
Bisindole Alkaloids from the Alstonia Species: Recent Isolation, Bioactivity, Biosynthesis, and Synthesis †




Abstract
:1. Introduction
2. Occurrence and Isolation of Bisindoles from Alstonia Species

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bisindoles | Types of Monomeric Units Present in Bisindoles | Alstonia Species | Morphology and References |
|---|---|---|---|
| (+)-Alstomacroline 1 | Macroline-sarpagine | A. scholaris, A. glaucescens, and A. macrophylla extracts | Leaves, stem-bark, and root-bark [19,20] |
| A. macrophylla | Bark [18] | ||
| (+)-Alstomacrophylline 2 | Macroline-macroline | A. macrophylla | Bark [19] |
| A. scholaris, A. glaucescens, and A. macrophylla extracts | Leaves, stem-bark, and root-bark [20] | ||
| (−) Alstonisidine 3 | Ajmaline-macroline | A. muelleriana | Bark [21,22] |
| (−)-Angustilongine E 6 | Macroline-sarpagine | A. penangiana | Stem-bark [5] |
| (−)-Angustilongine F 7 | Macroline-sarpagine | A. penangiana | Stem-bark [5] |
| (+)-Angustilongine G 8 | Macroline-sarpagine | A. penangiana | Stem-bark [5] |
| (+)-Angustilongine H 9 | Macroline-sarpagine | A. penangiana | Stem-bark [5] |
| (+)-Angustilongine J 10 | Macroline-sarpagine | A. penangiana | Stem-bark [5] |
| (+)-Angustilongine K 11 | Macroline-sarpagine | A. penangiana | Stem-bark [5] |
| (−)-Angustilongine L 12 | Macroline-pleiocarpamine | A. penangiana | Stem-bark [5] |
| (−)-Anhydromacralstonine 27 | Macroline-macroline | A. angustifolia | Stem-bark [28] |
| (+)-Des-N’a-Methylanhydromacralstonine 30 | Macroline-macroline | A. muelleriana | Bark [20] |
| A. angustifolia | Stem-bark [33] | ||
| A. muelleriana | Leaves, stem-bark and root-bark [20,35] | ||
| (−)-Lumusidine A 14 | Macroline-macroline | A. macrophylla | Stem-bark [18] |
| (−)-Lumusidine B 15 | Macroline-macroline | A. macrophylla | Stem-bark [18] |
| (−)-Lumusidine C 16 | Macroline-macroline | A. macrophylla | Stem-bark [18] |
