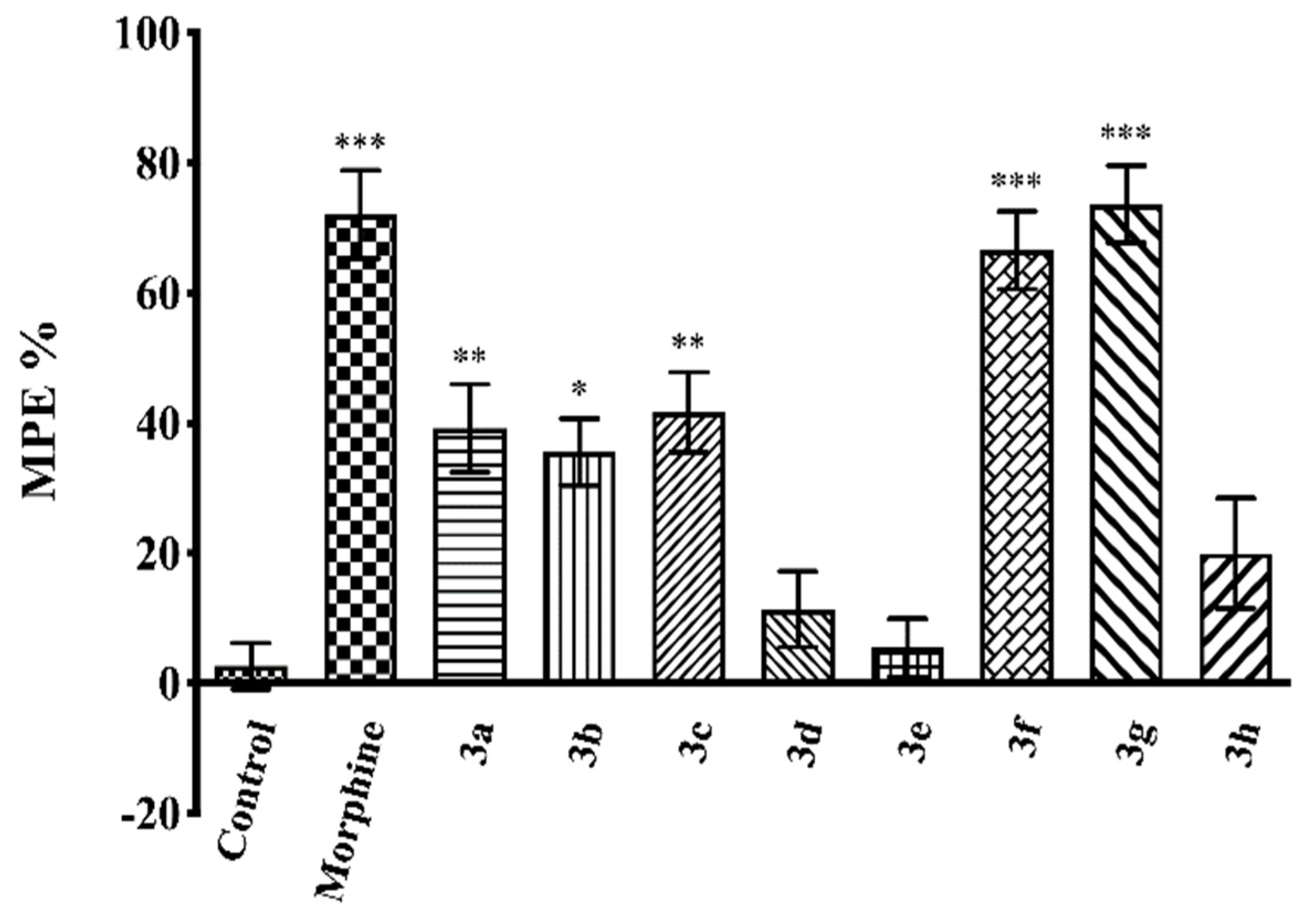
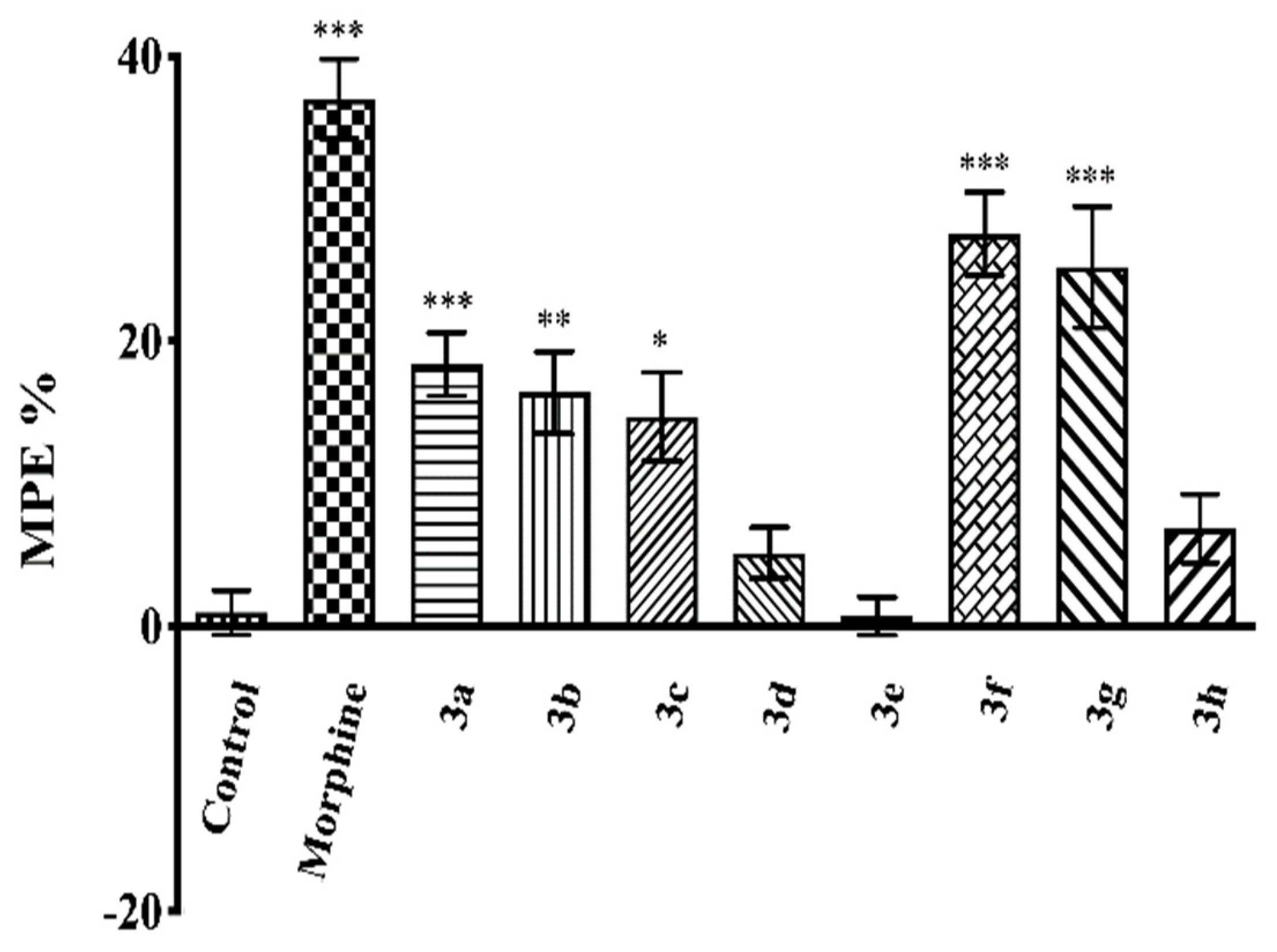
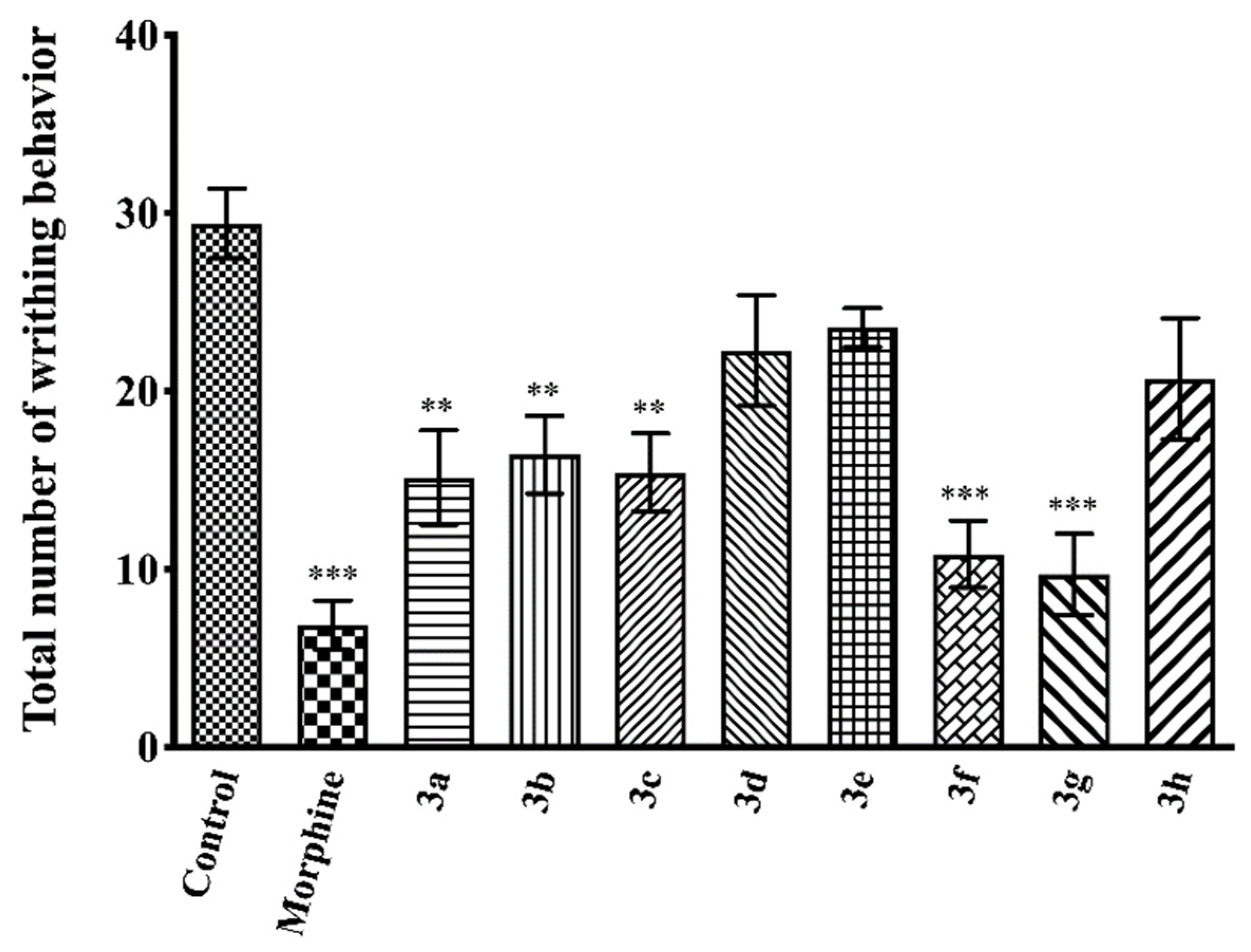
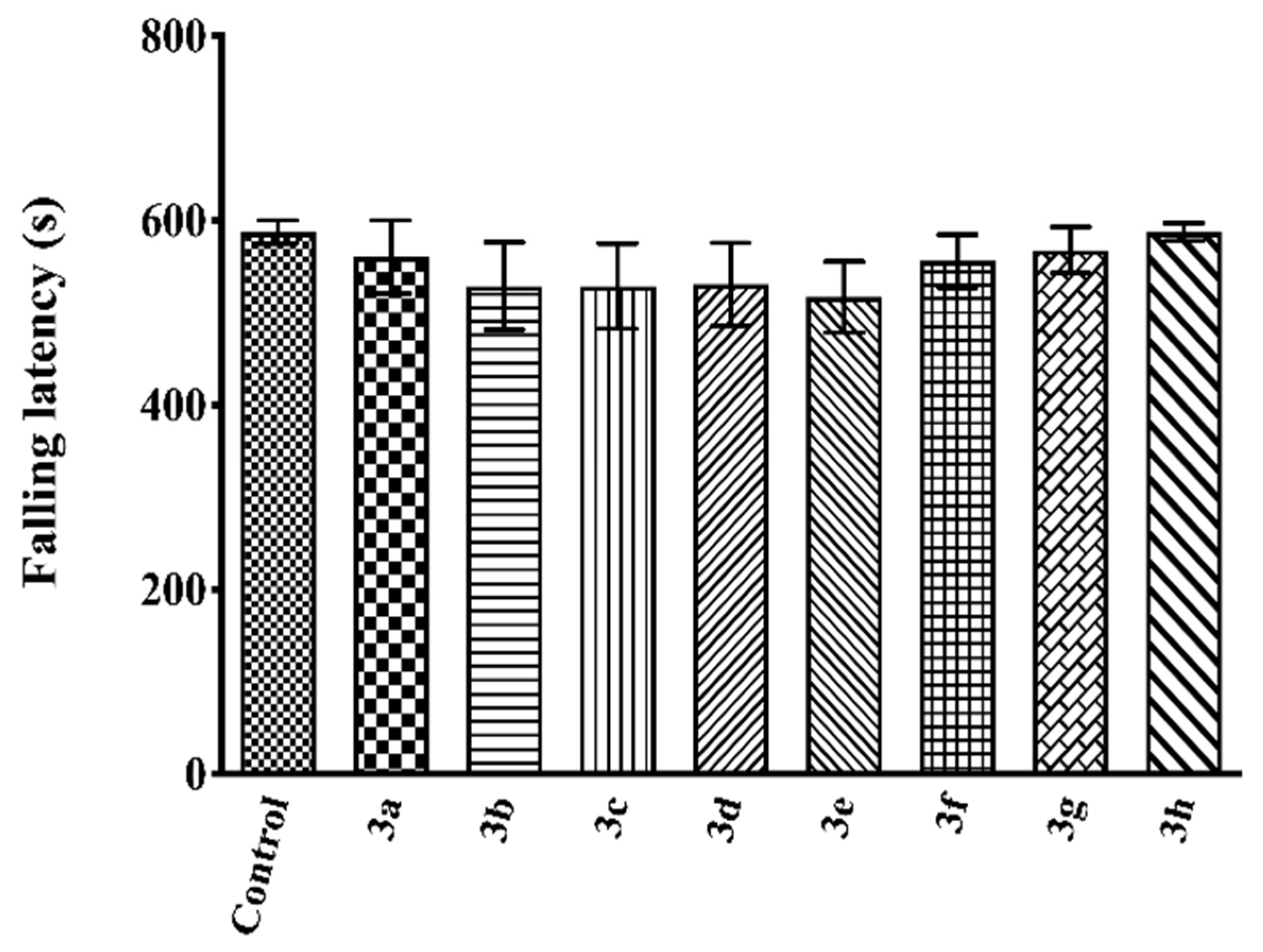
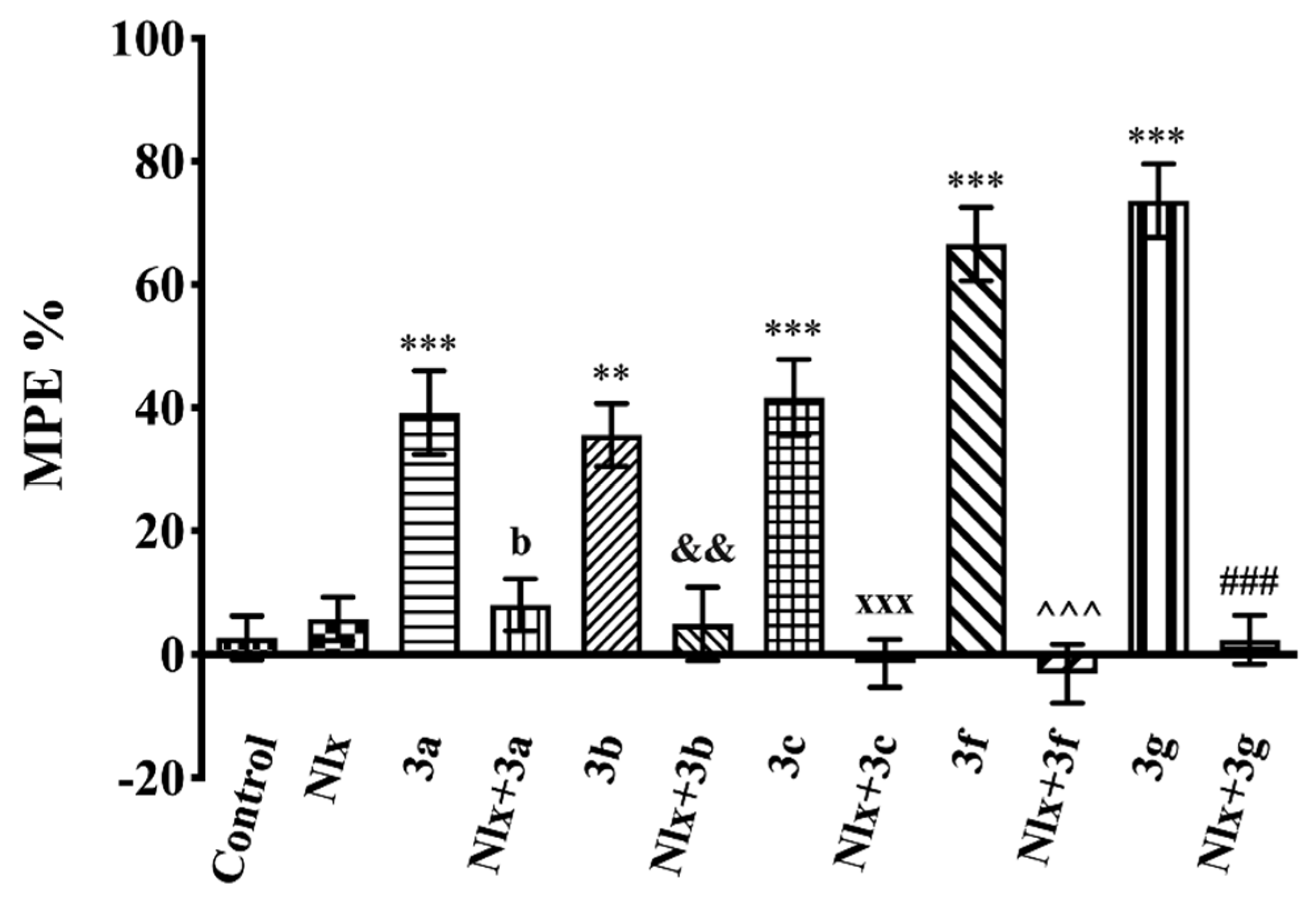
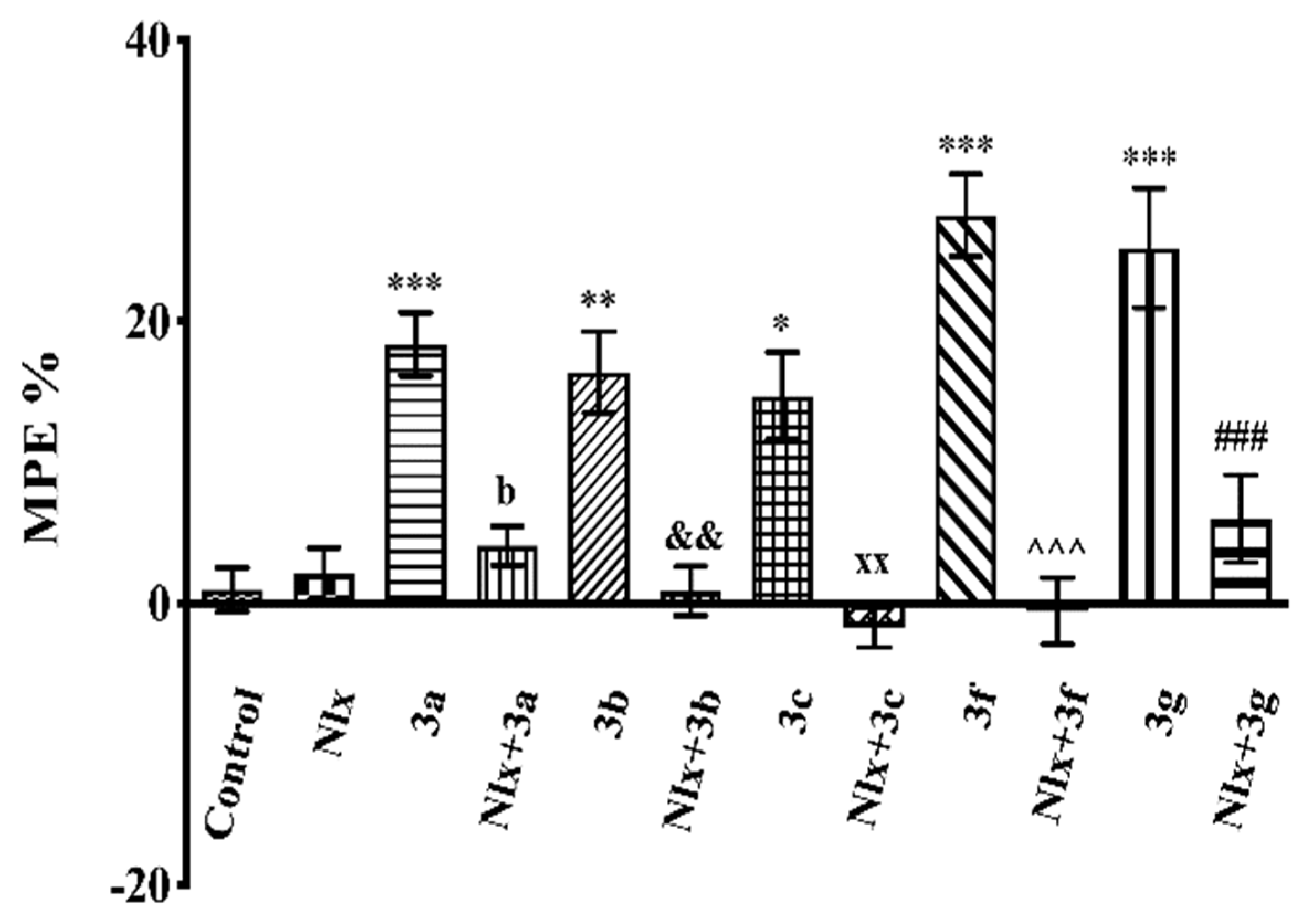
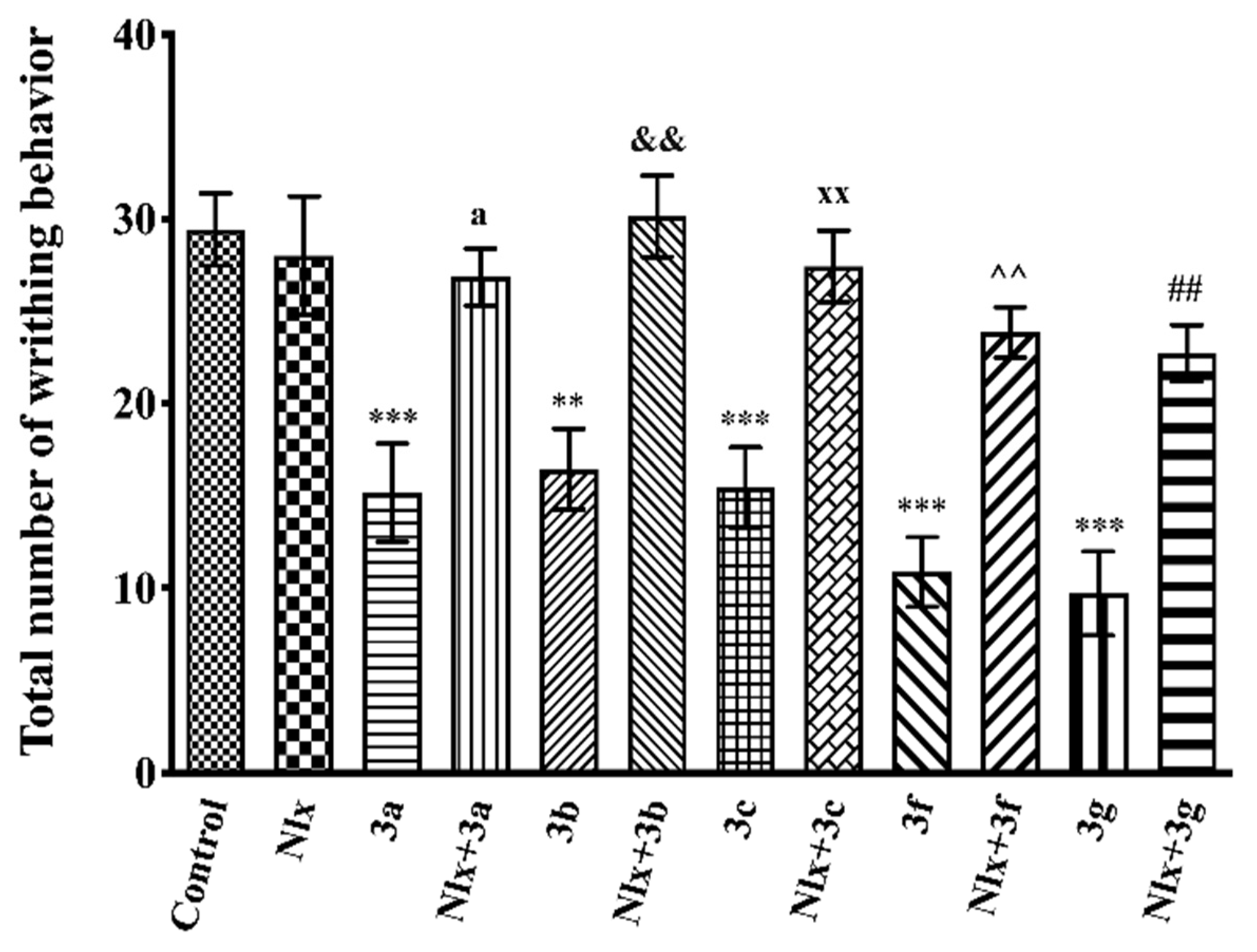
2.3. Pharmacology
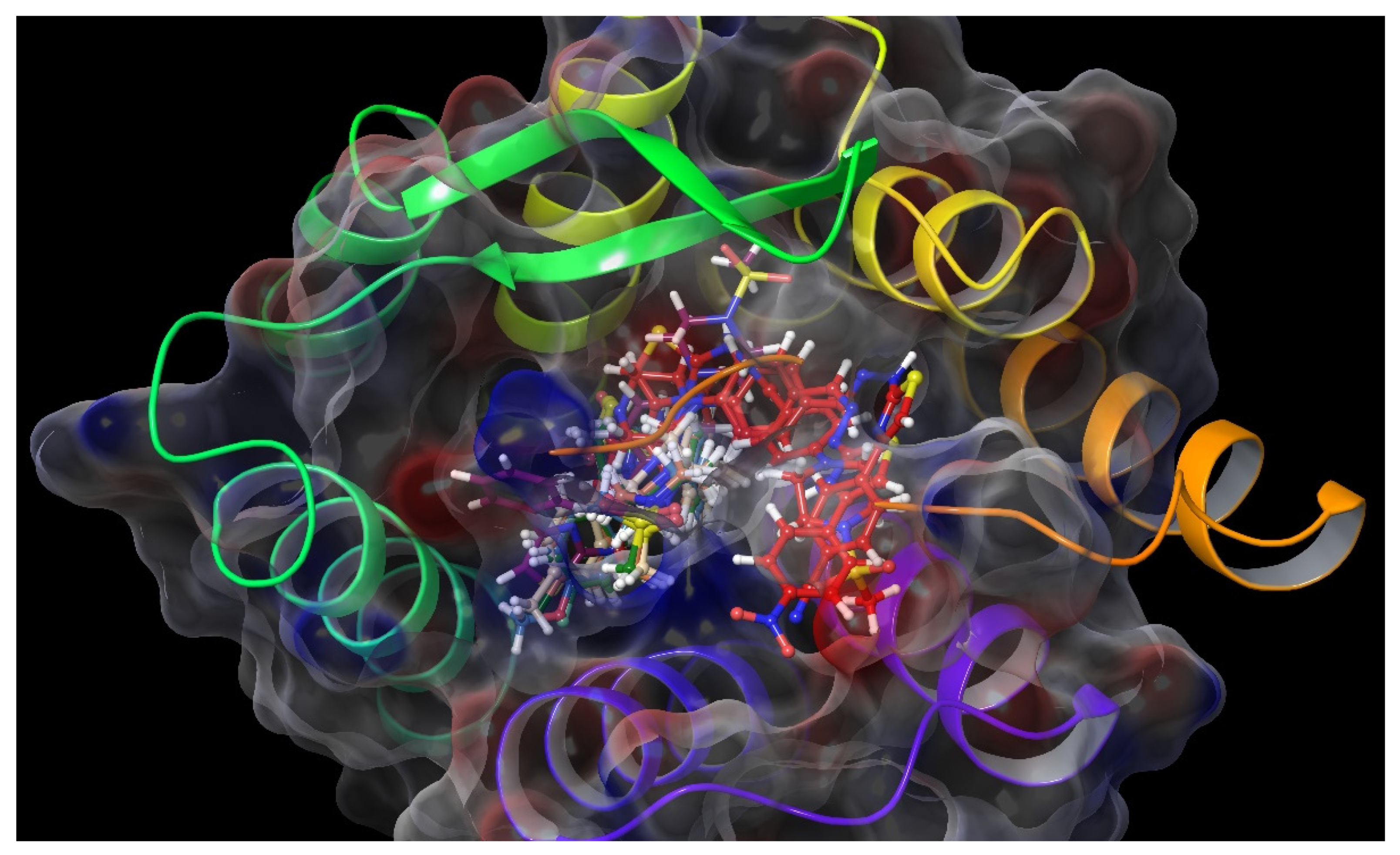
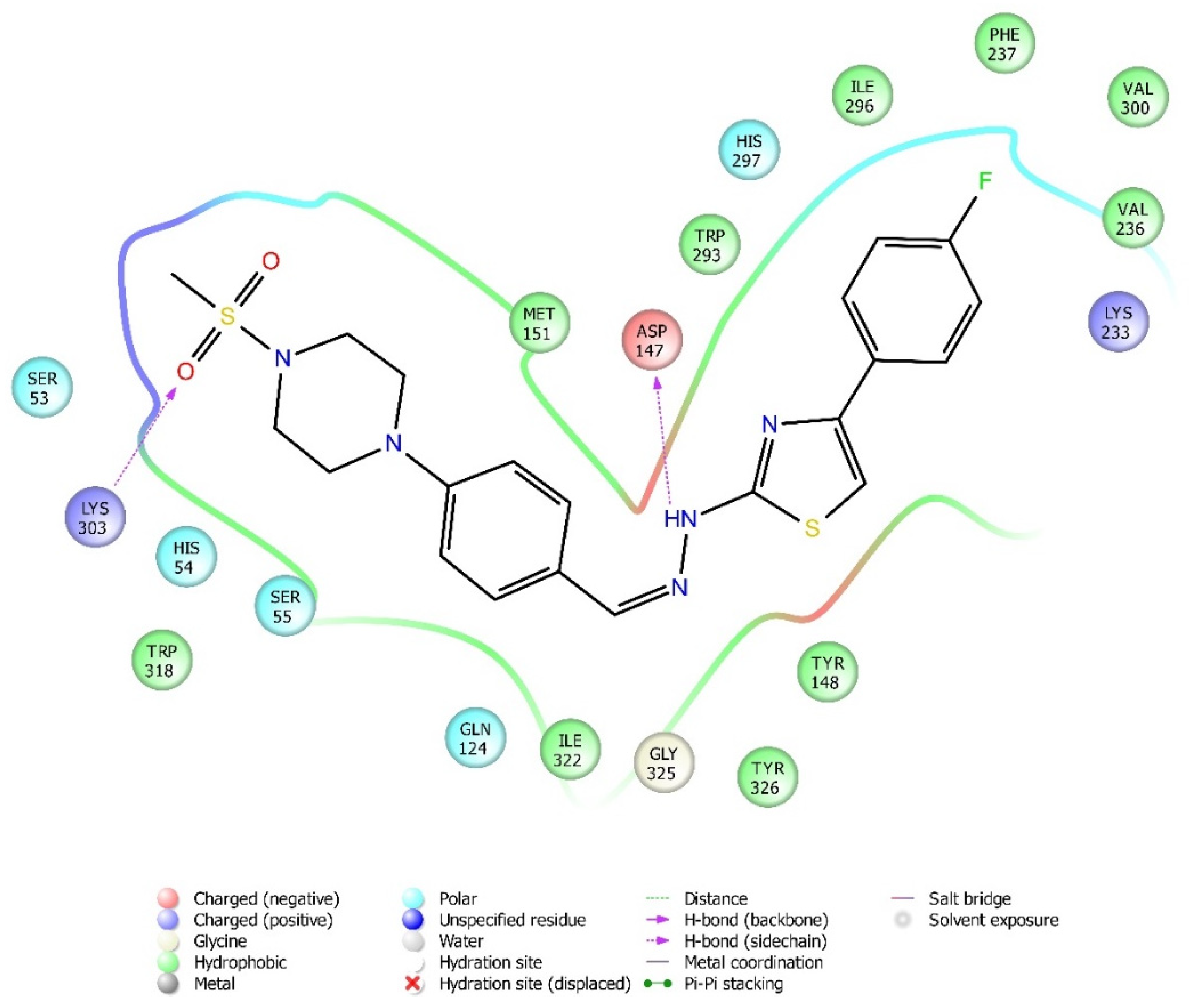
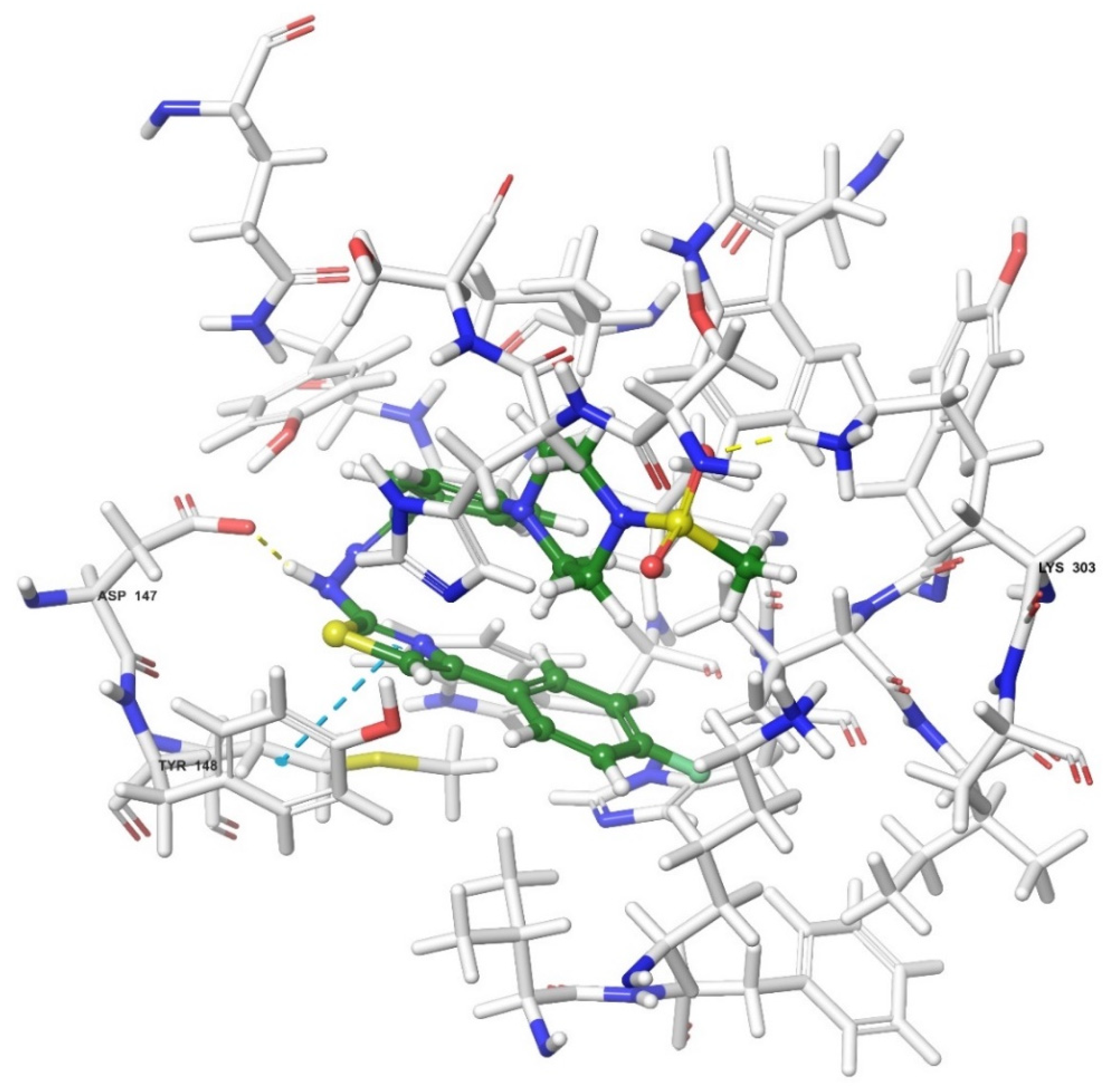
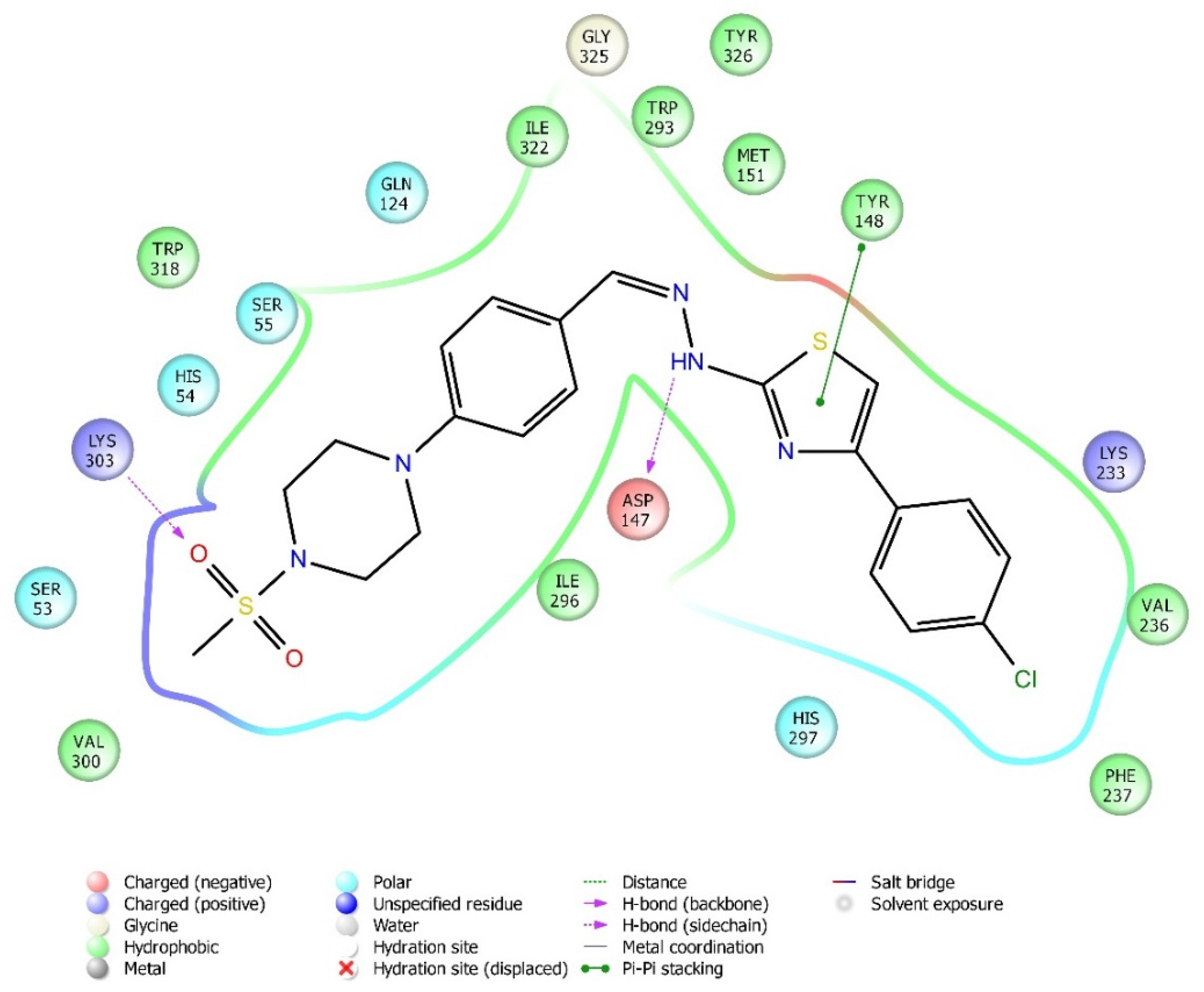
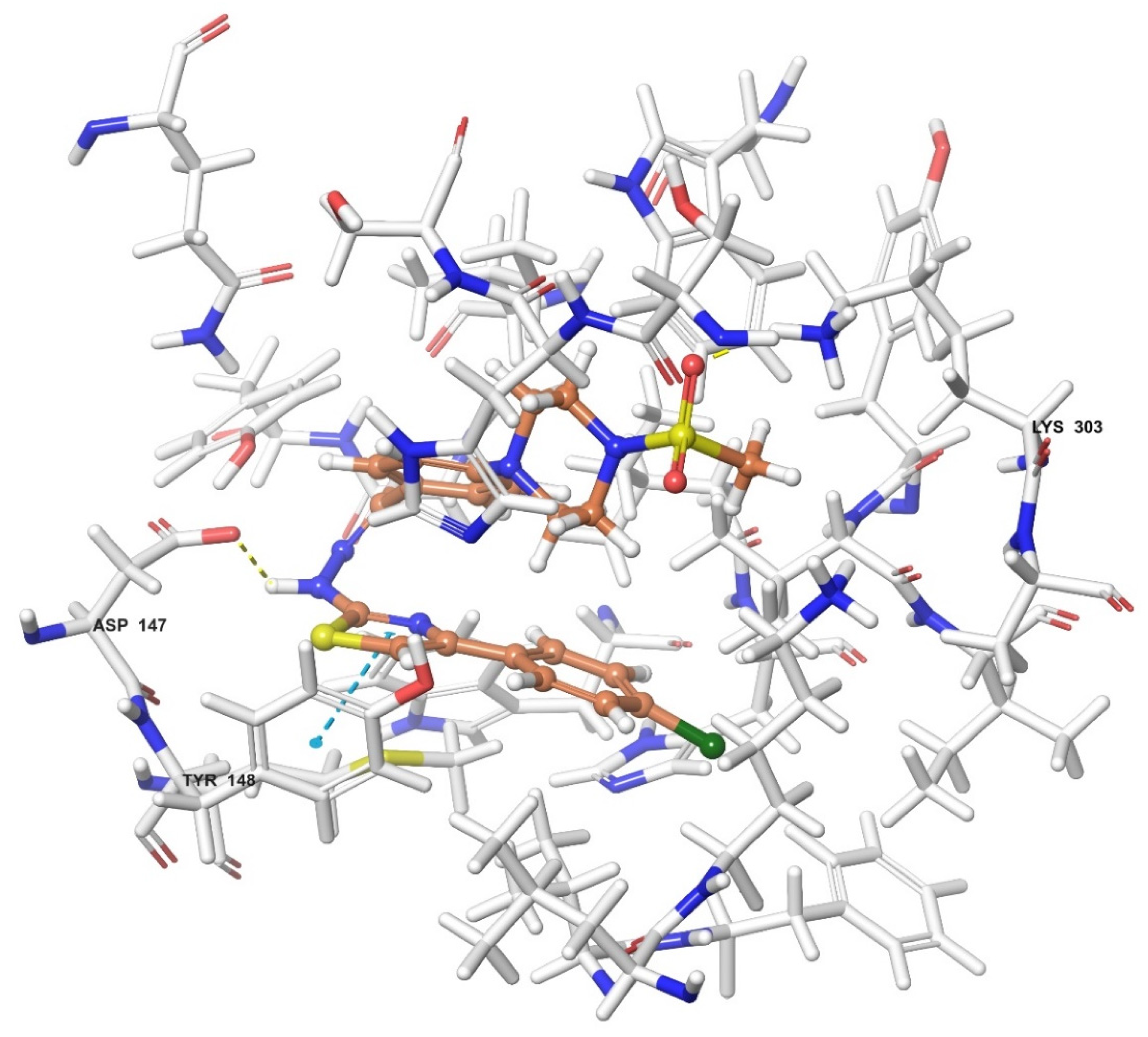
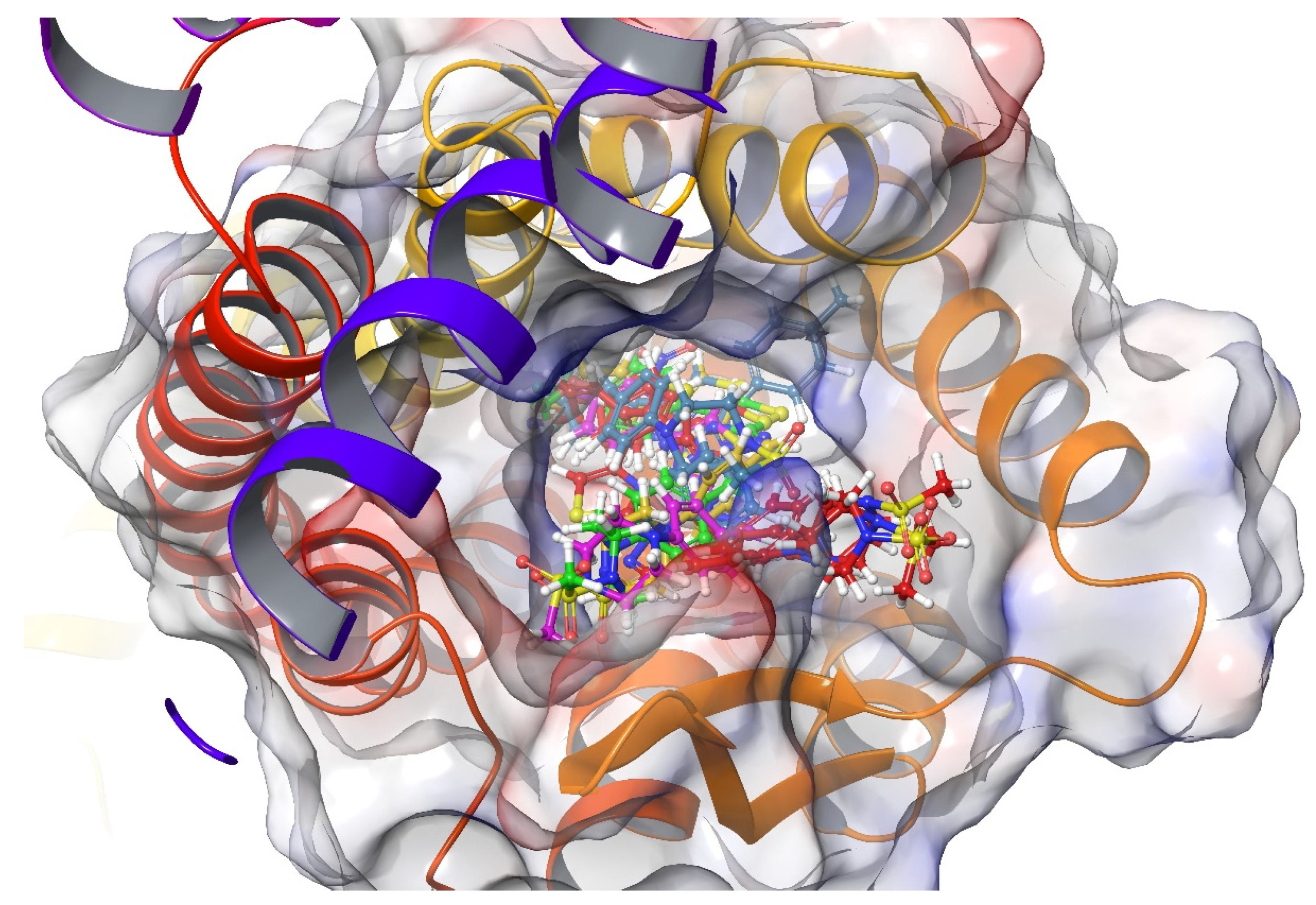
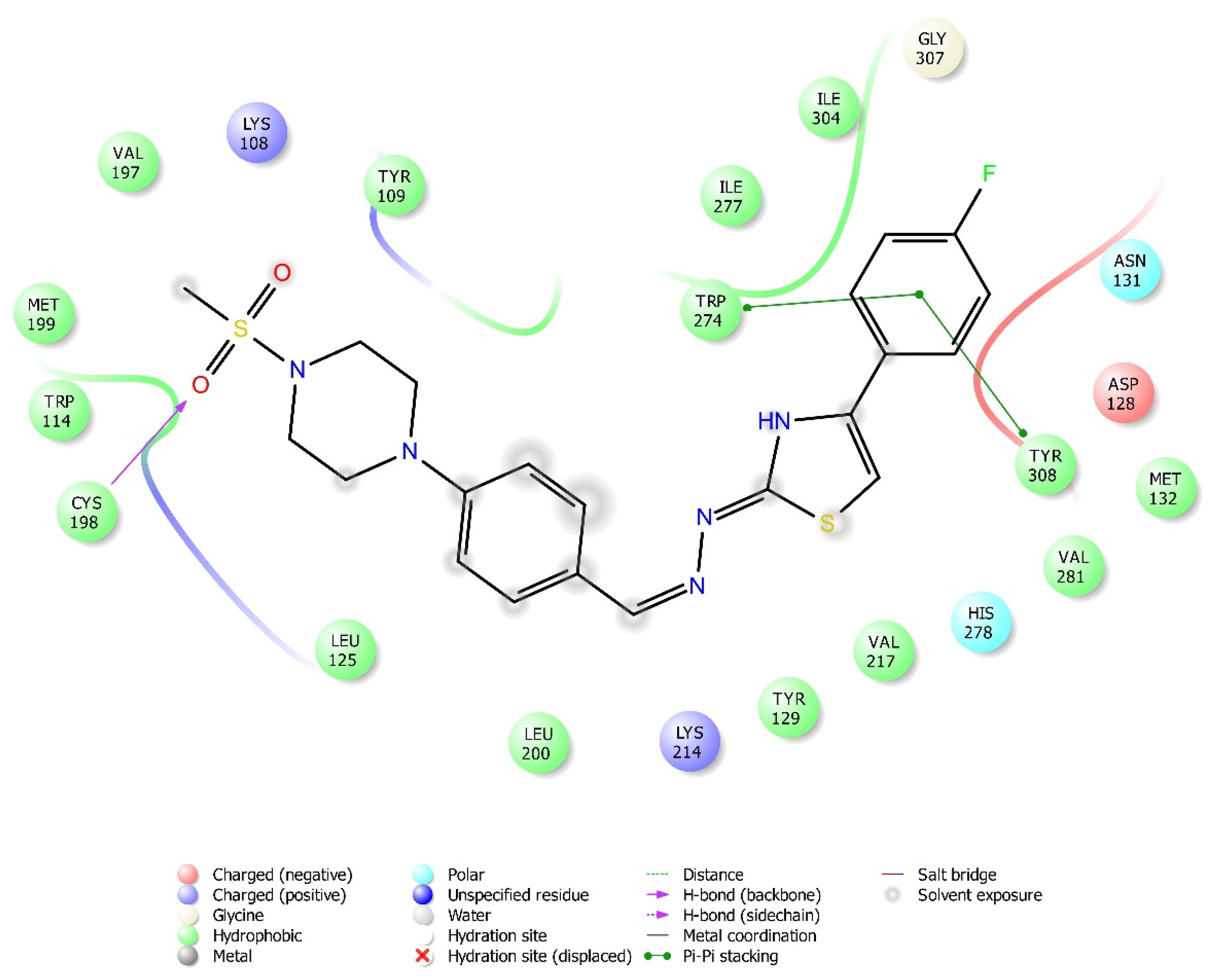
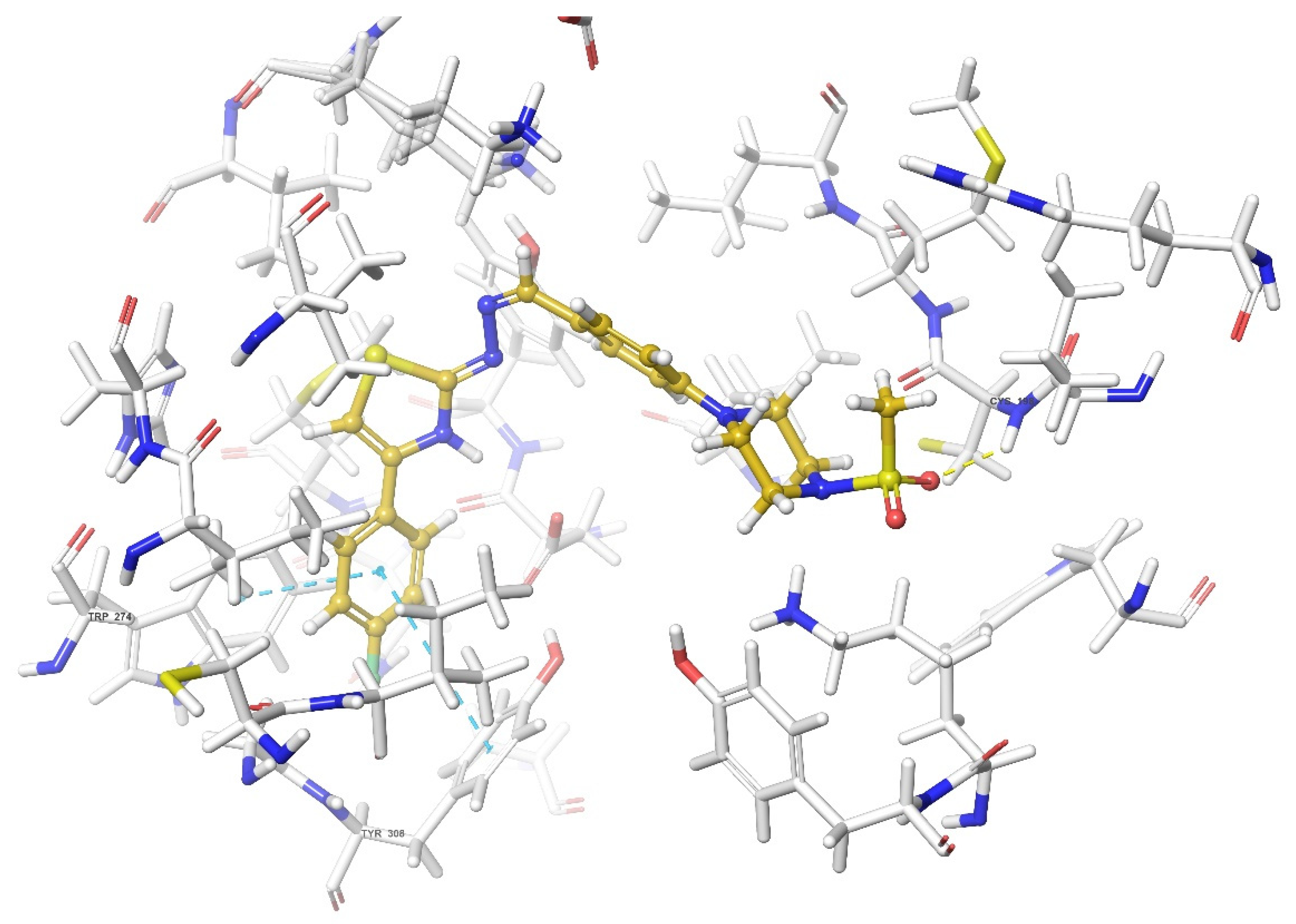
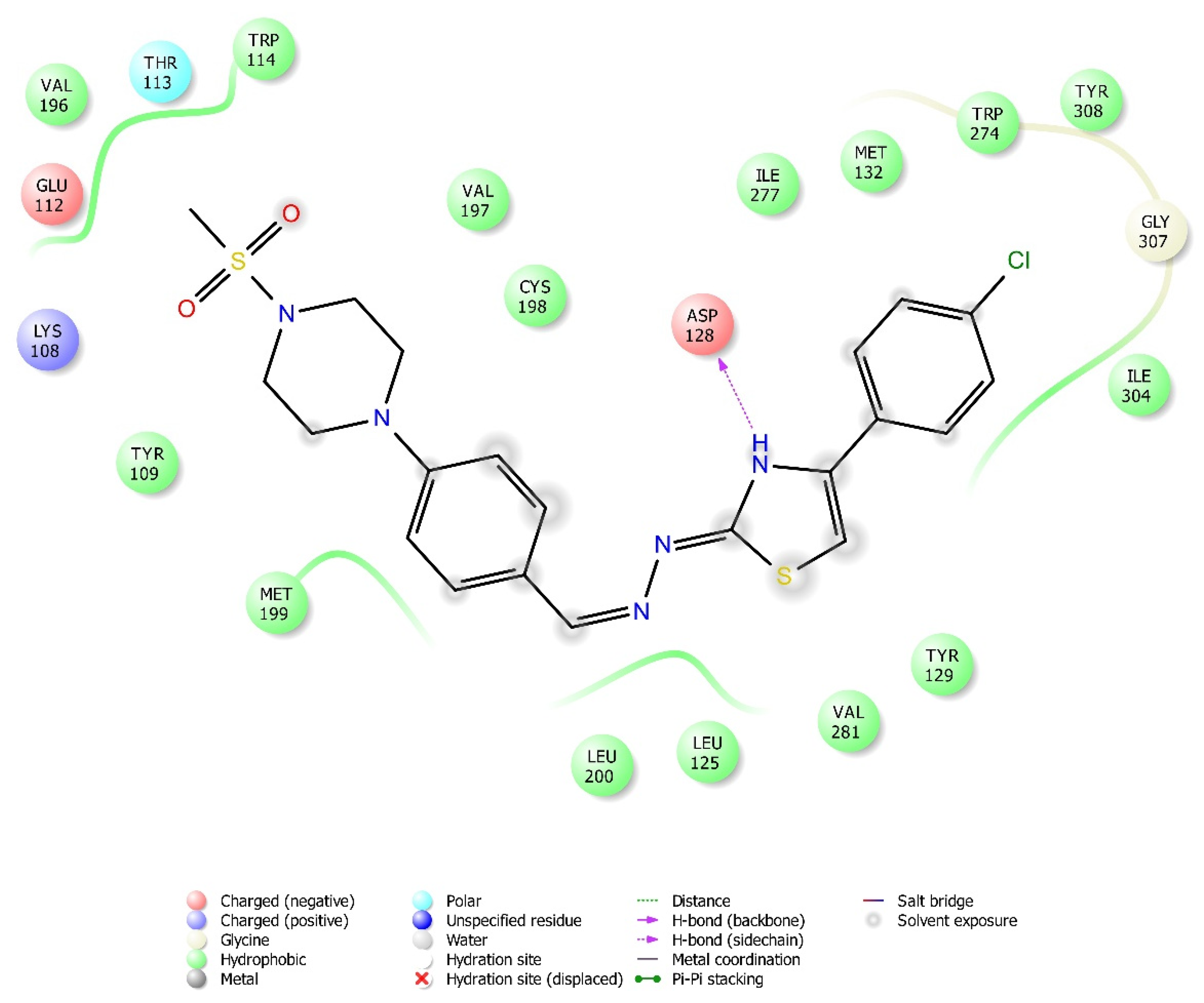
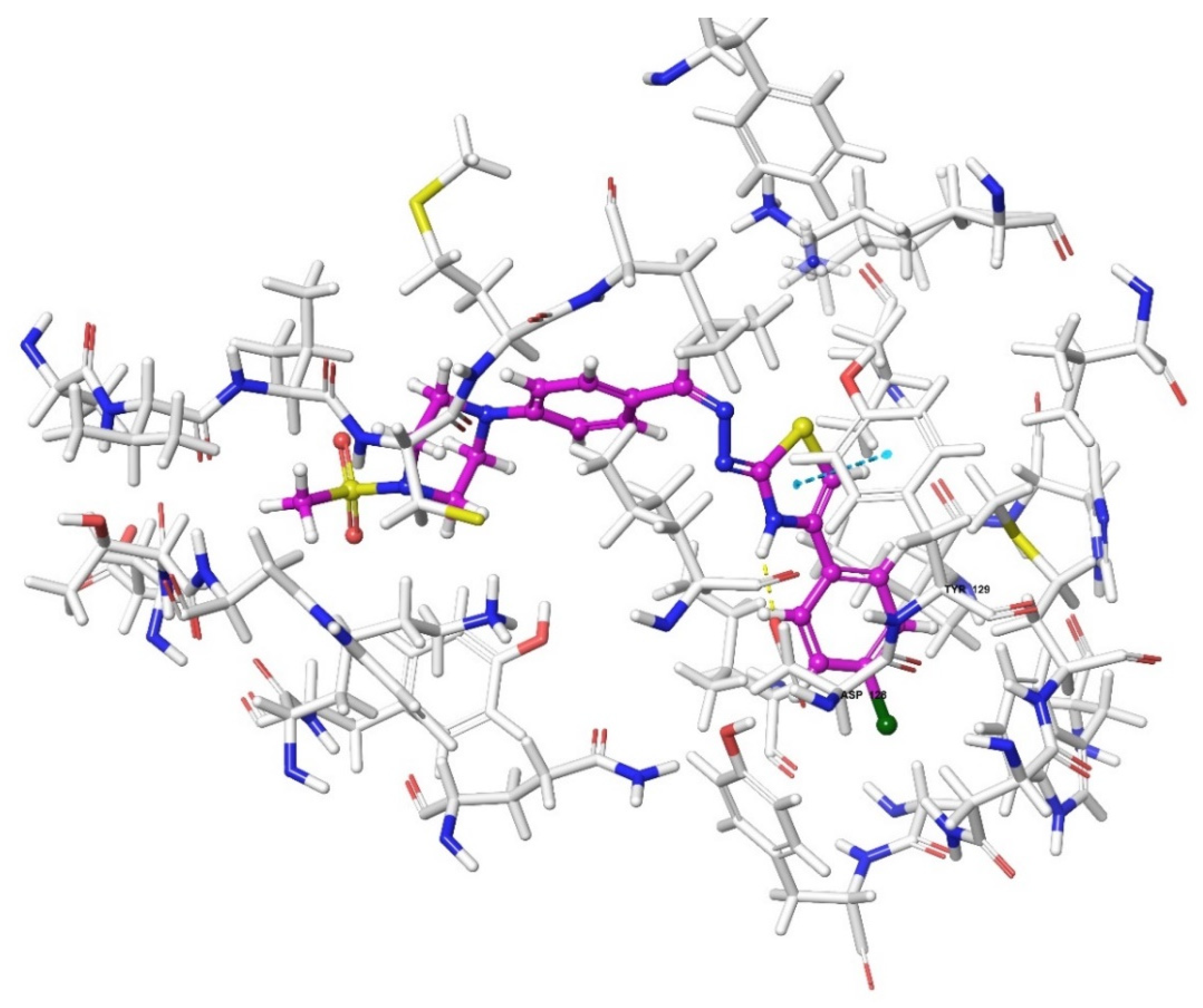
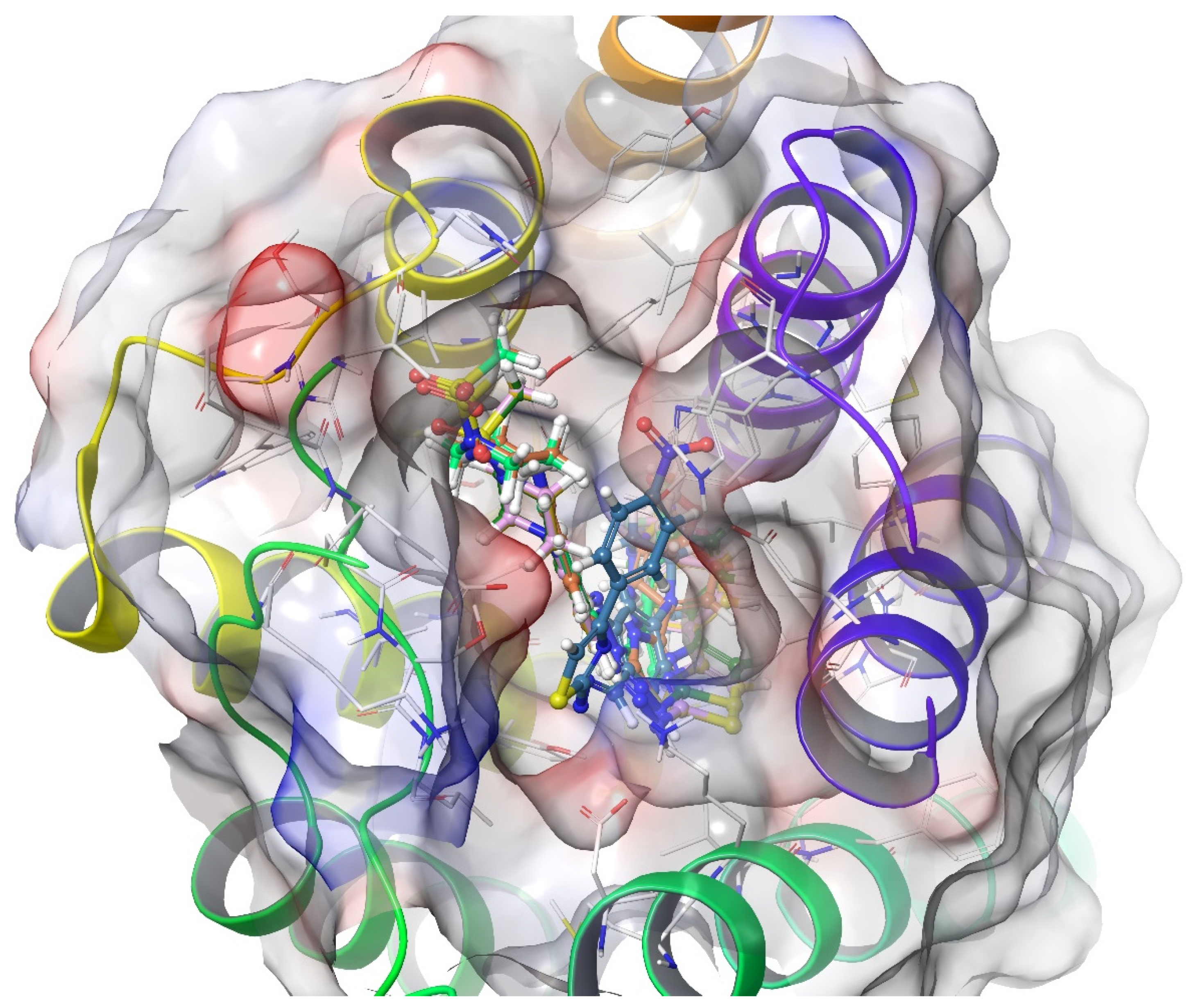
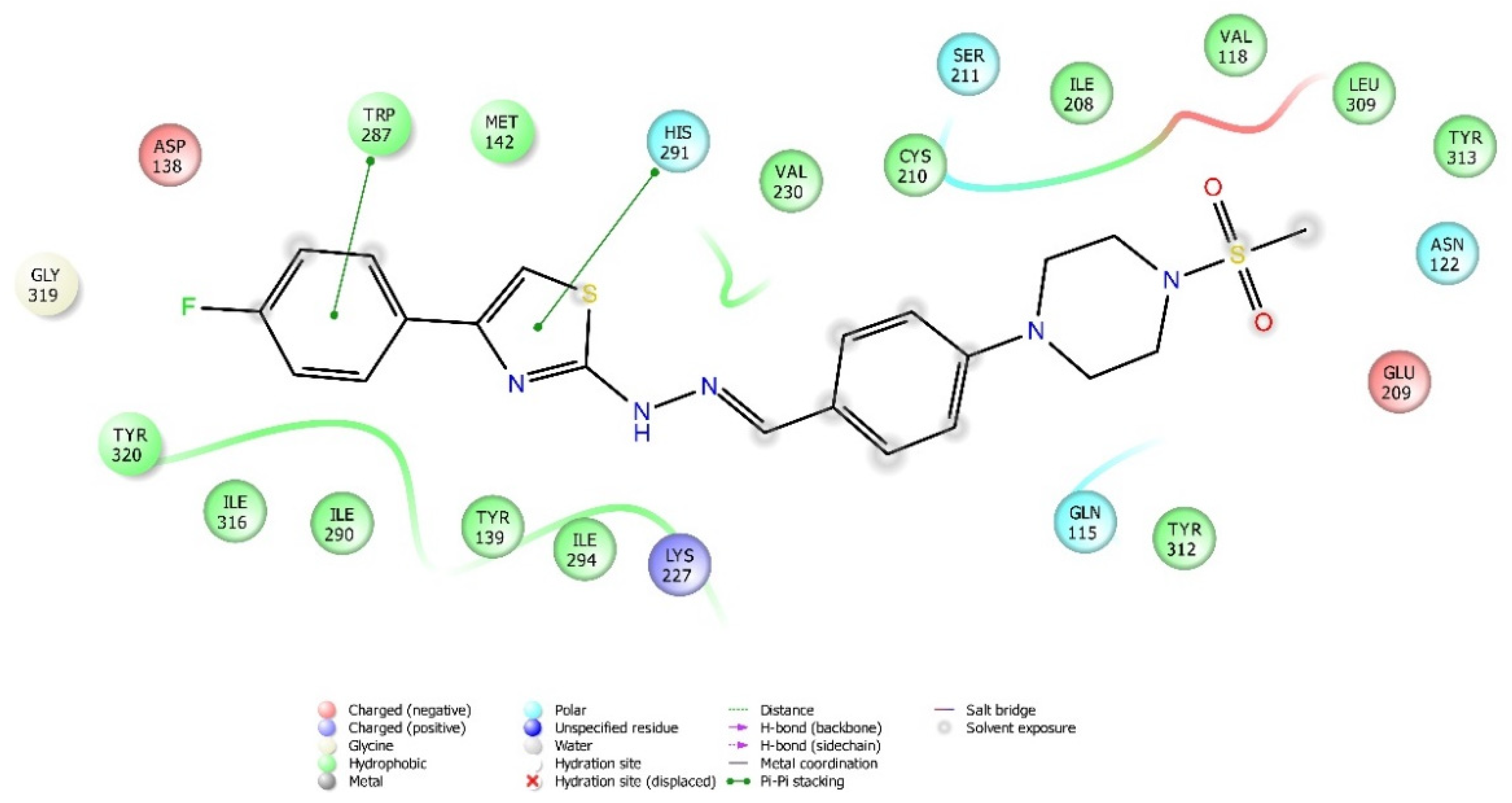
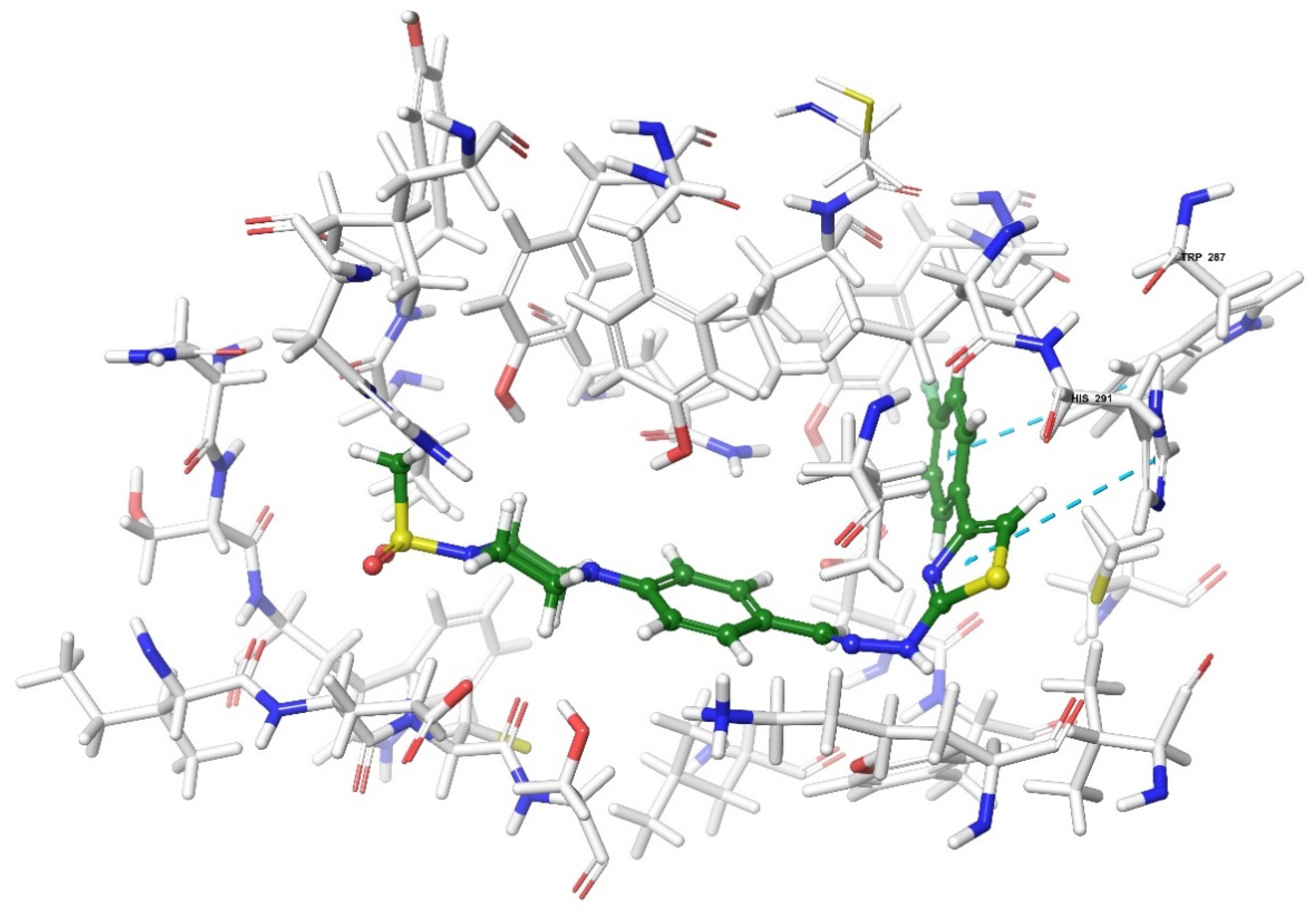
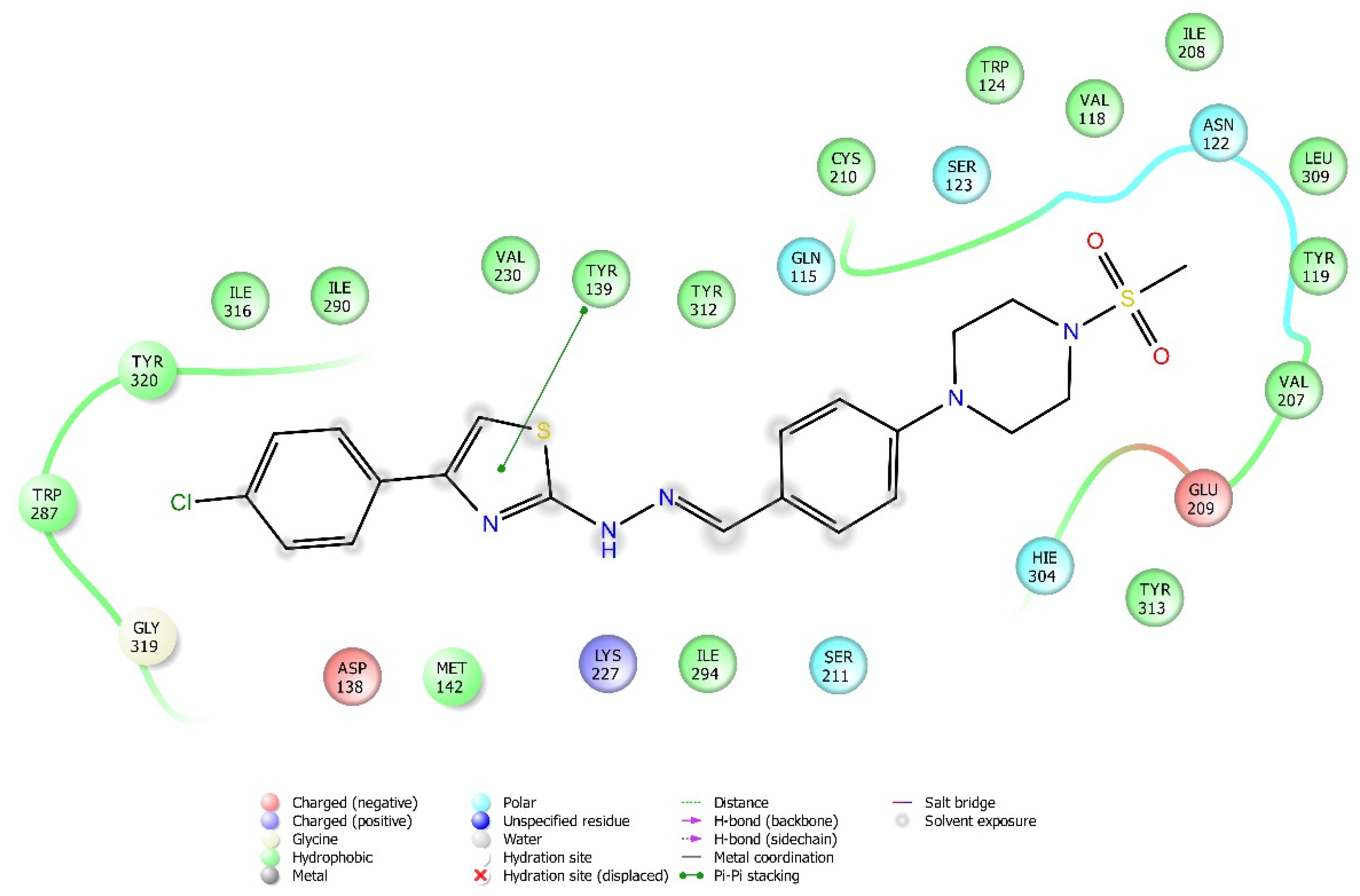
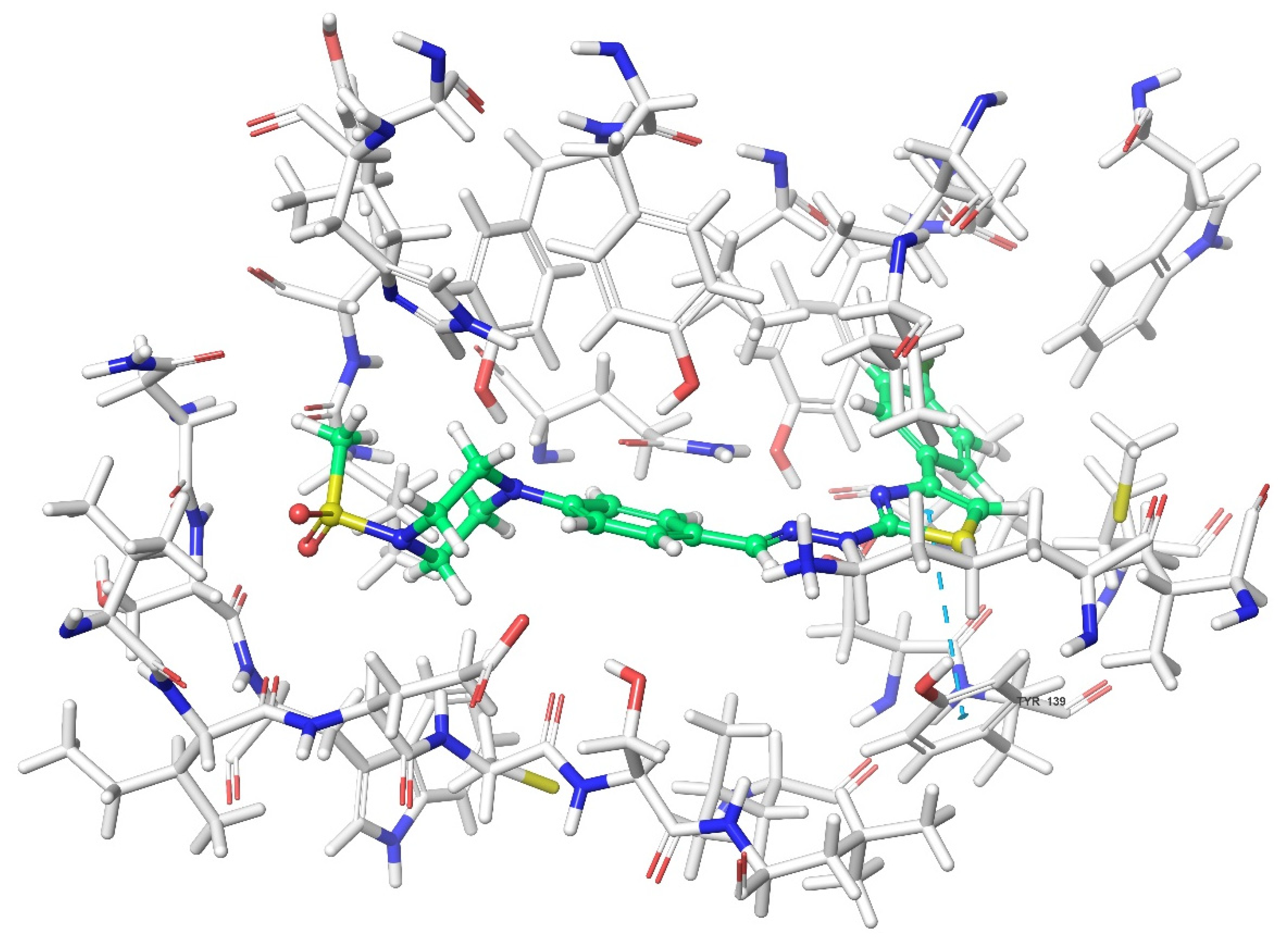
2.4. Molecular Docking Studies
3. Discussion
Future Directions
4. Materials and Methods
4.1. Chemicals
4.2. Chemistry
4.2.1. Synthesis of 4-(4-(methylsulphonyl)piperazin-1-yl)benzaldehyde (1)
4.2.2. Synthesis of 2-(4-(4-(methylsulphonyl)piperazin-1-yl)benzylidene)hydrazine-1-carbothioamide (2)
4.2.3. General Procedure for the Synthesis of Target Compounds (3a–3h)
1-Methylsulphonyl-4-(4-{[2-(4-phenyl-1,3-thiazol-2-yl)hydrazinylidene]methyl}phenyl)piperazine (3a)
1-Methylsulphonyl-4-[{2-[4-(4-methylphenyl)-1,3-thiazol-2-yl]hydrazinylidene}methyl]piperazine (3b)
1-Methylsulphonyl-4-[{2-[4-(4-methoxyphenyl)-1,3-thiazol-2-yl]hydrazinylidene}methyl]piperazine (3c)
1-Methylsulphonyl-4-[{2-[4-(4-cyanophenyl)-1,3-thiazol-2-yl]hydrazinylidene}methyl]piperazine (3d)
1-Methylsulphonyl-4-[{2-[4-(4-nitrophenyl)-1,3-thiazol-2-yl]hydrazinylidene}methyl]piperazine (3e)
1-Methylsulphonyl-4-[{2-[4-(4-fluorophenyl)-1,3-thiazol-2-yl]hydrazinylidene}methyl]piperazine (3f)
1-Methylsulphonyl-4-[{2-[4-(4-chlorophenyl)-1,3-thiazol-2-yl]hydrazinylidene}methyl]piperazine (3g)
1-Methylsulphonyl-4-[{2-[4-(4-trifluoromethylphenyl)-1,3-thiazol-2-yl]hydrazinylidene}methyl]piperazine (3h)
4.3. Pharmacology
4.3.1. Animals
4.3.2. Administration of the Test Compounds
4.3.3. Evaluation of the Antinociceptive Activity
Tail-Clip Test
Hot-Plate Test
Acetic Acid-Induced Writhing Test
Mechanistic Studies
4.3.4. Evaluation of the Motor Activity
Rota-Rod Test
4.3.5. Statistical Analysis
