
Large Stabilization Effects by Intramolecular Beryllium Bonds in Ortho-Benzene Derivatives

 ,
,  ,
,
Abstract
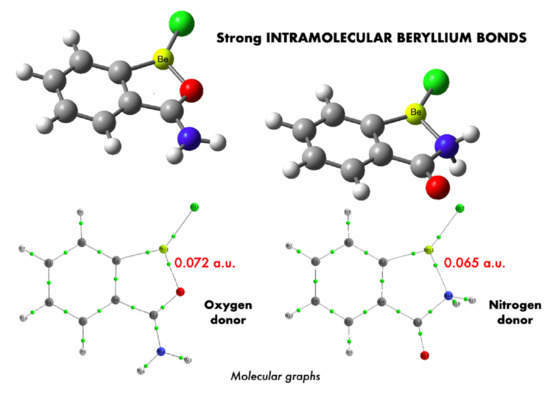
:
1. Introduction
2. Computational Details
3. Results and Discussion
3.1. Stabilization Gained by Beryllium Bond Formation
3.2. Nature of the Intramolecular Beryllium Bond
3.3. Isodesmic Reactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Van der Waals, J.D. On the Continuity of the Gaseous and Liquid States. Ph.D. Thesis, Hoogeschool te Leiden, Leiden, The Netherlands. Edited by J. S. Rowlinson. Dover Phoenix Ed.: New York, NY, USA, 2004.
- Muller-Dethlefs, K.; Hobza, P. Noncovalent interactions: A challenge for experiment and theory. Chem. Rev. 2000, 100, 143–167. [Google Scholar] [CrossRef]
- Dill, K.A. Dominant forces in protein folding. Biochemistry 1990, 29, 7133–7155. [Google Scholar] [CrossRef] [PubMed]
- Newberry, R.W.; Raines, R.T. Secondary Forces in Protein Folding. ACS Chem. Biol. 2019, 14, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.A.; Lee, T.H.; Xu, S.; Park, S.Y.; Kim, T.; Song, S.; Nguyen, T.L.; Ko, S.J.; Hwang, S.; Kim, J.Y.; et al. Interplay of Intramolecular Noncovalent Coulomb Interactions for Semicrystalline Photovoltaic Polymers. Chem. Mater. 2015, 27, 5997–6007. [Google Scholar] [CrossRef]
- Pauling, L. The Shared-Electron Chemical Bond. Proc. Nat. Acad. Sci. USA 1929, 14, 359–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauling, L. The Nature of the Chemical Bond; Cornell University Press: Ithaca, NY, USA, 1939. [Google Scholar]
- Pauling, L. The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry, 3rd ed.; Cornell University Press: Ithaca, NY, USA, 1960. [Google Scholar]
- Alkorta, I.; Elguero, J.; Frontera, A. Not only hydrogen bonds: Other noncovalent interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef]
- Yao, W.B.; Crabtree, R.H. An eta(1)-aldehyde complex and the role of hydrogen bonding in its conversion to an eta(1)-imine complex. Inorg. Chem. 1996, 35, 3007–3011. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Foces-Foces, C. Dihydrogen bonds (A-H ··· H-B). Chem. Commun. 1996, 14, 1633–1634. [Google Scholar] [CrossRef]