| (−)-Lumusidine D 17 | Macroline-macroline | A. macrophylla | Stem-bark [18] |
| (+)-Lumutinine A 18 | Macroline-macroline | A. macrophylla | Stem-bark [26] |
| (−)-Lumutinine B 19 | Macroline-macroline | A. macrophylla | Stem-bark [26] |
| (+)-Lumutinine C 20 | Macroline-sarpagine | A. macrophylla | Stem-bark [26] |
| (+)-Lumutinine D 19 | Macroline-sarpagine | A. macrophylla | Stem-bark [26] |
| (+)-Lumutinine E 21 | Macroline-sarpagine | A. angustifolia | Stem-bark [28] |
| (+)-Macralstonidine 23 | Macroline-sarpagine | A. macrophylla | Bark [29,30] |
| A. somersentenis | Bark [29] | ||
| A. spectabilis | Bark [31] | ||
| (+)-Macralstonine 24 | Macroline-macroline | A. scholaris, A. glaucescens, and A. macrophylla extracts | Leaves, stem-bark and root-bark [19] |
| A. macrophylla | [29,30,32,33,34] | ||
| A. angustifolia | [36] | ||
| A. muelleriana | [35] | ||
| A. glabriflora Mgf. | [31] | ||
| (+)-O-Acetylmacralstonine 25 | Macroline-macroline | A. scholaris, A. glaucescens, and A. macrophylla extracts | [20] |