4.4. Molecular Docking Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ruel, H.L.; Steagall, P.V. Adjuvant Analgesics in Acute Pain Management. Vet. Clin. N. Am. Small Anim. Pract. 2019, 49, 1127–1141. [Google Scholar] [CrossRef]
- Harirforoosh, S.; Asghar, W.; Jamali, F. Adverse effects of nonsteroidal antiinflammatory drugs: An update of gastrointestinal, cardiovascular and renal complications. J. Pharm. Pharm. Sci. 2013, 16, 821–847. [Google Scholar] [CrossRef] [Green Version]
- Imam, M.Z.; Kuo, A.; Ghassabian, S.; Smith, M.T. Progress in understanding mechanisms of opioid-induced gastrointestinal adverse effects and respiratory depression. Neuropharmacology 2018, 131, 238–255. [Google Scholar] [CrossRef]
- Jaen, J.C.; Wise, L.D.; Caprathe, B.W.; Tecle, H.; Bergmeier, S.; Humblet, C.C.; Heffner, T.G.; Meltzer, L.T.; Pugsley, T.A. 4-(1,2,5,6-Tetrahydro-1-alkyl-3-pyridinyl)-2-thiazolamines: A novel class of compounds with central dopamine agonist properties. J. Med. Chem. 1990, 33, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Perez-Lloret, S.; Rey, M.V.; Ratti, L.; Rascol, O. Pramipexole for the treatment of early Parkinson’s disease. Expert Rev. Neurother. 2011, 11, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Storch, A.; Burkhardt, K.; Ludolph, A.C.; Schwarz, J. Protective Effects of Riluzole on Dopamine Neurons. J. Neurochem. 2008, 75, 2259–2269. [Google Scholar] [CrossRef]
- Das Neves, A.M.; Berwaldt, G.A.; Avila, C.T.; Goulart, T.B.; Moreira, B.C.; Ferreira, T.P.; Soares, M.S.P.; Pedra, N.S.; Spohr, L.; De Souza, A.A.A.; et al. Synthesis of thiazolidin-4-ones and thiazinan-4-ones from 1-(2-aminoethyl)pyrrolidine as acetylcholinesterase inhibitors. J. Enzym. Inhib. Med. Chem. 2019, 35, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Verma, P.K.; Khatkar, A. Anticonvulsant and Neurological Profile of Benzothiazoles: A Mini-Review. Central Nerv. Syst. Agents Med. Chem. 2015, 15, 11–16. [Google Scholar] [CrossRef]
- Jin, Q.; Fu, Z.; Guan, L.; Jiang, H. Syntheses of Benzo[d]Thiazol-2(3H)-One Derivatives and Their Antidepressant and Anticonvulsant Effects. Mar. Drugs 2019, 17, 430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tikhonova, T.A.; Rassokhina, I.V.; Kondrakhin, E.A.; Fedosov, M.A.; Bukanova, J.V.; Rossokhin, A.V.; Sharonova, I.N.; Kovalev, G.I.; Zavarzin, I.V.; Volkova, Y.A. Development of 1,3-thiazole analogues of imidazopyridines as potent positive allosteric modulators of GABAA receptors. Bioorg. Chem. 2020, 94, 103334. [Google Scholar] [CrossRef]