- Yáñez, M.; Sanz, P.; Mó, O.; Alkorta, I.; Elguero, J. Beryllium Bonds, do they exist? J. Chem. Theor. Comput. 2009, 5, 2763–2771. [Google Scholar] [CrossRef]
- Yang, X.; Li, Q.Z.; Cheng, J.B.; Li, W.Z. A new interaction mechanism of LiNH2 with MgH2: Magnesium bond. J. Mol. Model. 2013, 19, 247–253. [Google Scholar] [CrossRef]
- Costa, P.; Mieres-Perez, J.; Ozkan, N.; Sander, W. Activation of the B-F Bond by Diphenylcarbene: A Reversible 1,2-Fluorine Migration between Boron and Carbon. Angew. Chem. Int. Ed. 2017, 56, 1760–1764. [Google Scholar] [CrossRef]
- Grabowski, S.J. Boron and other Triel Lewis Acid Centers: From Hypovalency to Hypervalency. Chemphyschem 2014, 15, 2985–2993. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Zahn, S.; Frank, R.; Hey-Hawkins, E.; Kirchner, B. Pnicogen Bonds: A New Molecular Linker? Chem. A Eur. J. 2011, 17, 6034–6038. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. The Pnicogen Bond in Review: Structures, Binding Energies, Bonding Properties, and Spin-Spin Coupling Constants of Complexes Stabilized by Pnicogen Bonds. In Noncovalent Forces; Scheiner, S., Ed.; Springer International Publisher: Cham, Germany, 2015; pp. 191–263. [Google Scholar]
- Wang, W.Z.; Ji, B.M.; Zhang, Y. Chalcogen Bond: A Sister Noncovalent Bond to Halogen Bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Resnati, G. Halogen bonding: A paradigm in supramolecular chemistry. Chem. Eur. J. 2001, 7, 2511–2519. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other s-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Montero-Campillo, M.M.; Alkorta, I.; Elguero, J. Fostering the Basic Instinct of Boron in Boron-Beryllium Interactions. J. Phys. Chem. A 2018, 122, 3313–3319. [Google Scholar] [CrossRef] [PubMed]