| (+)-O-Methylmacralstonine 26 | Macroline-macroline | A. scholaris, A. glaucescens, and A. macrophylla extracts | [20] |
| (+)-Macrocarpamine 31 | Macroline-pleiocarpamine | A. scholaris, A. glaucescens, and A. macrophylla extracts | Leaves, stem-bark, and root-bark [20] |
| A. angustifolia | Stem-bark [28] | ||
| 10-Methoxy macrocarpamine 34 | Macroline-pleiocarpamine | A. angustifolia | Leaves [41] |
| 10-Methoxy macrocarpamine 4′-N-oxide 35 | Macroline-pleiocarpamine | A. angustifolia | Leaves [41] |
| (−)-Perhentidine A 36 | Macroline-macroline | A. macrophylla and A. angustifolia | Stem-bark [28,33] |
| (−)-Perhentidine B 37 | Macroline-macroline | A. macrophylla and A. angustifolia | Stem-bark [33] |
| (−)-Perhentidine C 38 | Macroline-macroline | A. macrophylla and A. angustifolia | Stem-bark [28,33] |
| (−)-Perhentinine 39 | Macroline-macroline | A. macrophylla | Stem-bark [18] |
| A. angustifolia | Leaves [18] | ||
| A. macrophylla | Leaves [18] | ||