- Shaquiquzzaman, M.; Verma, G.; Marella, A.; Akhter, M.; Akhtar, W.; Khan, M.F.; Tasneem, S.; Alam, M.M. Piperazine scaffold: A remarkable tool in generation of diverse pharmacological agents. Eur. J. Med. Chem. 2015, 102, 487–529. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; Asin, K.E.; Artigas, F. Vortioxetine, a novel antidepressant with multimodal activity: Review of preclinical and clinical data. Pharmacol. Ther. 2015, 145, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Thore, S.N.; Gupta, S.V.; Baheti, K.G. Docking, synthesis, and pharmacological investigation of novel substituted thiazole derivatives as non-carboxylic, anti-inflammatory, and analgesic agents. Med. Chem. Res. 2013, 22, 3802–3811. [Google Scholar] [CrossRef]
- Kalkhambkar, R.; Kulkarni, G.; Shivkumar, H.; Rao, R.N. Synthesis of novel triheterocyclic thiazoles as anti-inflammatory and analgesic agents. Eur. J. Med. Chem. 2007, 42, 1272–1276. [Google Scholar] [CrossRef] [PubMed]
- Raj, K.V.; Narayana, B.; Ashalatha, B.; Kumari, N.S.; Sarojini, B. Synthesis of some bioactive 2-bromo-5-methoxy-N′-[4-(aryl)-1,3-thiazol-2-yl]benzohydrazide derivatives. Eur. J. Med. Chem. 2007, 42, 425–429. [Google Scholar] [CrossRef]
- Ochi, T.; Motoyama, Y.; Goto, T. The analgesic effect profile of FR122047, a selective cyclooxygenase-1 inhibitor, in chemical nociceptive models. Eur. J. Pharmacol. 2000, 391, 49–54. [Google Scholar] [CrossRef]
- Saravanan, G.; Alagarsamy, V.; Prakash, C.R.; Dinesh-Kumar, P.; Selvam, T.P. Synthesis of novel thiazole derivatives as analgesic agents. Am. J. Pol. Sci. 2011, 1, 134–138. [Google Scholar]
- Salem, M.A.; Thabet, H.K.H.; Helal, M.H.; Abdelaal, A.S.; Ammar, Y.A. Synthesis and pharmacological evaluation of some pyrazoles, thiazolopyrimidine, triazolopyrimidine, pyridone and 2-iminochromene containing naproxenoyl moiety as NSAIDs. Chem. Sci. J. 2011, 2, 32. [Google Scholar]
- Venkateshwarlu, E.; Rao, J.V.; Umasankar, K.; Dheeraj, G. Study of anti-inflammatory, analgesic and antipyretic activity of novel isatin derivatives. Asian J. Pharm. Clin. Res. 2012, 5, 187–190. [Google Scholar]