- Martín-Sómer, A.; Montero-Campillo, M.M.; Mó, O.; Yáñez, M.; Alkorta, I.; Elguero, J. Some Interesting Features of Non-Covalent Interactions. Croat. Chem. Acta 2014, 87, 291–306. [Google Scholar] [CrossRef]
- Hurtado, M.; Yáñez, M.; Herrero, R.; Guerrero, A.; Dávalos, J.Z.; Abboud, J.-L.M.; Khater, B.; Guillemin, J.C. The Ever-Surprising Boron Chemistry. Enhanced Acidity of Phosphine-boranes. Chem. Eur. J. 2009, 15, 4622–4629. [Google Scholar] [CrossRef] [PubMed]
- Martín-Fernández, C.; Montero-Campillo, M.M.; Alkorta, I.; Yáñez, M.; Mó, O.; Elguero, J. Large Proton-Affinity Enhancements Triggered by Noncovalent Interactions. Chem. Eur. J. 2018, 24, 1971–1977. [Google Scholar] [CrossRef]
- Brea, O.; Alkorta, I.; Corral, I.; Mó, O.; Yáñez, M.; Elguero, J. Intramolecular Beryllium Bonds. Further Insights into Resonance Asistance Phenomena. In Intermolecular Interactions in Crystals: Fundamentals of Crystal Engineering; Novoa, J.J., Ed.; The Royal Society of Chemistry: London, UK, 2018. [Google Scholar]
- Sanz, P.; Montero-Campillo, M.M.; Mó, O.; Yáñez, M.; Alkorta, I.; Elguero, J. Intramolecular magnesium bonds in malonaldehyde-like systems: A critical view of the resonance-assisted phenomena. Theor. Chem. Acc. 2018, 137, 97. [Google Scholar] [CrossRef] [Green Version]
- Foti, M.C.; Barclay, L.R.C.; Ingold, K.U. The role of hydrogen bonding on the H-atom-donating abilities of catechols and naphthalene diols and on a previously overlooked aspect of their infrared spectra. J. Am. Chem. Soc. 2002, 124, 12881–12888. [Google Scholar] [CrossRef]
- Kjaergaard, H.G.; Howard, D.L.; Schofield, D.P.; Robinson, T.W.; Ishiuchi, S.; Fujii, M. OH- and CH-stretching overtone spectra of catechol. J. Phys. Chem. A 2002, 106, 258–266. [Google Scholar] [CrossRef]
- Hunt, N.T.; Turner, A.R.; Wynne, K. Inter- and intramolecular hydrogen bonding in phenol derivatives: A model system for poly-L-tyrosine. J. Phys. Chem. B 2005, 109, 19008–19017. [Google Scholar] [CrossRef]