| (+)-Perhentisine A 40 | Macroline-sarpagine | A. angustifolia | Stem-bark [28] |
| (−)-Perhentisine B 41 | Macroline-sarpagine | A. angustifolia | Stem-bark [28] |
| (+)-Perhentisine C 42 | Macroline-sarpagine | A. angustifolia | Stem-bark [28] |
| A. muelleriana | Leaves and stem-bark [37] | ||
| (+)-Villalstonidine A 47 | Macroline-pleiocarpamine | A. angustifolia | Stem-bark [28] |
| (+)-Villalstonidine B 48 | Macroline-pleiocarpamine | A. angustifolia | Stem-bark [28] |
| A. macrophylla | Stem-bark [27] | ||
| (+)-Villalstonidine C 49 | Macroline-pleiocarpamine | A. angustifolia | Stem-bark [28] |
| (+)-Villalstonidine D 50 | Macroline-pleiocarpamine | A. angustifolia | Stem-bark [28] |
| (+)-Villalstonidine E 51 | Macroline-pleiocarpamine | A. angustifolia | Stem-bark [28] |
| (+)-Villalstonidine F 52 | Macroline-pleiocarpamine | A. macrophylla | Stem-bark [27] |
| (+)-Villalstonine 43 | Macroline-pleiocarpamine | A. angustifolia | Leaves and stem-bark [28,36,39,41] |