- Martins, D.F.; Rosa, A.O.; Gadotti, V.M.; Mazzardo-Martins, L.; Nascimento, F.P.; Egea, J.; Lopez, M.G.; Santos, A.R. The Antinociceptive Effects of AR-A014418, a Selective Inhibitor of Glycogen Synthase Kinase-3 Beta, in Mice. J. Pain 2011, 12, 315–322. [Google Scholar] [CrossRef]
- Yoon, S.-Y.; Woo, J.; Park, J.-O.; Choi, E.-J.; Shin, H.-S.; Roh, D.-H.; Kim, K.-S. Intrathecal RGS4 Inhibitor, CCG50014, Reduces Nociceptive Responses and Enhances Opioid-Mediated Analgesic Effects in the Mouse Formalin Test. Anesthesia Analg. 2015, 120, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Salam, O.M.; El-Batran, S. Pharmacological Investigation of Trimetazidine in Models of Inflammation, Pain and Gastric Injury in Rodents. Pharmacology 2005, 75, 122–132. [Google Scholar] [CrossRef]
- Biancalani, C.; Giovannoni, M.P.; Pieretti, S.; Cesari, N.; Graziano, A.; Vergelli, C.; Cilibrizzi, A.; Di Gianuario, A.; Colucci, M.; Mangano, G.; et al. Further Studies on Arylpiperazinyl Alkyl Pyridazinones: Discovery of an Exceptionally Potent, Orally Active, Antinociceptive Agent in Thermally Induced Pain. J. Med. Chem. 2009, 52, 7397–7409. [Google Scholar] [CrossRef]
- Kam, Y.L.; Rhee, H.-K.; Rhim, H.; Back, S.K.; Na, H.S.; Choo, H.-Y.P. Synthesis and T-type calcium channel blocking activity of novel diphenylpiperazine compounds, and evaluation of in vivo analgesic activity. Bioorg. Med. Chem. 2010, 18, 5938–5944. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, G.; Xu, X.; Liu, B.-F.; Li, J.; Zhang, G. Design, Synthesis and Biological Activity Evaluation of Arylpiperazine Derivatives for the Treatment of Neuropathic Pain. Molecules 2011, 16, 5785–5806. [Google Scholar] [CrossRef]
- Chae, E.; Yi, H.; Choi, Y.; Cho, H.; Lee, K.; Moon, H. Synthesis and pharmacological evaluation of carbamic acid 1-phenyl-3-(4-phenyl-piperazine-1-yl)-propyl ester derivatives as new analgesic agents. Bioorg. Med. Chem. Lett. 2012, 22, 2434–2439. [Google Scholar] [CrossRef]
- Papadopoulou, C.; Geronikaki, A.; Hadjipavlou-Litina, D. Synthesis and biological evaluation of new thiazolyl/benzothiazolyl-amides, derivatives of 4-phenyl-piperazine. Il Farm. 2005, 60, 969–973. [Google Scholar] [CrossRef]
- Ahmadi, A.; Khalili, M.; Nafarie, A.; Yazdani, A.; Nahri-Niknafs, B. Synthesis and anti-inflammatory effects of new piperazine and ethanolamine derivatives of H(1)-antihistaminic drugs. Mini Rev. Med. Chem. 2012, 12, 1282–1292. [Google Scholar] [CrossRef]