- Varfolomeev, M.A.; Abaidullina, D.I.; Gainutdinova, A.Z.; Solomonov, B.N. FTIR study of H-bonds cooperativity in complexes of 1,2-dihydroxybenzene with proton acceptors in aprotic solvents: Influence of the intramolecular hydrogen bond. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2010, 77, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Vedernikova, I.; Proynov, E.; Salahub, D.; Haemers, A. Local atomic and orbital reactivity indices from density functional calculations for hydrogen-bonded 1,2-dihydroxybenzene. Int. J. Quantum Chem. 2000, 77, 161–173. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Intramolecular hydrogen bonds in ortho-substituted hydroxybenzenes and in 8-susbtituted 1-hydroxynaphthalenes: Can a methyl group be an acceptor of hydrogen bonds? J. Phys. Chem. A 2001, 105, 10462–10467. [Google Scholar] [CrossRef]
- Zhang, H.Y.; Sung, Y.M.; Wang, X.L. Substituent effects on O-H bond dissociation enthalpies and ionization potentials of catechols: A DFT study and its implications in the rational design of phenolic antioxidants and elucidation of structure-Activity relationships for flavonoid antioxidants. Chem. Eur. J. 2003, 9, 502–508. [Google Scholar] [CrossRef]
- Mandado, M.; Graña, A.M.; Mosquera, R.A. Do 1,2-ethanediol and 1,2-dihydroxybenzene present intramolecular hydrogen bond? Phys. Chem. Chem. Phys. 2004, 6, 4391–4396. [Google Scholar] [CrossRef]
- Gu, J.D.; Wang, J.; Leszczynski, J.; Xie, Y.M.; Schaefer III, H.F. To stack or not to stack: Performance of a new density functional for the uracil and thymine dimers. Chem. Phys. Lett. 2008, 459, 164–166. [Google Scholar] [CrossRef]
- Frisch, M.J.T.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Keith, T.A. AIMAll (Version 19.10.12); TK Gristmill Software: Overland Park, KS, USA, 2019. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Clarendon Press: Oxford, UK, 1990. [Google Scholar]