| A. macrophylla | [41] | ||
| A. villosa | [41] | ||
| A. verticillosa | [41] | ||
| A. somersentensis | [41] | ||
| Villalstonine N(4)-oxide 44 | Macroline-pleiocarpamine | A. angustifolia | Stem-bark [28] |
| A. scholaris, A. glaucescens, and A. macrophylla extracts | Leaves, stem-bark, and root-bark [20,30,32,38] | ||
| (+)-10-Methoxy villalstonine 45 | Macroline-pleiocarpamine | A. angustifolia | Leaves [41] |
| 10-Methoxy villalstonine 4′-N-oxide 46 | Macroline-pleiocarpamine | A. angustifolia | Leaves [41] |
3. Bioactivity of Bisindole Alkaloids from Alstonia Species
4. Biosynthesis
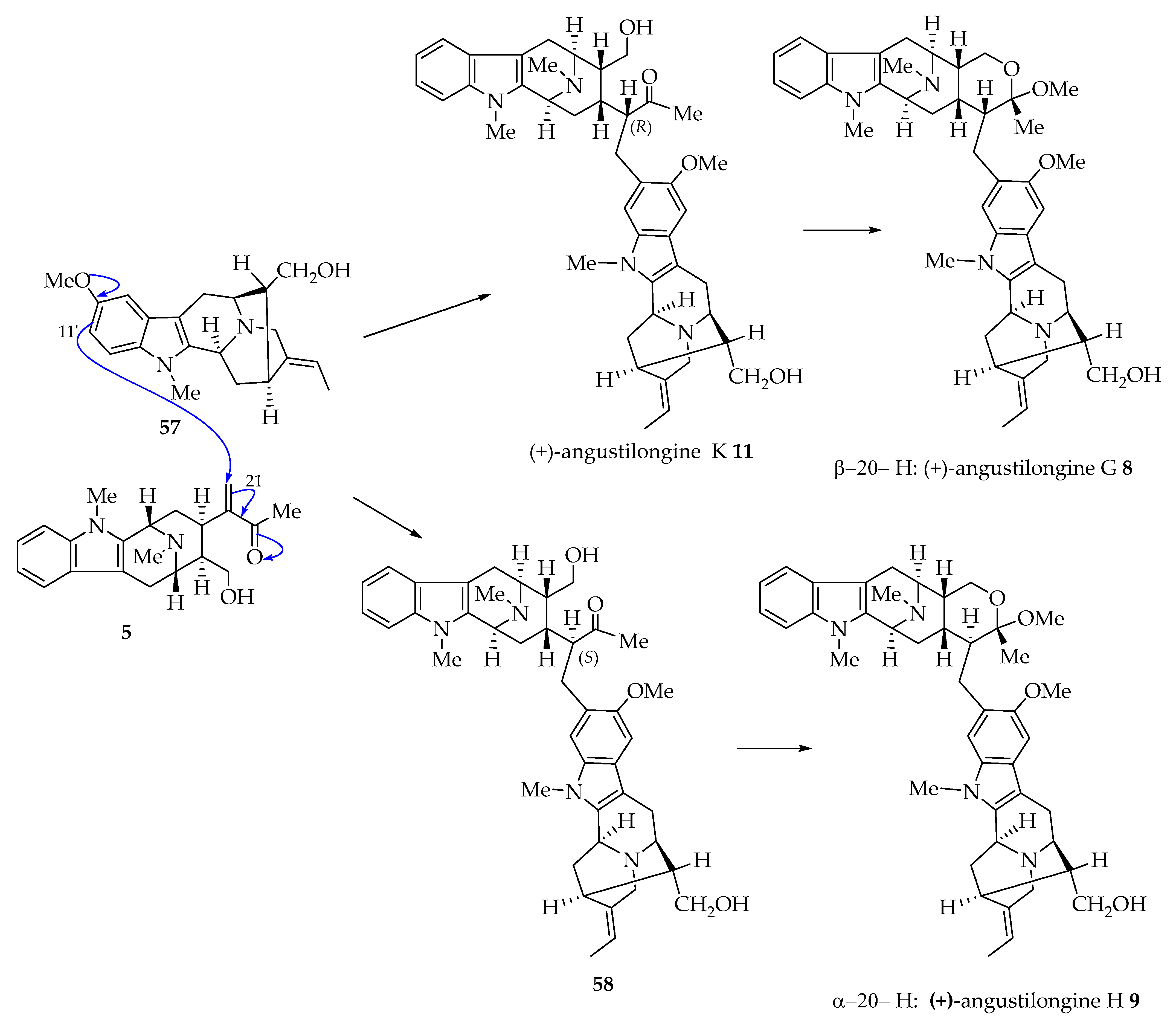
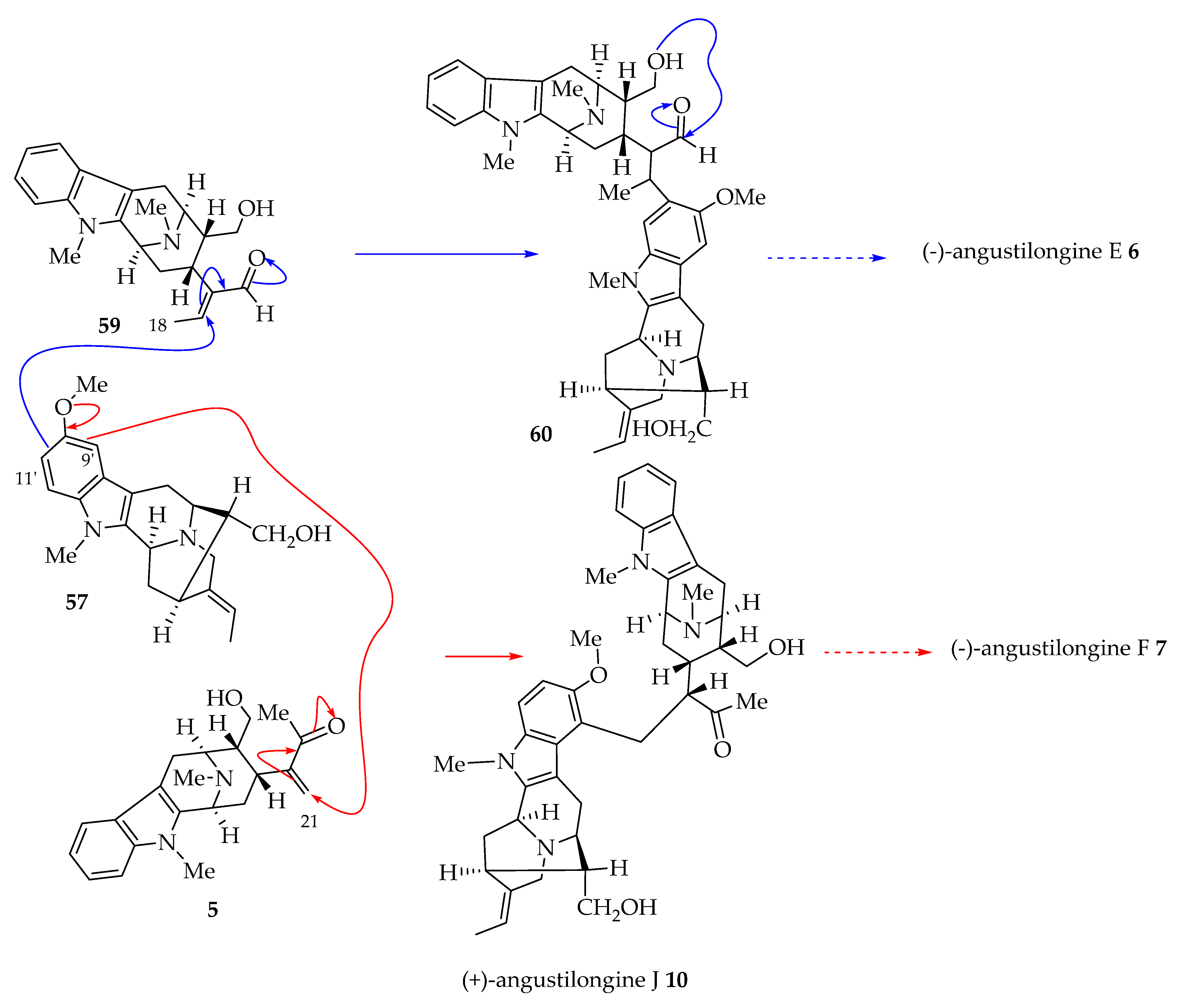
4.1. Proposed Biogenetic Pathway toward Angustilongines (6–9)
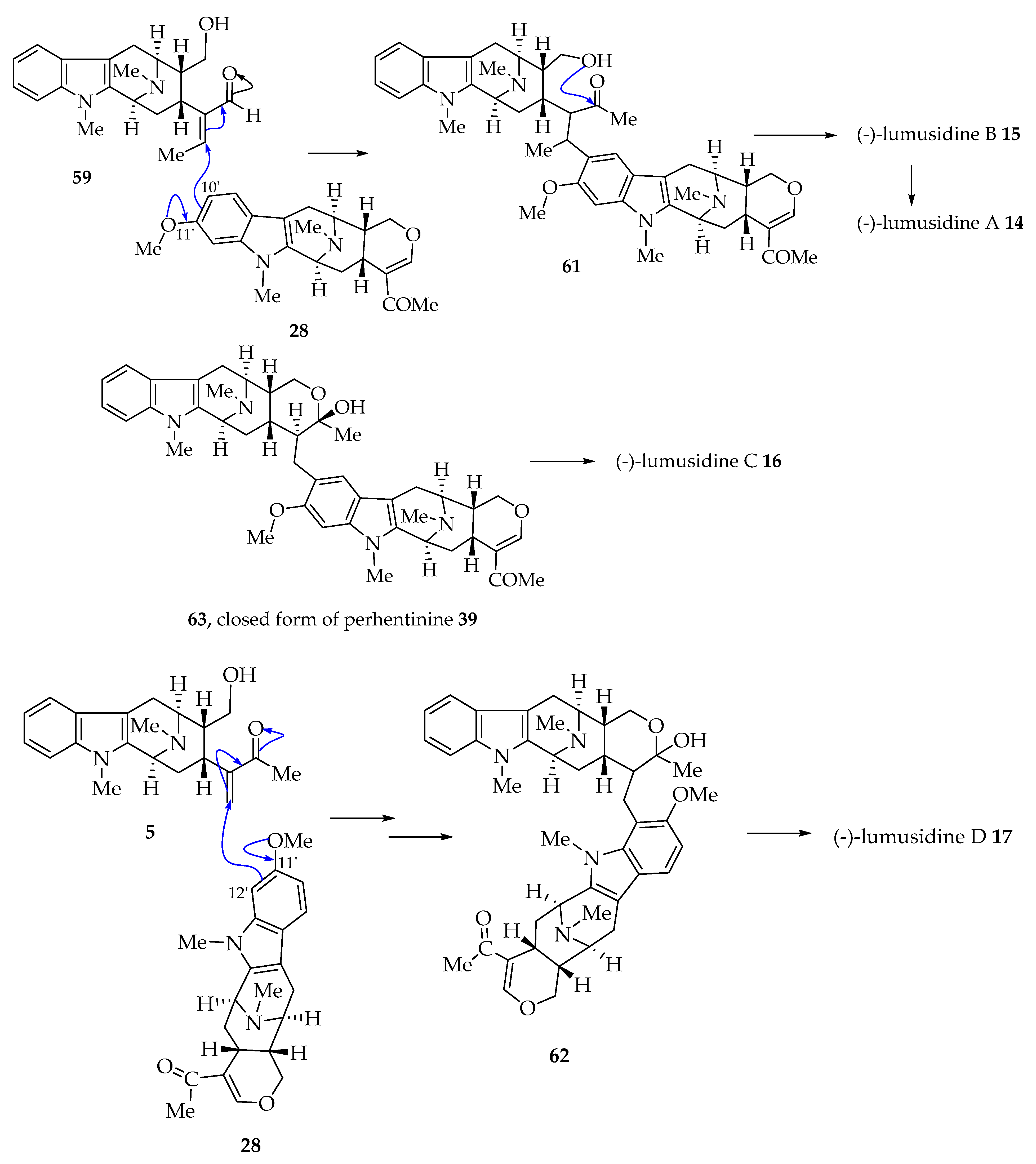
4.2. Plausible Biogenetic Pathway to (-)-Lumusidines A-D (14–17)
4.3. Proposed Biogenetic Pathway to Lumutinines A–D (16–19)
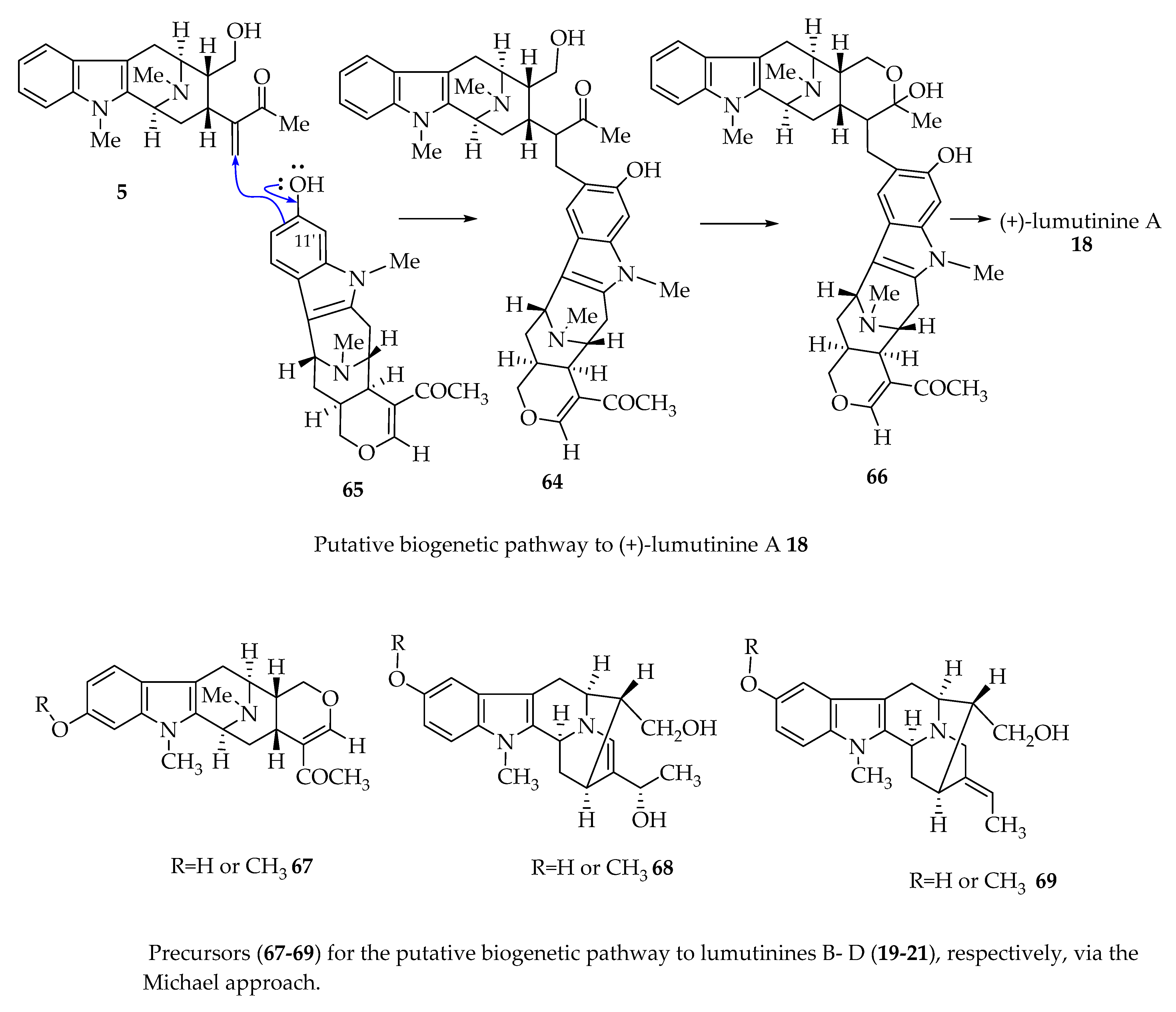
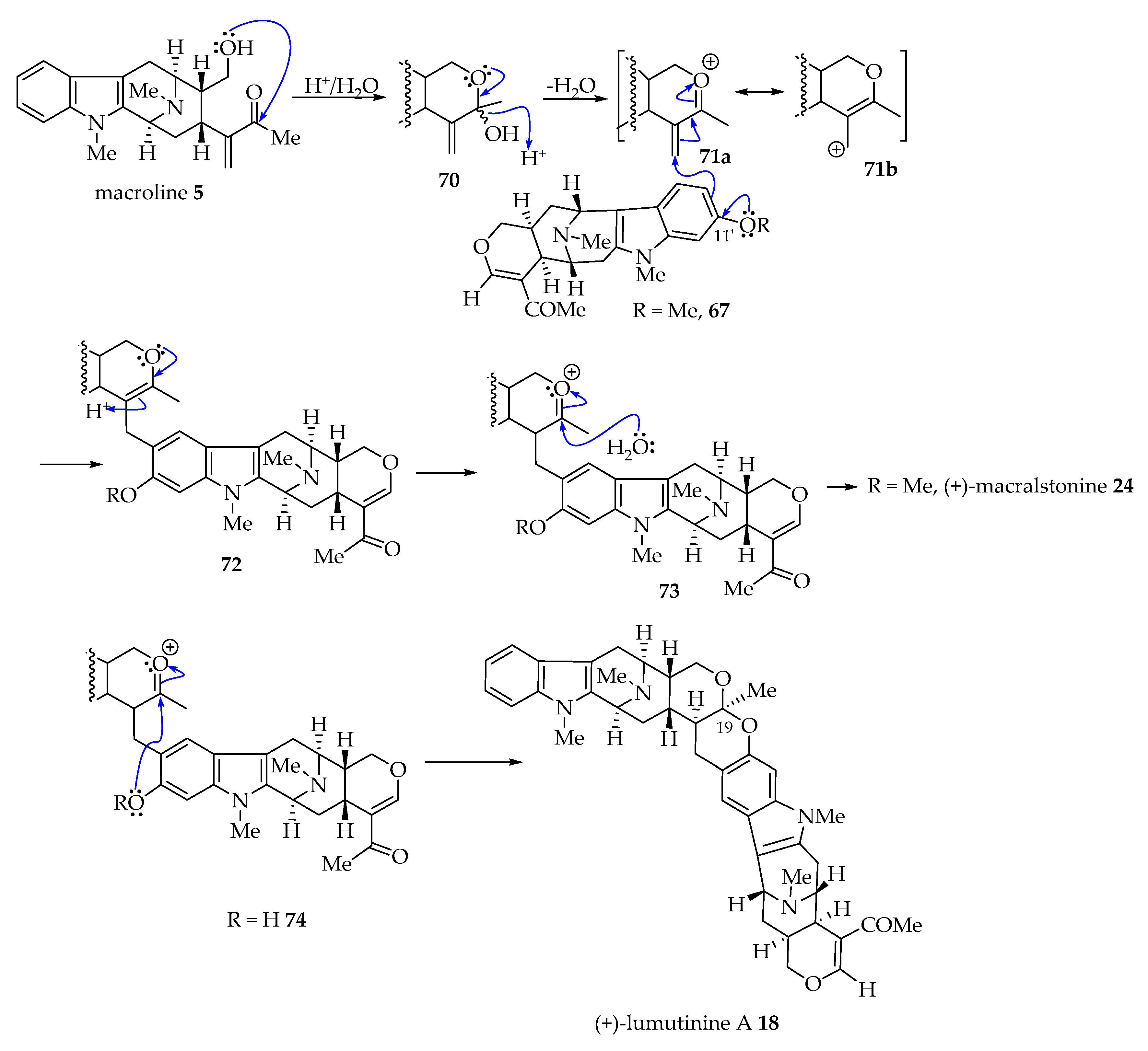