- Salat, K.; Moniczewski, A.; Salat, R.; Janaszek-Mańkowska, M.; Filipek, B.; Malawska, B.; Więckowski, K. Analgesic, anticonvulsant and antioxidant activities of 3-[4-(3-trifluoromethyl-phenyl)-piperazin-1-yl]-dihydrofuran-2-one dihydrochloride in mice. Pharmacol. Biochem. Behav. 2012, 101, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Metcalf, M.D.; Yekkirala, A.S.; Powers, M.D.; Kitto, K.F.; Fairbanks, C.A.; Wilcox, G.L.; Portoghese, P.S. The δ Opioid Receptor Agonist SNC80 Selectively Activates Heteromeric μ-δ Opioid Receptors. ACS Chem. Neurosci. 2012, 3, 505–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, S.P.; Kong, Q.H.; Li, Y.L.; Pan, C.L.; Yu, J.; Cui, B.Q.; Wang, Y.F.; Wang, G.L.; Zhou, P.L.; Wang, L.L.; et al. The opioid receptor triple agonist DPI-125 produces analgesia with less respiratory depression and reduced abuse liability. Acta Pharmacol. Sin. 2017, 38, 977–989. [Google Scholar] [CrossRef] [Green Version]
- Van De Waterbeemd, H.; Gifford, E. ADMET in silico modelling: Towards prediction paradise? Nat. Rev. Drug Discov. 2003, 2, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into µ-opioid receptor activation. Nat. Cell Biol. 2015, 524, 315–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenalti, G.; Giguere, P.M.; Katritch, V.; Huang, X.-P.; Thompson, A.A.; Cherezov, V.; Roth, B.L.; Stevens, R.C. Molecular control of δ-opioid receptor signalling. Nat. Cell Biol. 2014, 506, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, T.; Majumdar, S.; Zaidi, S.A.; Ondachi, P.; McCorvy, J.D.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 2018, 172, 55–67.e15. [Google Scholar] [CrossRef] [Green Version]
- Jarończyk, M.; Lipiński, P.F.J.; Dobrowolski, J.C.; Sadlej, J. The FMO analysis of the molecular interaction of fentanyl derivatives with the μ-opioid receptor. Chem. Pap. 2017, 71, 1429–1443. [Google Scholar] [CrossRef]
- Le Bars, D.; Gozariu, M.; Cadden, S.W. Animal models of nociception. Pharmacol. Rev. 2001, 53, 597–652. [Google Scholar] [PubMed]
- Wong, C.H.; Dey, P.; Yarmush, J.; Wu, W.-H.; Zbuzek, V.K. Nifedipine-Induced Analgesia After Epidural Injection in Rats. Anesthesia Analg. 1994, 79, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Gabra, B.H.; Sirois, P. Beneficial effect of chronic treatment with the selective bradykinin B1 receptor antagonists, R-715 and R-954, in attenuating streptozotocin-diabetic thermal hyperalgesia in mice. Peptides 2003, 24, 1131–1139. [Google Scholar] [CrossRef]