- Boto, R.A.; Peccati, F.; Laplaza, R.; Quan, C.; Carbone, A.; Piquemal, J.P.; Maday, Y.; Contreras-García, J. Fast, Robust, and Quantitative Analysis of Noncovalent Interactions. J. Chem. Theory Comput. 2020, 16, 4150–4158. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Song, J.W.; Tsuneda, T.; Sato, T.; Hirao, K. Calculations of alkane energies using long-range corrected DFT combined with intramolecular van der waals correlation. Org. Lett. 2010, 12, 1440–1443. [Google Scholar] [CrossRef]
- Snitsiriwat, S.; Bozzelli, J.W. Thermochemical properties for isooctane and carbon radicals: Computational study. J. Phys. Chem. A 2013, 117, 421–429. [Google Scholar] [CrossRef]
- Sánchez-Sanz, G.; Alkorta, I.; Trujillo, C.; Elguero, J. Intramolecular pnicogen interactions in PHF-(CH2) n-PHF (n=2-6) systems. ChemPhysChem 2013, 14, 1656–1665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezei, P.D.; Csonka, G.I.; Ruzsinszky, A.; Kállay, M. Construction and Application of a New Dual-Hybrid Random Phase Approximation. J. Chem. Theory Comput. 2015, 11, 4615–4626. [Google Scholar] [CrossRef] [PubMed]
- Channar, P.A.; Saeed, A.; Larik, F.A.; Flörke, U.; El-Seedi, H.; Rodríguez Pirani, L.S.; Erben, M.F. An intramolecular 1,5-chalcogen bond on the conformational preference of carbonyl thiocarbamate species. New J. Chem. 2020, 44, 5243–5253. [Google Scholar] [CrossRef]
- Kovács, A.; Hargittai, I. Hydrogen-bonding interactions of the trifluoromethyl group: 2-Trifluoromethylvinyl alcohol. Int. J. Quant. Chem. 1997, 62, 645–652. [Google Scholar] [CrossRef]
- Howard, S.T.; Abernethy, C.D. Intramolecular C-H...Ccarbene Hydrogen Bonds and Competing Interactions in Monoprotonated Tripodal Carbenes. J. Comput. Chem. 2004, 25, 649–659. [Google Scholar] [CrossRef]
- Sanz, P.; Mó, O.; Yáñez, M.; Elguero, J. Bonding in Tropolone, 2-Aminotropone and Aminotroponimine. No Evidence of Resonance Assisted Hydrogen Bond (RAHB) Effects. Chem. Eur. J. 2008, 14, 4225–4232. [Google Scholar] [CrossRef]
- Rosado, M.T.S.; Jesus, A.J.L.; Reva, I.D.; Fausto, R.; Redinha, J.S. Conformational cooling dynamics in matrix-isolated 1,3-butanediol. J. Phys. Chem. A 2009, 113, 7499–7507. [Google Scholar] [CrossRef] [Green Version]