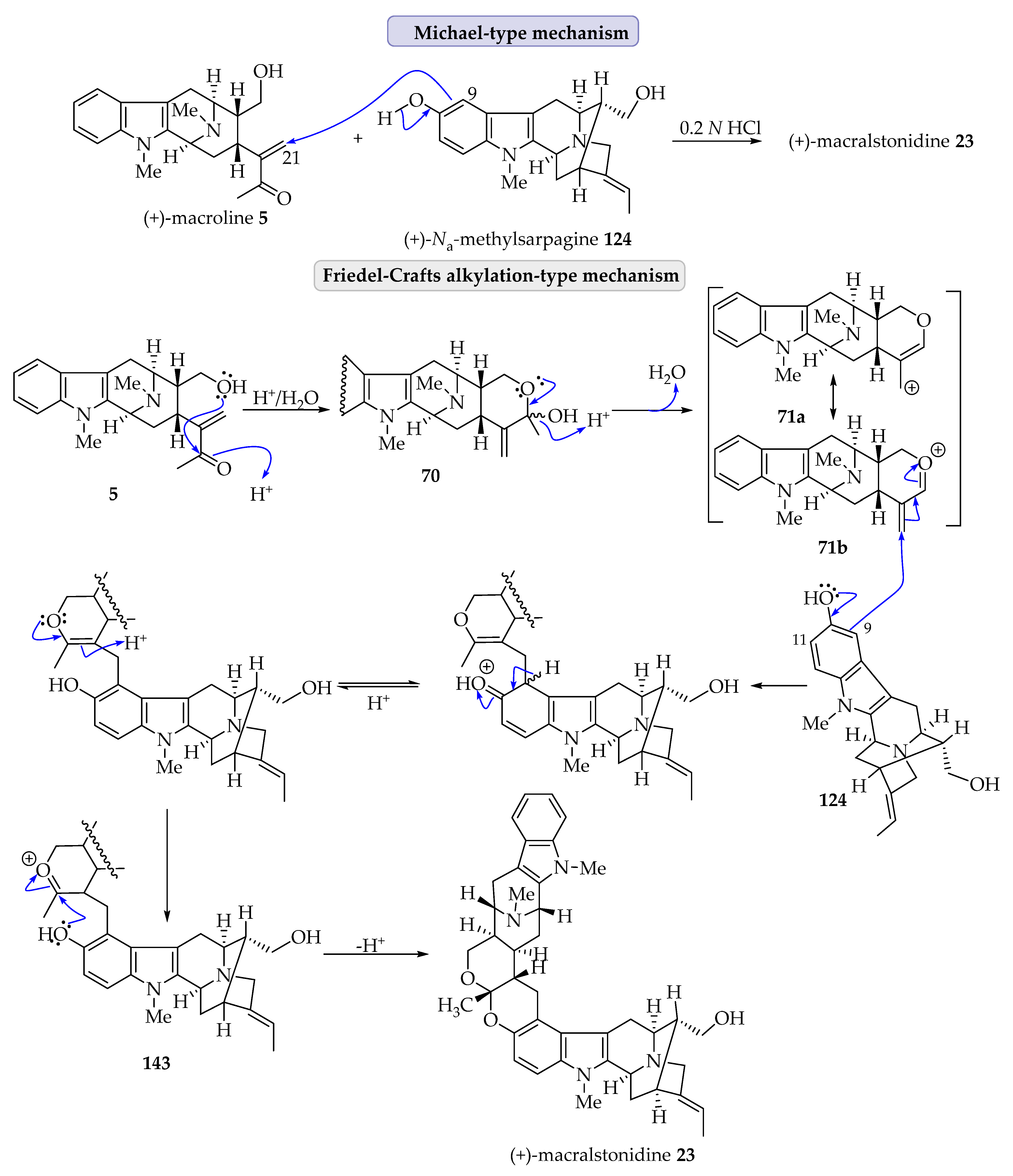
4.4. Possible Alternative Mechanism of Bisindole Formation of (+)-Lumutinine A 16 and (+)-Macralstonine 24 as Representative Examples via a Friedel–Crafts Alkylation Process as Suggested by Fukuyama
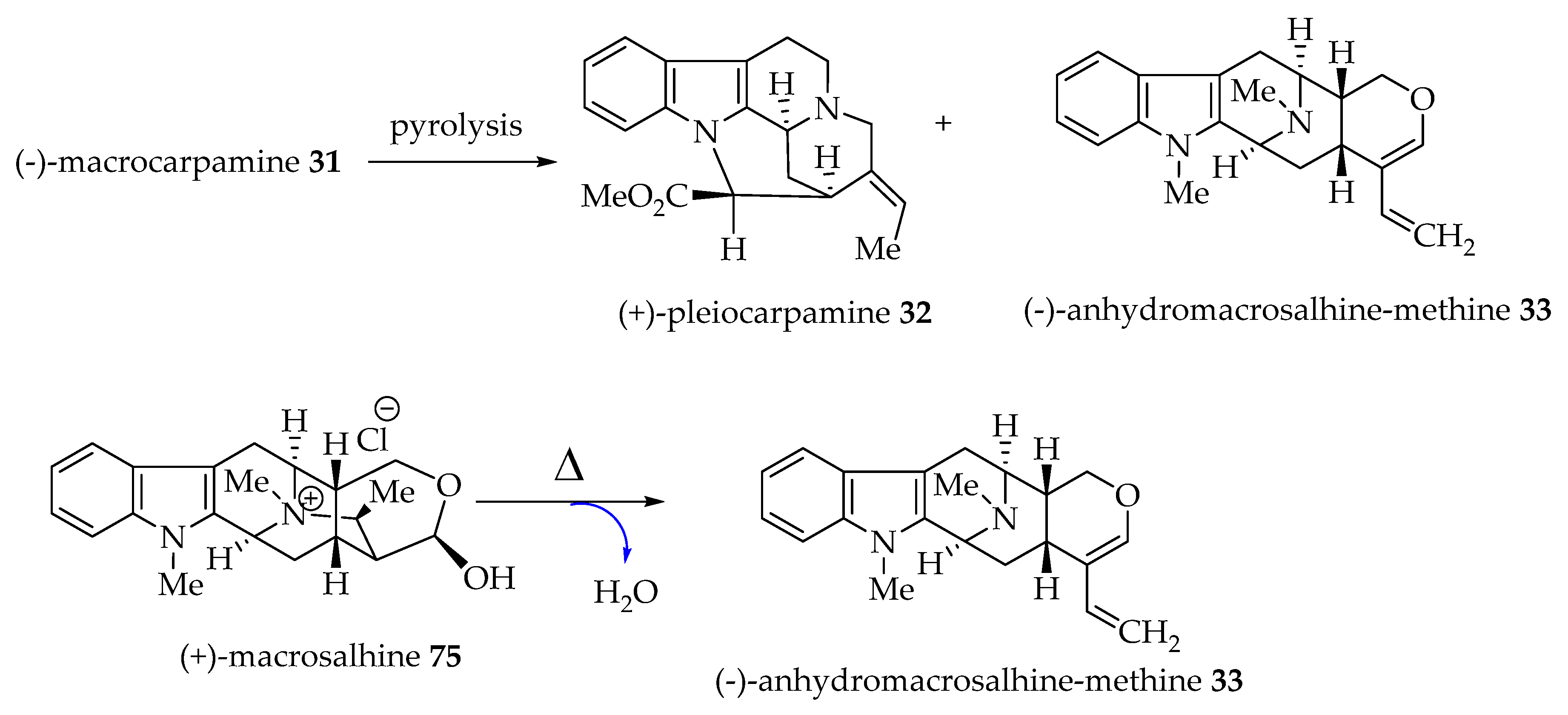
4.5. Proposed Biogenetic Pathway to (-)-Macrocarpamine 31
5. Synthesis of Representative Monomeric Units of Bisindole Alkaloids from Alstonia Species Discussed Herein, Required for Bisindole Synthesis
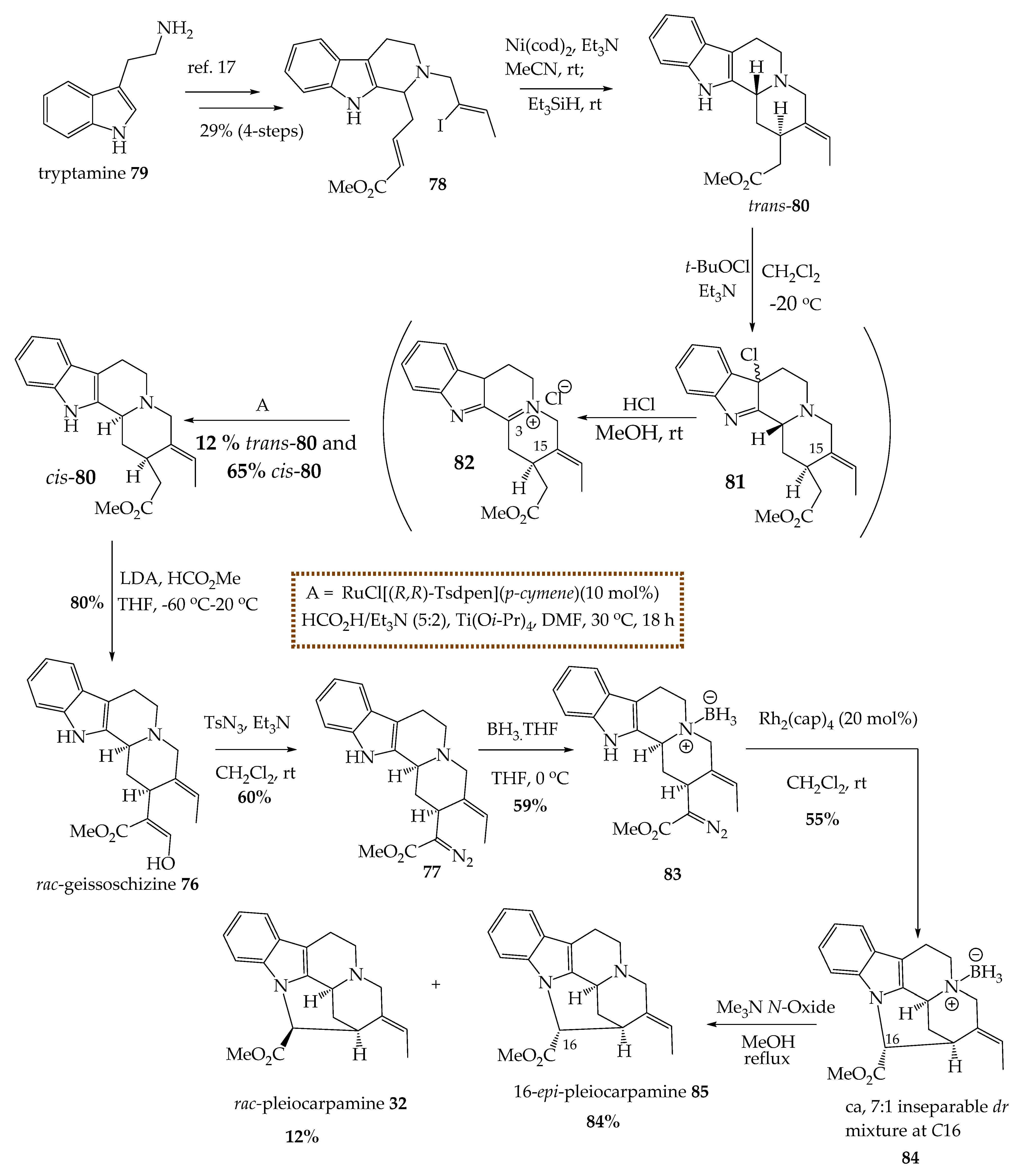
5.1. The Total Synthesis of (±)-Pleiocarpamine 32
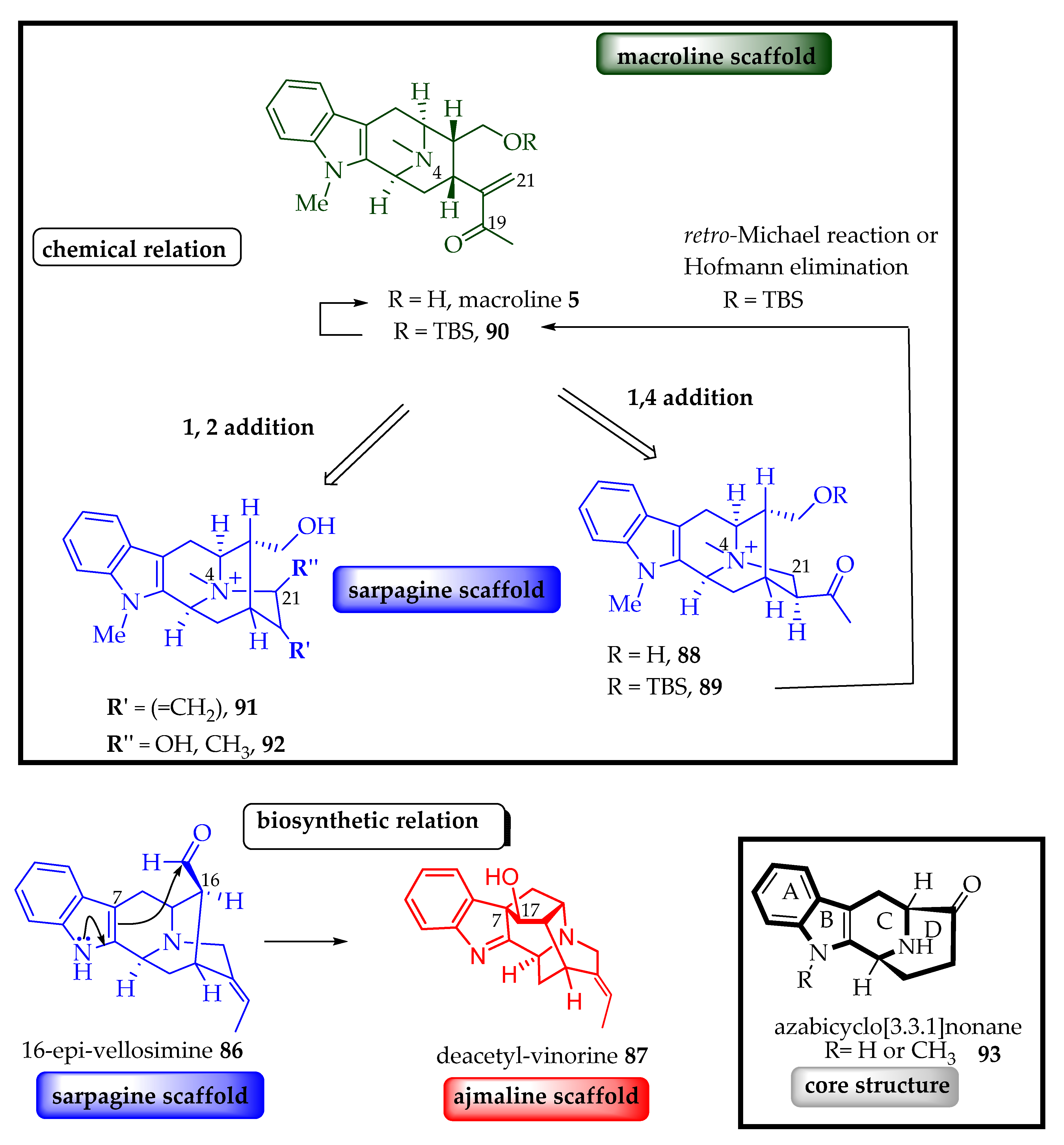
5.2. Biosynthetic Relations among the Sarpagine/Macroline/Ajmaline Family of Alkaloids
5.3. Synthesis of Monomeric Macroline Units Employed for the Synthesis of Bisindole Alkaloids
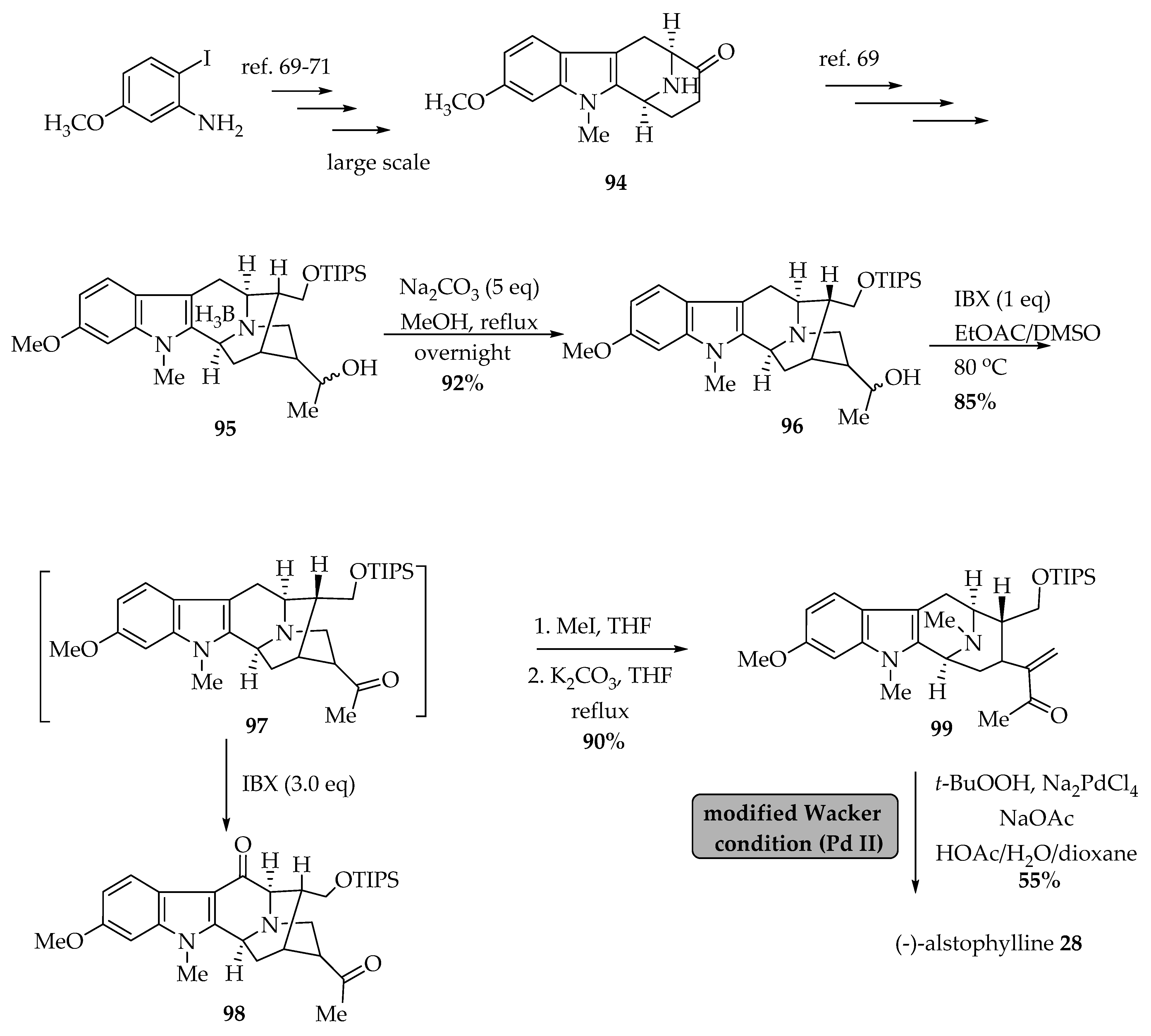
5.3.1. An Improved Total Synthesis of (-)-Alstophylline 28
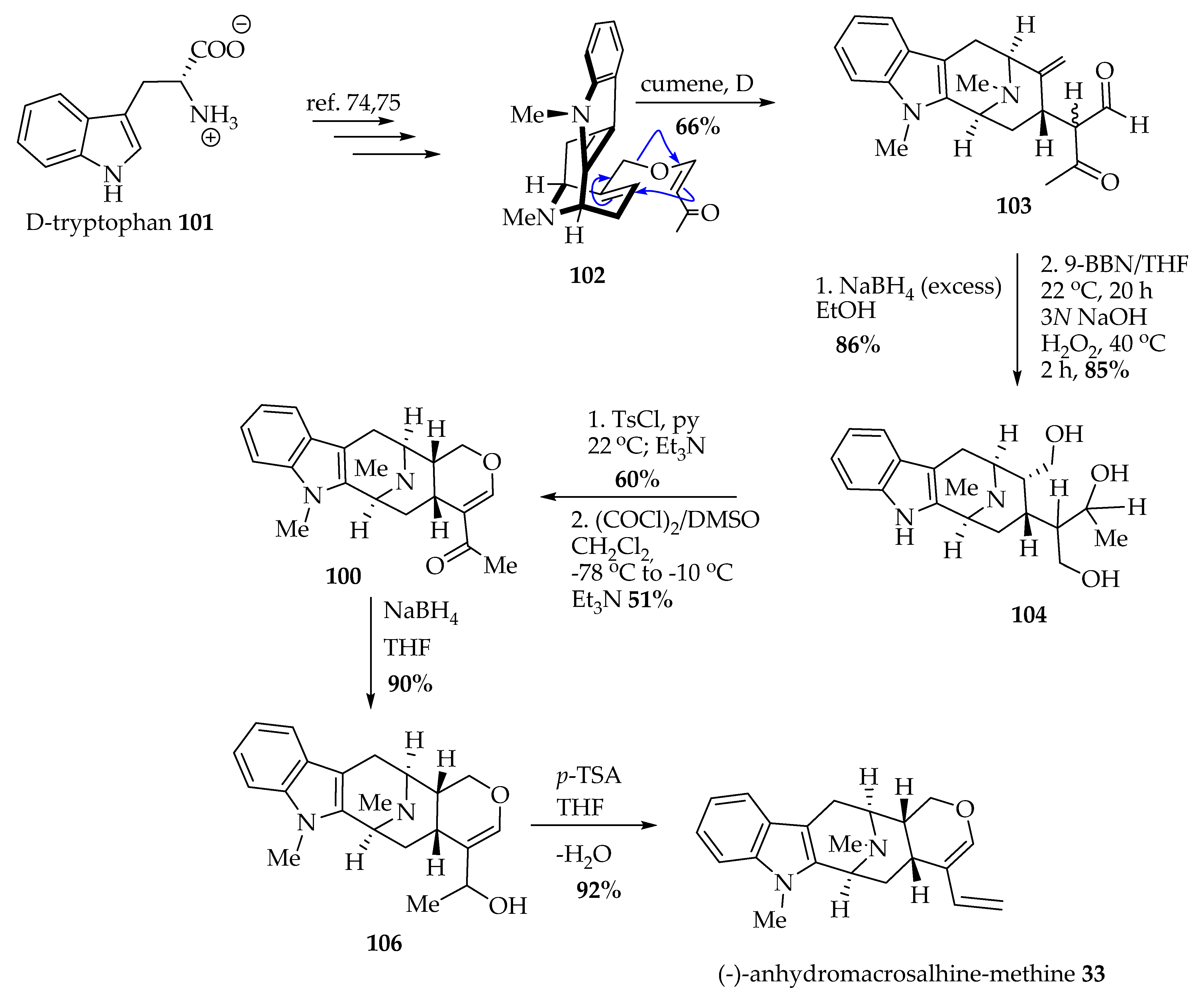