- Kasap, M.; Can, Ö.D. Opioid system mediated anti-nociceptive effect of agomelatine in mice. Life Sci. 2016, 163, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Özkay, Ü.D.; Can, Ö.D. Anti-nociceptive effect of vitexin mediated by the opioid system in mice. Pharmacol. Biochem. Behav. 2013, 109, 23–30. [Google Scholar] [CrossRef]
- De Souza, M.; Pereira, M.; Ardenghi, J.; Mora, T.; Bresciani, L.; Yunes, R.; Monache, F.D.; Cechinel-Filho, V. Filicene obtained from Adiantum cuneatum interacts with the cholinergic, dopaminergic, glutamatergic, GABAergic, and tachykinergic systems to exert antinociceptive effect in mice. Pharmacol. Biochem. Behav. 2009, 93, 40–46. [Google Scholar] [CrossRef]
- Ayumi, R.R.; Mossadeq, W.M.S.; Zakaria, Z.A.; Bakhtiar, M.T.; Kamarudin, N.; Hisamuddin, N.; Talib, M.; Sabar, A.M. Antinociceptive Activity of Asiaticoside in Mouse Models of Induced Nociception. Planta Med. 2020, 86, 548–555. [Google Scholar] [CrossRef]
- Ishola, I.O.; Akindele, A.J.; Adeyemi, O.O. Analgesic and anti-inflammatory activities of Cnestis ferruginea Vahl ex DC (Connaraceae) methanolic root extract. J. Ethnopharmacol. 2011, 135, 55–62. [Google Scholar] [CrossRef]
- Park, S.-H.; Sim, Y.-B.; Kang, Y.-J.; Kim, S.-S.; Kim, C.-H.; Kim, S.-J.; Seo, J.-Y.; Lim, S.-M.; Suh, H.-W. Hop Extract Produces Antinociception by Acting on Opioid System in Mice. Korean J. Physiol. Pharmacol. 2012, 16, 187–192. [Google Scholar] [CrossRef] [Green Version]
- Coelho, L.P.; Reis, P.A.; De Castro, F.L.; Gayer, C.R.M.; Lopes, C.D.S.; Silva, M.C.D.C.E.; Sabino, K.C.D.C.; Todeschini, A.R.; Coelho, M.G.P. Antinociceptive properties of ethanolic extract and fractions of Pterodon pubescens Benth. seeds. J. Ethnopharmacol. 2005, 98, 109–116. [Google Scholar] [CrossRef]
- Argoff, C. Mechanisms of pain transmission and pharmacologic management. Curr. Med. Res. Opin. 2011, 27, 2019–2031. [Google Scholar] [CrossRef] [PubMed]
- Millan, M.J. Descending control of pain. Prog. Neurobiol. 2002, 66, 355–474. [Google Scholar] [CrossRef]
- Liaras, K.; Fesatidou, M.; Geronikaki, A. Thiazoles and Thiazolidinones as COX/LOX Inhibitors. Molecules 2018, 23, 685. [Google Scholar] [CrossRef] [Green Version]