- Jesus, A.J.L.; Redinha, J.S. Conformational study of charged cyclohexyldiamines and their gas phase acid-base properties. Struct. Chem. 2011, 22, 999–1006. [Google Scholar] [CrossRef]
- Zhang, L.; Li, D. An insight into intramolecular blue-shifting C–H⋯π hydrogen bonds in 1,3-hexadien-5-yne and its halogen-substituted derivatives. Chem. Phys. 2019, 518, 58–68. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Houk, K.N. Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem. Int. Ed. 2017, 56, 10070–10086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, A.; Kollman, P.; Rothenberg, S.; McKelvey, J. Hydrogen Bonding Ability of the Amide Group. J. Am. Chem. Soc. 1974, 96, 3794–3800. [Google Scholar] [CrossRef]
- Emsley, J.; Hoyte, O.P.A.; Overill, R.E. Ab Initio Calculations on the Very Strong Hydrogen Bond of the Biformate Anion and Comparative Esterification Studies. J. Am. Chem. Soc. 1978, 100, 3303–3306. [Google Scholar] [CrossRef]
- Ewbank, J.D.; Klimkowski, V.J.; Siam, K.; Schäfer, L. Conformational analysis of the methyl ester of alanine by gas electron diffraction and Ab initio geometry optimization. J. Mol. Struct. 1987, 160, 275–285. [Google Scholar] [CrossRef]
- Wu, J.; Lebrilla, C.B. Gas-Phase Basicities and Sites of Protonation of Glycine Oligomers (GLYn; n = 1–5). J. Am. Chem. Soc. 1993, 115, 3270–3275. [Google Scholar] [CrossRef]
- Mikshiev, V.Y.; Pozharskii, A.F.; Filarowski, A.; Novikov, A.S.; Antonov, A.S.; Tolstoy, P.M.; Vovk, M.A.; Khoroshilova, O.V. How Strong is Hydrogen Bonding to Amide Nitrogen? ChemPhysChem 2020, 21, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Oziminski, W.P. The kinetics of water-assisted tautomeric 1,2-proton transfer in azoles: A computational approach. Struct. Chem. 2016, 27, 1845–1854. [Google Scholar] [CrossRef] [Green Version]
- Mata, I.; Alkorta, I.; Molins, E.; Espinosa, E. Universal features of the electron density distribution in hydrogen- bonding regions: A comprehensive study involving H···X (X=H, C, N, O, F, S, Cl, π) interactions. Chem. Eur. J. 2010, 16, 2442–2452. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