- Yam, M.F.; Loh, Y.C.; Tan, C.S.; Adam, S.K.; Manan, N.A.; Basir, R. General Pathways of Pain Sensation and the Major Neurotransmitters Involved in Pain Regulation. Int. J. Mol. Sci. 2018, 19, 2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friderichs, E. Analgesics: From Chemistry and Pharmacology to Clinical Application; Wiley: Hoboken, NJ, USA, 2002. [Google Scholar]
- Sarigol, D.; Uzgoren-Baran, A.; Tel, B.C.; Somuncuoglu, E.I.; Kazkayasi, I.; Ozadali-Sari, K.; Unsal-Tan, O.; Okay, G.; Ertan, M.; Tozkoparan, B. Novel thiazolo[3,2-b]-1,2,4-triazoles derived from naproxen with analgesic/anti-inflammatory properties: Synthesis, biological evaluation and molecular modeling studies. Bioorg. Med. Chem. 2015, 23, 2518–2528. [Google Scholar] [CrossRef]
- Kaplancikli, Z.A.; Turan-Zitouni, G.; Özdemir, A.; Can, Ö.D.; Chevallet, P. Synthesis and antinociceptive activities of some pyrazoline derivatives. Eur. J. Med. Chem. 2009, 44, 2606–2610. [Google Scholar] [CrossRef]
- D’Amour, F.E.; Smith, D.L. A method for determining loss of pain sensation. J. Pharmacol. Exp. Ther. 1941, 72, 74–79. [Google Scholar]
- Pavin, N.F.; Donato, F.; Cibin, F.W.; Jesse, C.R.; Schneider, P.H.; De Salles, H.D.; Soares, L.D.A.; Alves, D.; Savegnago, L. Antinociceptive and anti-hypernociceptive effects of Se-phenyl thiazolidine-4-carboselenoate in mice. Eur. J. Pharmacol. 2011, 668, 169–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koster, R.; Anderson, M.; Beer, E. Acetic acid for analgesic screening. Fed. Proc. 1959, 18, 412. [Google Scholar]
- Dunham, N.W.; Miya, T.S. A Note on a Simple Apparatus for Detecting Neurological Deficit in Rats and Mice. J. Am. Pharm. Assoc. 1957, 46, 208–209. [Google Scholar] [CrossRef]
- Schrödinger LLC. Maestro, Version 10.6; Schrödinger, LLC: New York, NY, USA, 2016. [Google Scholar]
- Schrödinger LLC. Schrödinger Suite; Schrödinger, LLC: New York, NY, USA, 2016. [Google Scholar]
- Schrödinger LLC. LigPrep, Version 3.8; Schrödinger, LLC: New York, NY, USA, 2016. [Google Scholar]
- Schrödinger LLC. Glide, Version 7.1; Schrödinger, LLC: New York, NY, USA, 2016. [Google Scholar]

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | R | MW | TPSA | logP | AHB | DHB | MV | Vio |
|---|---|---|---|---|---|---|---|---|
| 3a | -H | 444.58 | 77.90 | 3.33 | 7 | 1 | 379.00 | 0 |
| 3b | -CH3 | 455.61 | 77.90 | 3.77 | 7 | 1 | 395.56 | 0 |
| 3c | -OCH3 | 471.61 | 87.14 | 3.38 | 8 | 1 | 404.55 | 0 |
| 3d | -CN | 466.59 | 101.69 | 3.08 | 8 | 1 | 395.86 | 0 |
| 3e | -NO2 | 486.58 | 123.72 | 3.29 | 10 | 1 | 402.34 | 0 |
| 3f | -F | 459.57 | 77.90 | 3.49 | 7 | 1 | 383.94 | 0 |
| 3g | -Cl | 476.03 | 77.90 | 4.00 | 7 | 1 | 392.54 | 0 |
| 3h | -CF3 | 509.58 | 77.90 | 4.22 | 7 | 1 | 410.30 | 1 |
| Treatment | Protection % |
|---|---|
| Control | - |
| Morphine | 76.69 |
| 3a | 48.54 |
| 3b | 44.17 |
| 3c | 47.57 |
| 3d | 24.27 |
| 3e | 19.90 |
| 3f | 63.10 |
| 3g | 66.99 |
| 3h | 29.61 |
| Compound | Receptor * | H-Bond | π-π Interaction | Salt Bridge | π-Cation Interaction |
|---|---|---|---|---|---|
| 3a | MOR | His54 | |||
| DOR | Cys198 | Trp274, Tyr308 | |||
| KOR | Try139, Trp287 | ||||
| 3b | MOR | Lys303 | Tyr148 | ||
| DOR | Lys214, Asp128 | Tyr129 | |||
| KOR | Trp287 | ||||
| 3c | MOR | Lys303, Asp147 | Tyr148 | ||
| DOR | |||||
| KOR | |||||
| 3d | MOR | ||||
| DOR | |||||
| KOR | His291 | ||||
| 3e | MOR | Lys303 | |||
| DOR | His278 | ||||
| KOR | Try139 | Lys227, Glu297 | Lys227, Tyr312 | ||
| 3f | MOR | Lys303, Asp147 | |||
| DOR | Cys198 | Trp274, Tyr308 | |||
| KOR | His291, Trp287 | ||||
| 3g | MOR | Lys303, Asp147 | Tyr148 | ||
| DOR | Asp128 | ||||
| KOR | Tyr139 | ||||
| 3h | MOR | His54 | |||
| DOR | |||||
| KOR | His291 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yücel, N.T.; Osmaniye, D.; Kandemir, Ü.; Evren, A.E.; Can, Ö.D.; Demir Özkay, Ü. Synthesis and Antinociceptive Effect of Some Thiazole-Piperazine Derivatives: Involvement of Opioidergic System in the Activity. Molecules 2021, 26, 3350. https://doi.org/10.3390/molecules26113350
Yücel NT, Osmaniye D, Kandemir Ü, Evren AE, Can ÖD, Demir Özkay Ü. Synthesis and Antinociceptive Effect of Some Thiazole-Piperazine Derivatives: Involvement of Opioidergic System in the Activity. Molecules. 2021; 26(11):3350. https://doi.org/10.3390/molecules26113350
Chicago/Turabian StyleYücel, Nazlı Turan, Derya Osmaniye, Ümmühan Kandemir, Asaf Evrim Evren, Özgür Devrim Can, and Ümide Demir Özkay. 2021. "Synthesis and Antinociceptive Effect of Some Thiazole-Piperazine Derivatives: Involvement of Opioidergic System in the Activity" Molecules 26, no. 11: 3350. https://doi.org/10.3390/molecules26113350
APA StyleYücel, N. T., Osmaniye, D., Kandemir, Ü., Evren, A. E., Can, Ö. D., & Demir Özkay, Ü. (2021). Synthesis and Antinociceptive Effect of Some Thiazole-Piperazine Derivatives: Involvement of Opioidergic System in the Activity. Molecules, 26(11), 3350. https://doi.org/10.3390/molecules26113350