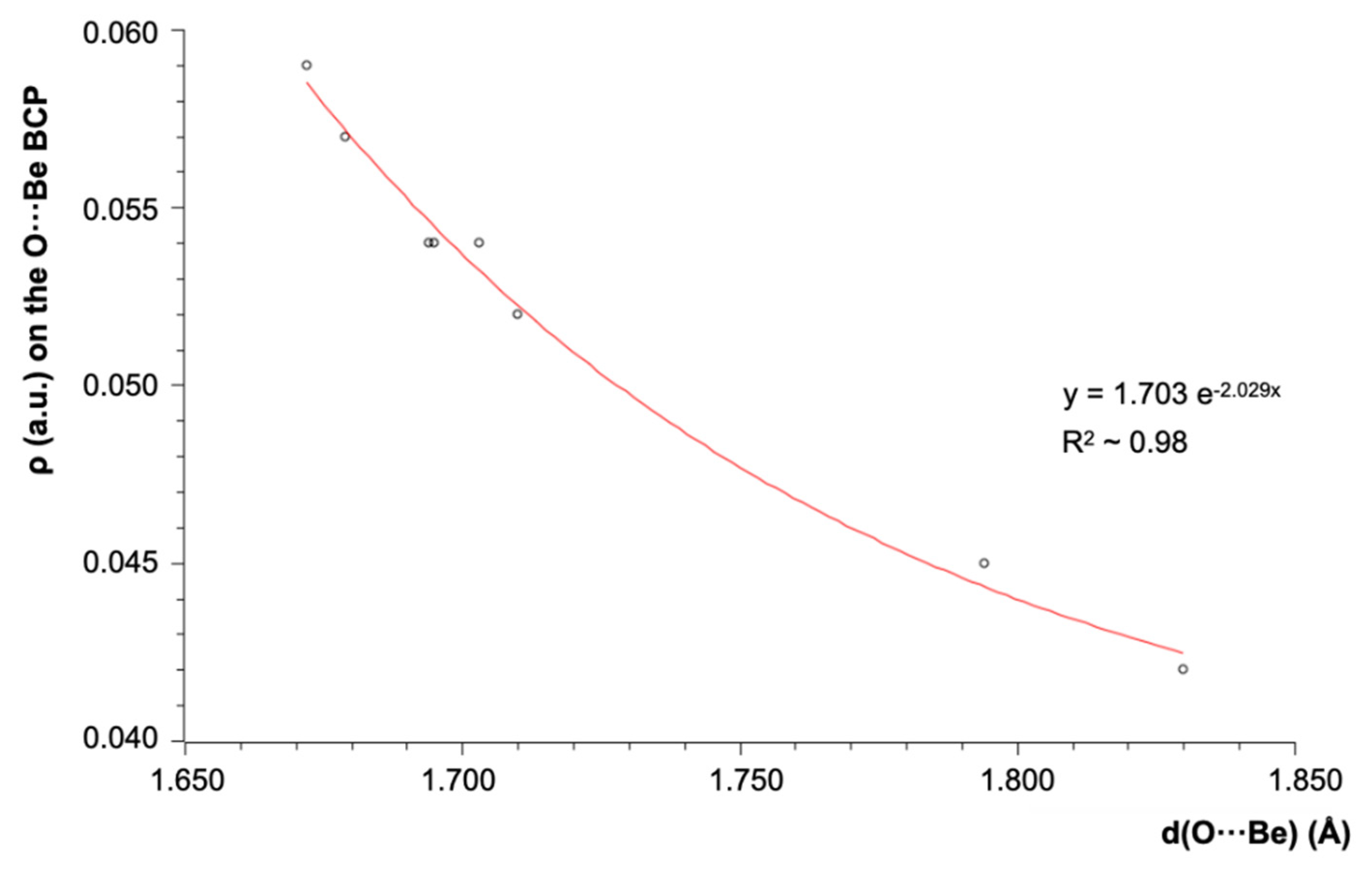{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ΔH1 | Acceptor | BeH | CH2BeH | C2H4BeH | BeF | CH2BeF | C2H4BeF | BeCl | CH2BeCl | C2H4BeCl |
| Donor | ||||||||||
| OH | 2.2 | 60.4 | 61.8 | 6.7 | 59.9 | 62.5 | 7.1 | 67.6 | 71.0 | |
| SH | 12.5 | 41.8 | 35.8 | 11.3 | 40.9 | 32.5 | 11.7 | 47.8 | 39.8 | |
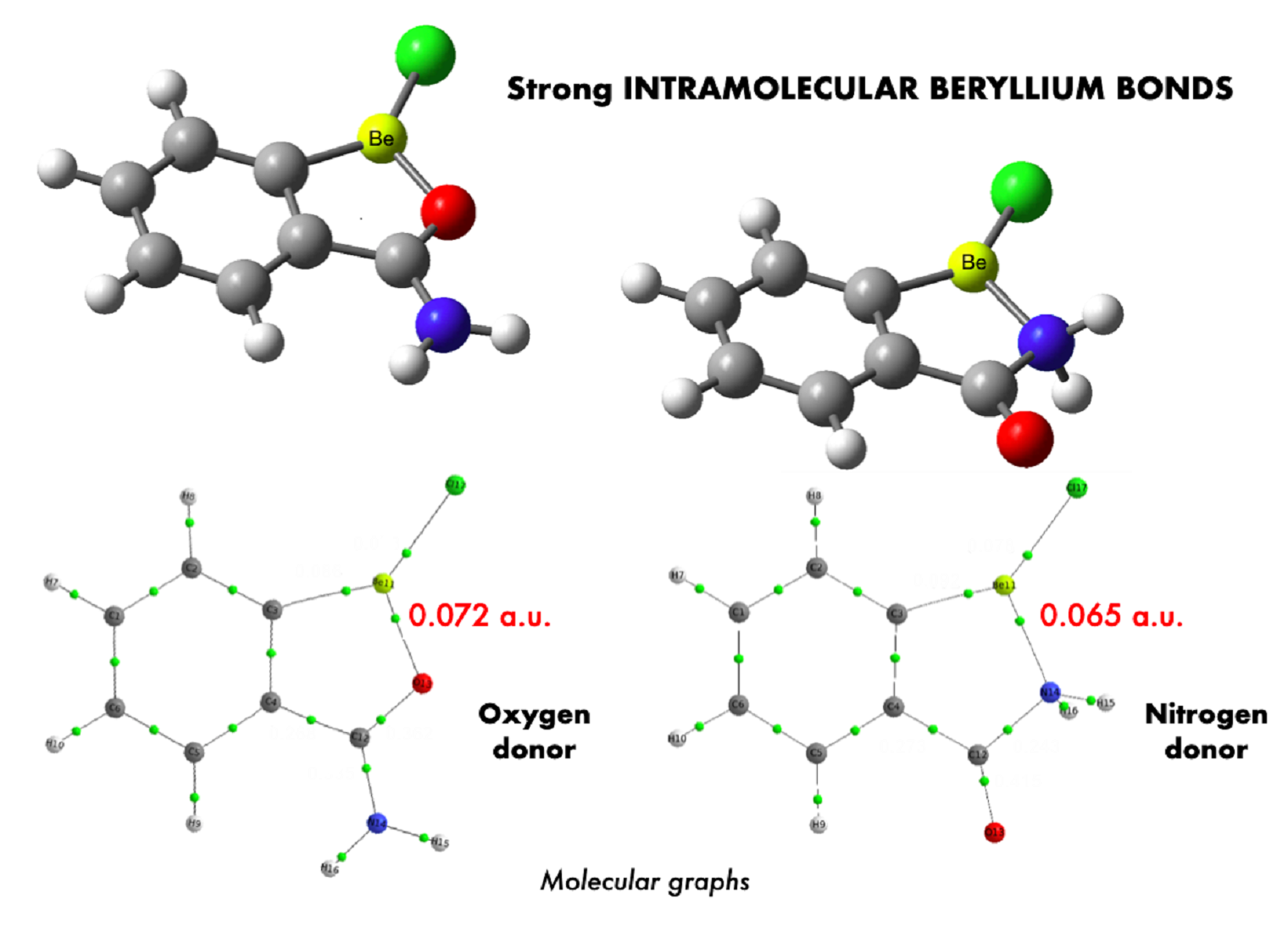
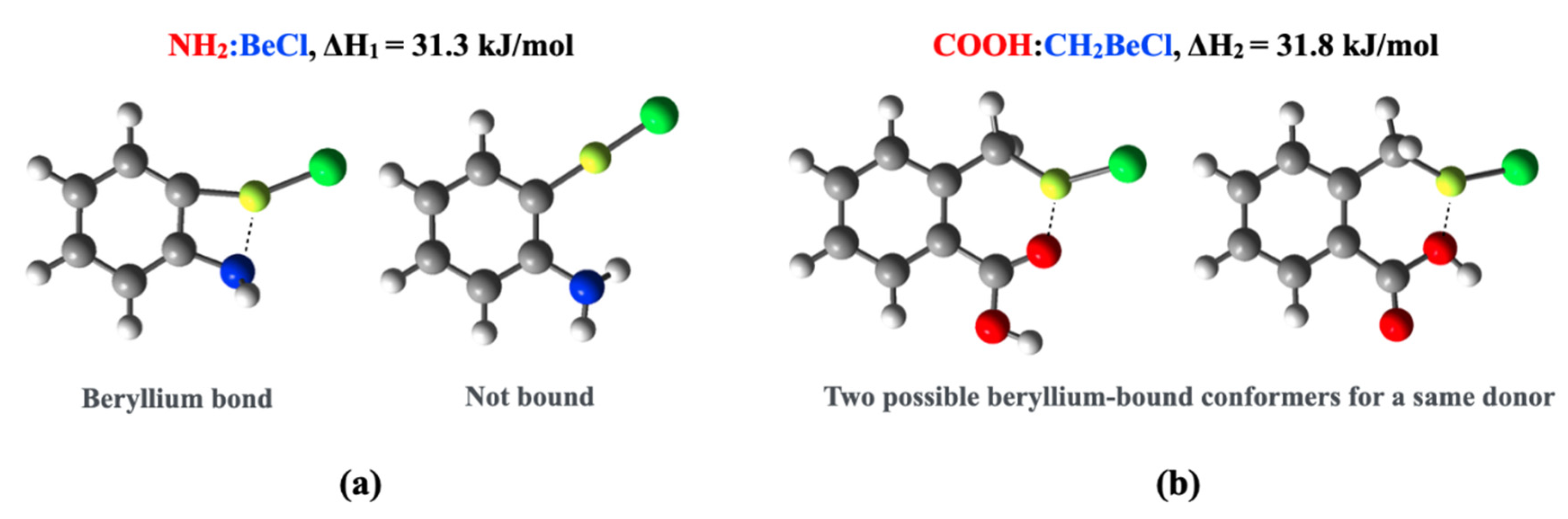
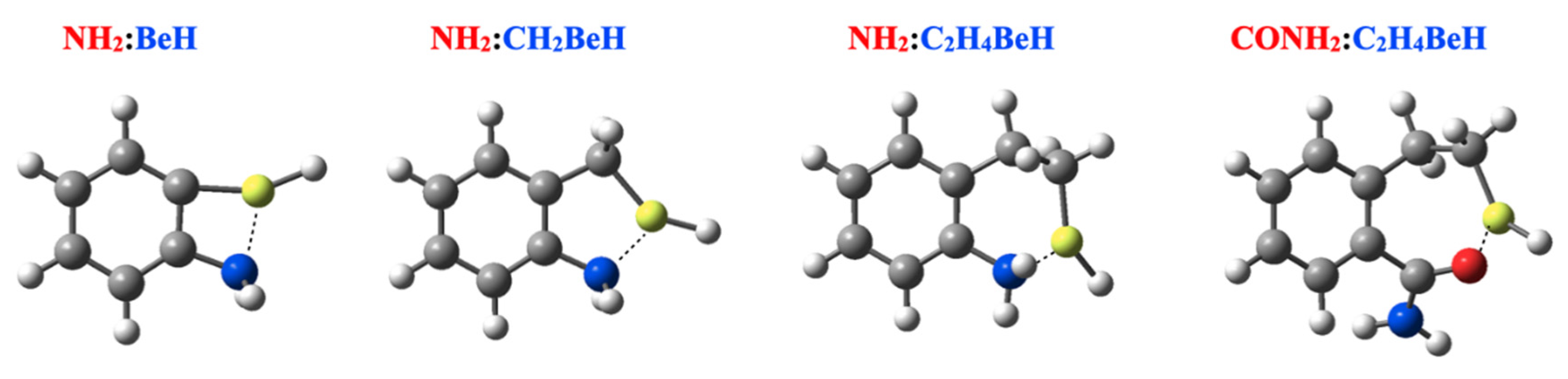
| NH2 | 23.1 | 69.6 | 74.5 | 26.8 | 73.6 | 74.3 | 31.3 | 81.0 | 87.0 | |
| PH2 | 4.4 | 21.8 | 26.4 | 1.6 | 20.2 | 21.9 | 1.4 | 26.3 | 29.9 | |
| ΔH2 | Acceptor | BeH | CH2BeH | C2H4BeH | BeF | CH2BeF | C2H4BeF | BeCl | CH2BeCl | C2H4BeCl |
| Donor | ||||||||||
| COOH | 14.8 | 28.0 | 25.2 | 19.0 | 30.6 | 24.7 | 18.8 | 31.8 | 27.9 | |
| CONH2 | 51.3 | 53.9 | 50.5 | 56.2 | 57.6 | 54.1 | 54.1 | 57.2 | 56.4 |
 | |||||||||
| BeH | CH2BeH | C2H4BeH | BeF | CH2BeF | C2H4BeF | BeCl | CH2BeCl | C2H4BeCl | |
| OH | 1.838 159.4 | 1.695 149.1 | 1.694 146.4 | 1.830 159.7 | 1.710 149.4 | 1.703 146.8 | 1.794 157.7 | 1.679 148.0 | 1.672 144.2 |
| SH | 2.727 168.9 | 2.241 149.2 | 2.246 147.1 | 2.648 166.4 | 2.290 149.9 | 2.311 148.1 | 2.487 160.6 | 2.228 145.9 | 2.232 143.3 |
| NH2 | 1.855 154.6 | 1.787 145.5 | 1.794 141.7 | 1.847 154.7 | 1.796 145.2 | 1.796 142.1 | 1.819 153.7 | 1.769 144.0 | 1.767 139.7 |
| PH2 | 3.007 172.5 | 2.310 146.4 | 2.280 142.6 | 2.987 171.3 | 2.365 147.8 | 2.353 144.2 | 2.984 170.1 | 2.285 143.4 | 2.266 139.9 |
| CO*OH COO*H | 1.697 143.3 1.690 148.8 | 1.650 137.0 1.674 144.8 | 1.690 138.6 1.740 144.9 | 1.694 143.3 1.701 149.0 | 1.652 138.0 1.681 145.2 | 1.693 138.0 1.742 145.4 | 1.675 141.6 1.676 147.2 | 1.631 135.7 1.685 142.8 | 1.663 135.8 1.710 141.1 |
| CO*NH2 CON*H2 | 1.660 141.5 1.773 144.4 | 1.635 136.5 1.779 140.4 | 1.660 135.8 1.828 142.2 | 1.657 141.3 1.779 144.5 | 1.633 136.8 1.787 140.7 | 1.662 135.1 1.832 142.3 | 1.639 139.8 1.751 142.7 | 1.613 140.7 1.761 138.4 | 1.636 133.2 1.797 139.3 |
| Interacting Pairs | Eiso | Eint | Edef | |
|---|---|---|---|---|
| OH | BeH | −6.1 | −86.6 | 80.5 |
| BeF | −9.0 | −99.1 | 90.1 | |
| BeCl | −10.6 | −103.9 | 93.3 | |
| NH2 | BeH | −49.7 | −128.1 | 78.4 |
| BeF | −53.6 | −143.0 | 89.3 | |
| BeCl | −59.2 | −148.2 | 89.0 | |
| SH | BeH | −13.7 | −29.3 | 15.7 |
| BeF | −14.3 | −37.4 | 23.0 | |
| BeCl | −15.7 | −53.1 | 37.4 | |
| PH2 | BeH | −7.4 | −11.8 | 4.4 |
| BeF | −7.9 | −13.2 | 5.3 | |
| BeCl | −8.4 | −13.5 | 5.2 | |
| COOH | BeH | −82.6 | −166.3 | 83.7 |
| −66.1 | −130.9 | 64.8 | ||
| BeF | −85.1 | −173.4 | 88.3 | |
| −64.3 | −129.7 | 65.4 | ||
| BeCl | −91.2 | −182.7 | 91.5 | |
| −70.4 | −141.4 | 71.0 | ||
| CONH2 | BeH | −111.2 | −198.1 | 86.9 |
| −59.5 | −168.4 | 109.0 | ||
| BeF | −114.1 | −206.6 | 92.5 | |
| −57.9 | −172.8 | 114.9 | ||
| BeCl | −122.2 | −216.9 | 94.7 | |
| −67.8 | −189.8 | 122.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
I-Ting, T.; Montero-Campillo, M.M.; Alkorta, I.; Elguero, J.; Yáñez, M. Large Stabilization Effects by Intramolecular Beryllium Bonds in Ortho-Benzene Derivatives. Molecules 2021, 26, 3401. https://doi.org/10.3390/molecules26113401
I-Ting T, Montero-Campillo MM, Alkorta I, Elguero J, Yáñez M. Large Stabilization Effects by Intramolecular Beryllium Bonds in Ortho-Benzene Derivatives. Molecules. 2021; 26(11):3401. https://doi.org/10.3390/molecules26113401
Chicago/Turabian StyleI-Ting, Tsai, M. Merced Montero-Campillo, Ibon Alkorta, José Elguero, and Manuel Yáñez. 2021. "Large Stabilization Effects by Intramolecular Beryllium Bonds in Ortho-Benzene Derivatives" Molecules 26, no. 11: 3401. https://doi.org/10.3390/molecules26113401
APA StyleI-Ting, T., Montero-Campillo, M. M., Alkorta, I., Elguero, J., & Yáñez, M. (2021). Large Stabilization Effects by Intramolecular Beryllium Bonds in Ortho-Benzene Derivatives. Molecules, 26(11), 3401. https://doi.org/10.3390/molecules26113401