5.3.2. Enantiospecific Total Synthesis of (-)-Anhydromacrosalhine-methine 33 via the Asymmetric Pictet–Spengler Reaction
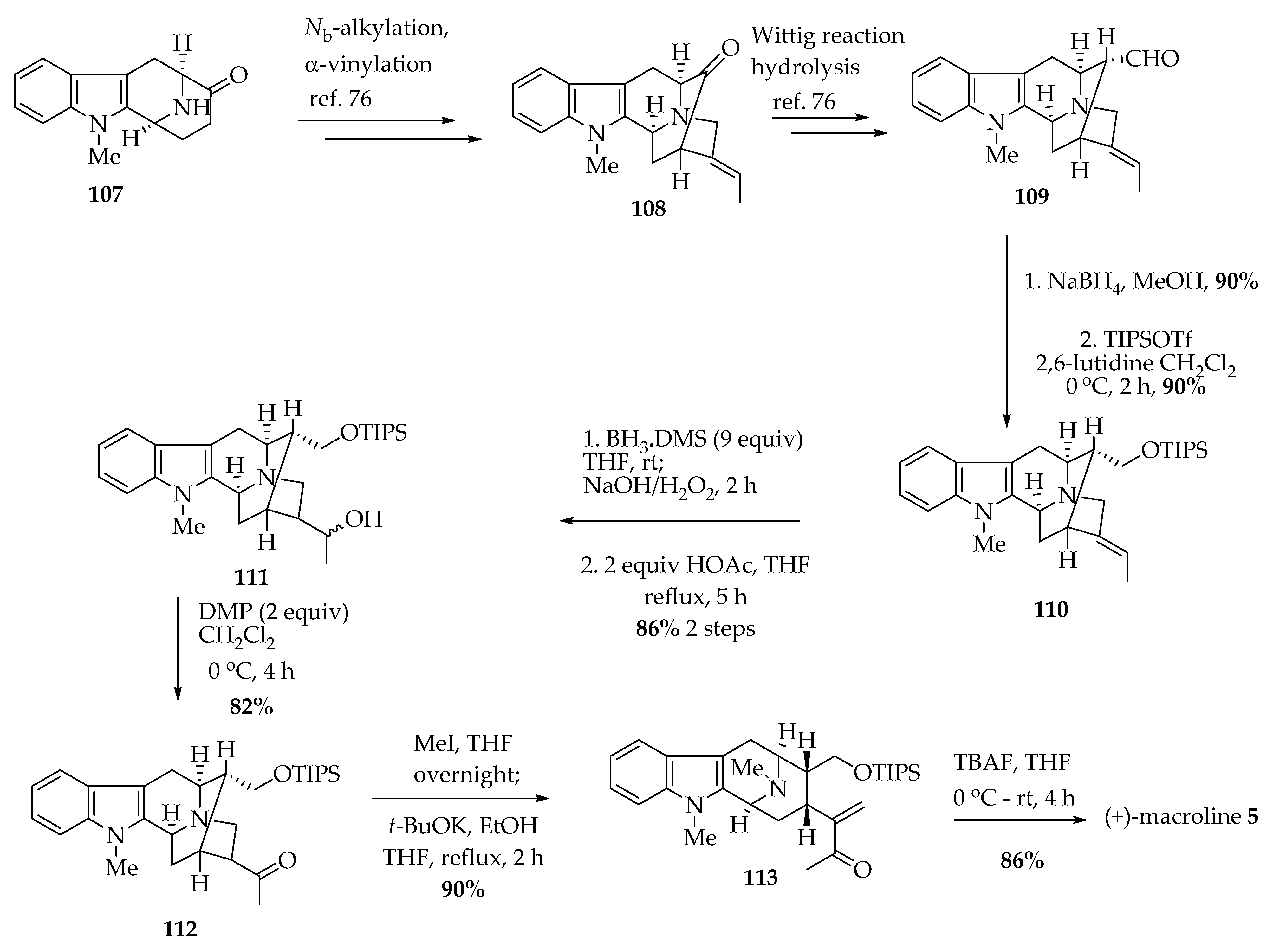
5.3.3. Improved Synthesis of (+)-Macroline 5
5.3.4. Stereospecific Access to the Macroline Core 5
5.3.5. Enantiomeric Total Synthesis of (+)-Na-Methylsarpagine 124
6. The Synthesis of Bisindole Alkaloids
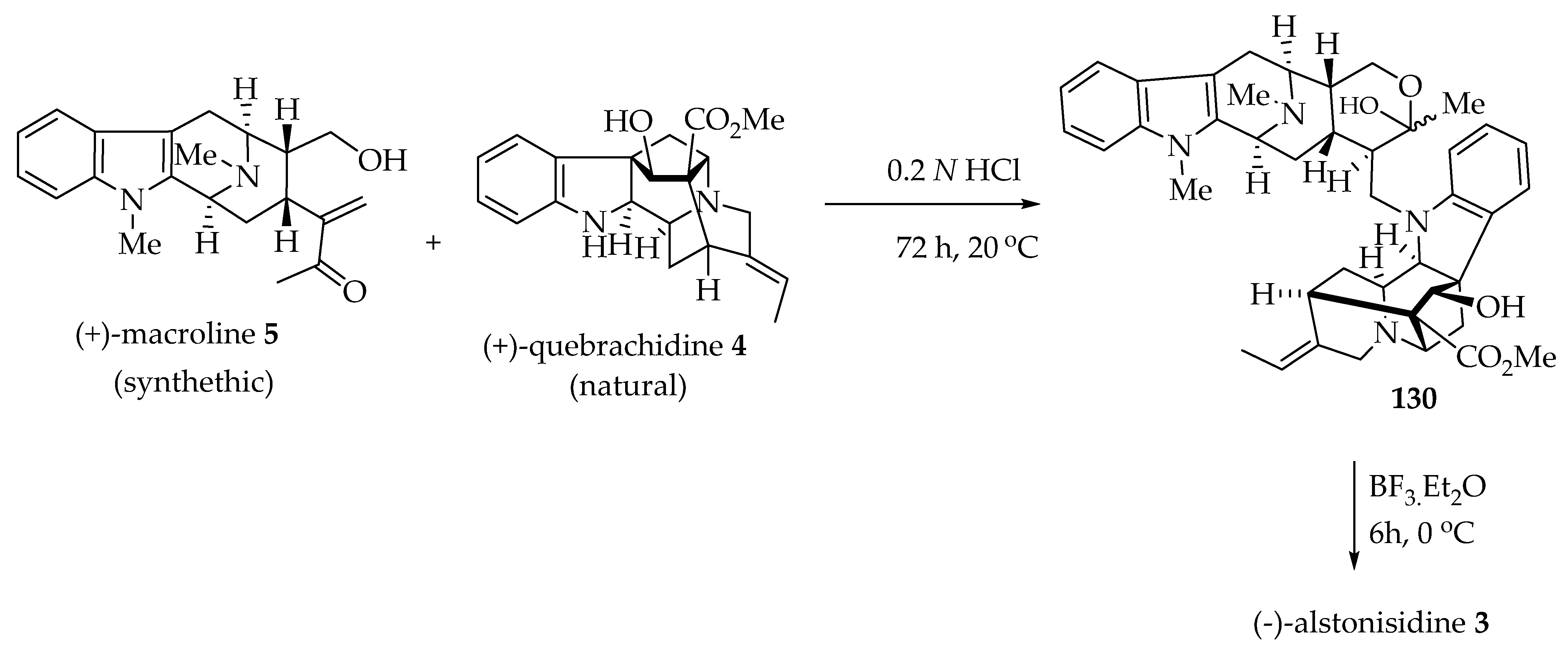
6.1. Biomimetic Partial Synthesis of (-)-Alstonisidine 3
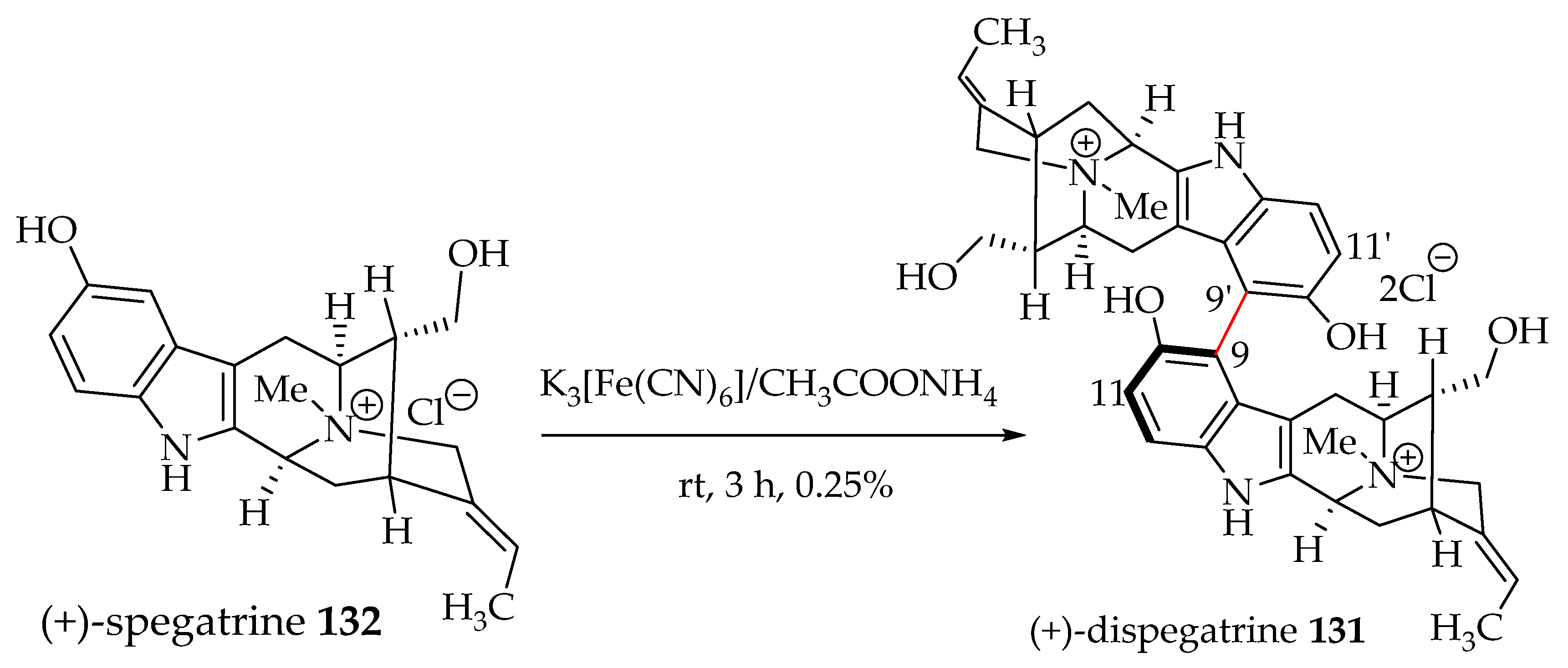
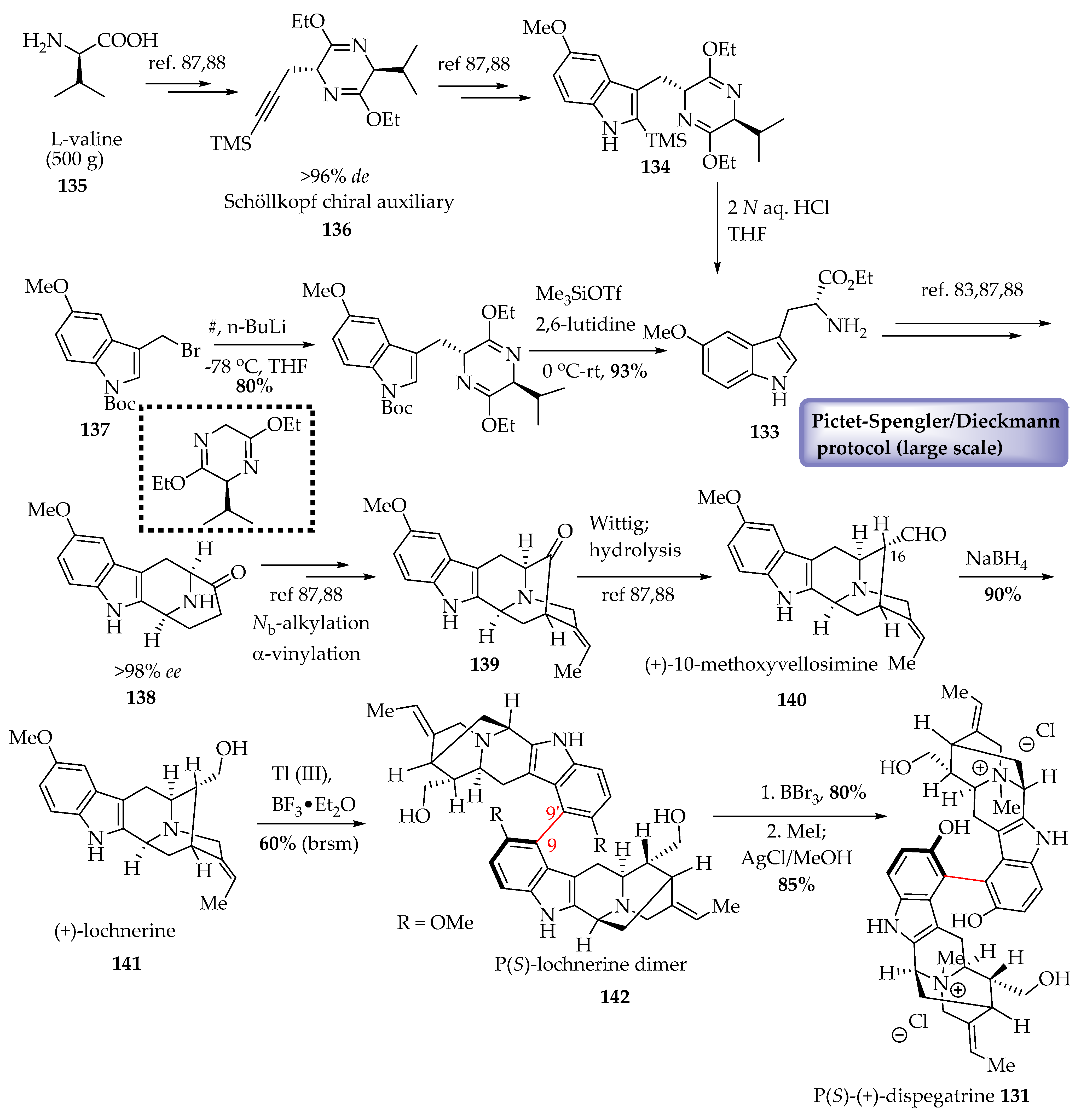
6.2. Biomimetic Partial Synthesis of (+)-Dispegatrine 131
Enantiospecific Total Synthesis of (+)-dispegatrine 131 in >98% ee
6.3. Biomimetic Total Synthesis of (+)-Macralstonidine 23
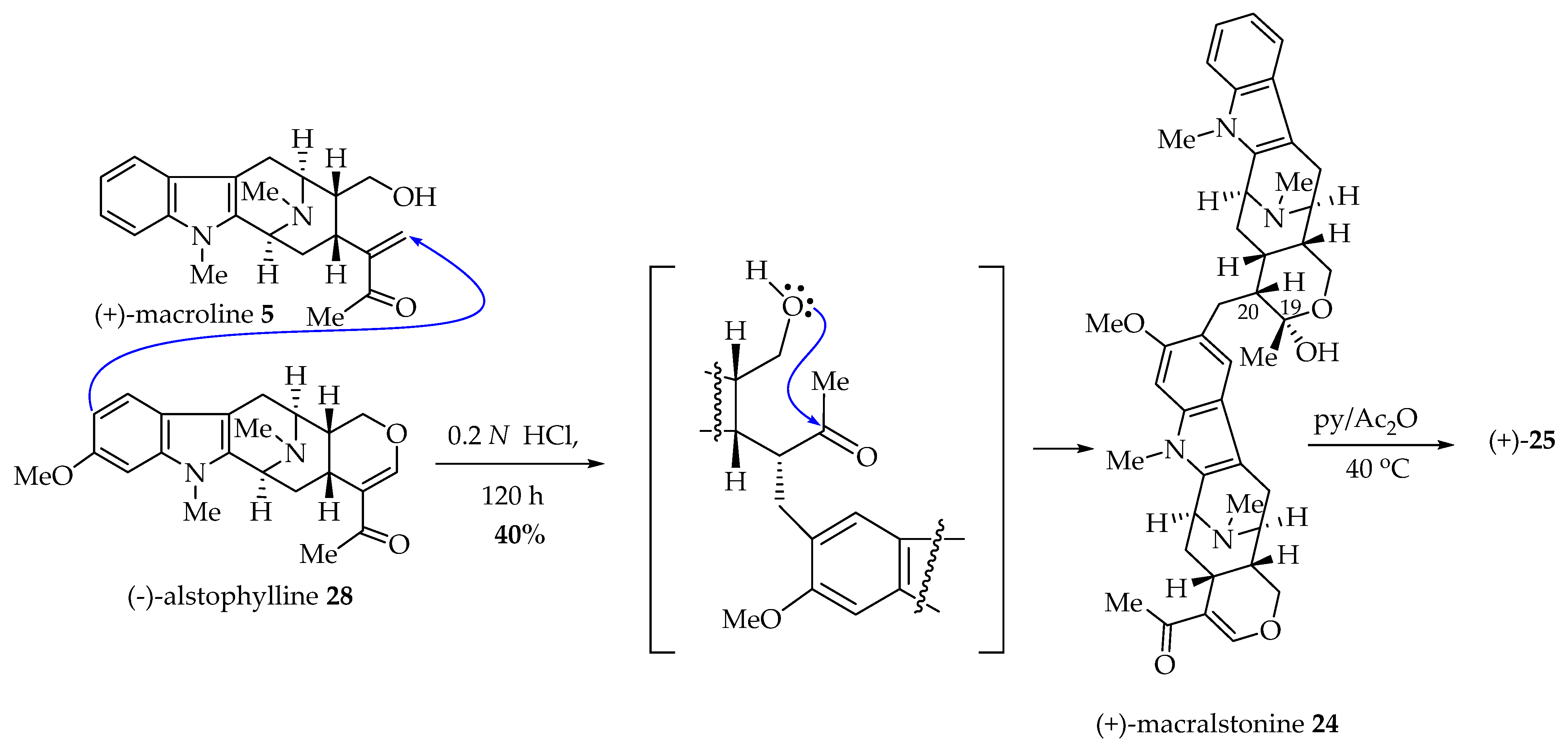
6.4. Biomimetic Total Synthesis of (+)-Macralstonine 24
6.5. Partial Synthesis of (-)-Macrocarpamine 31
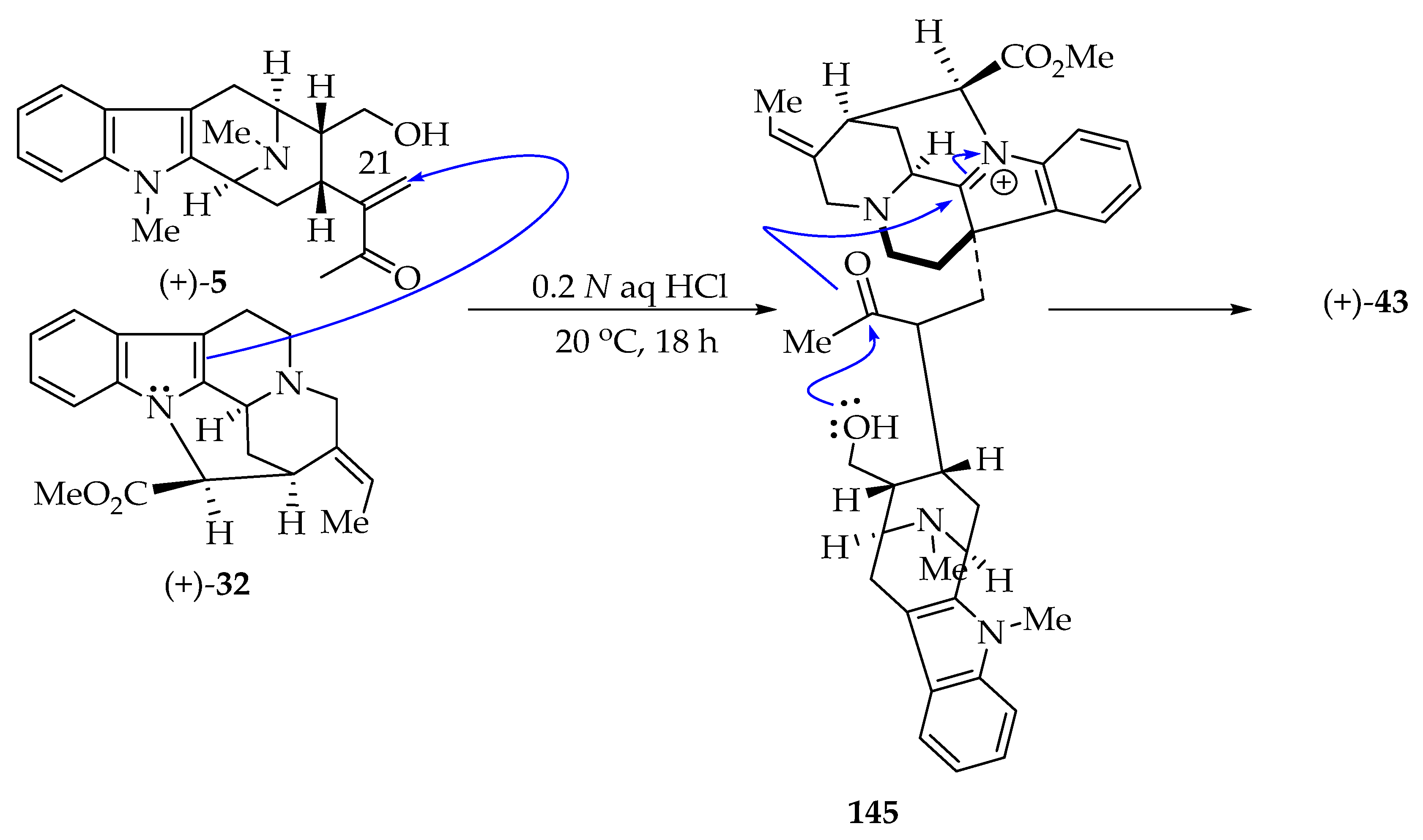
6.6. Partial Synthesis of (+)-Villalstonine 43
6.7. Improved Partial Synthesis of (+)-Villalstonine 43
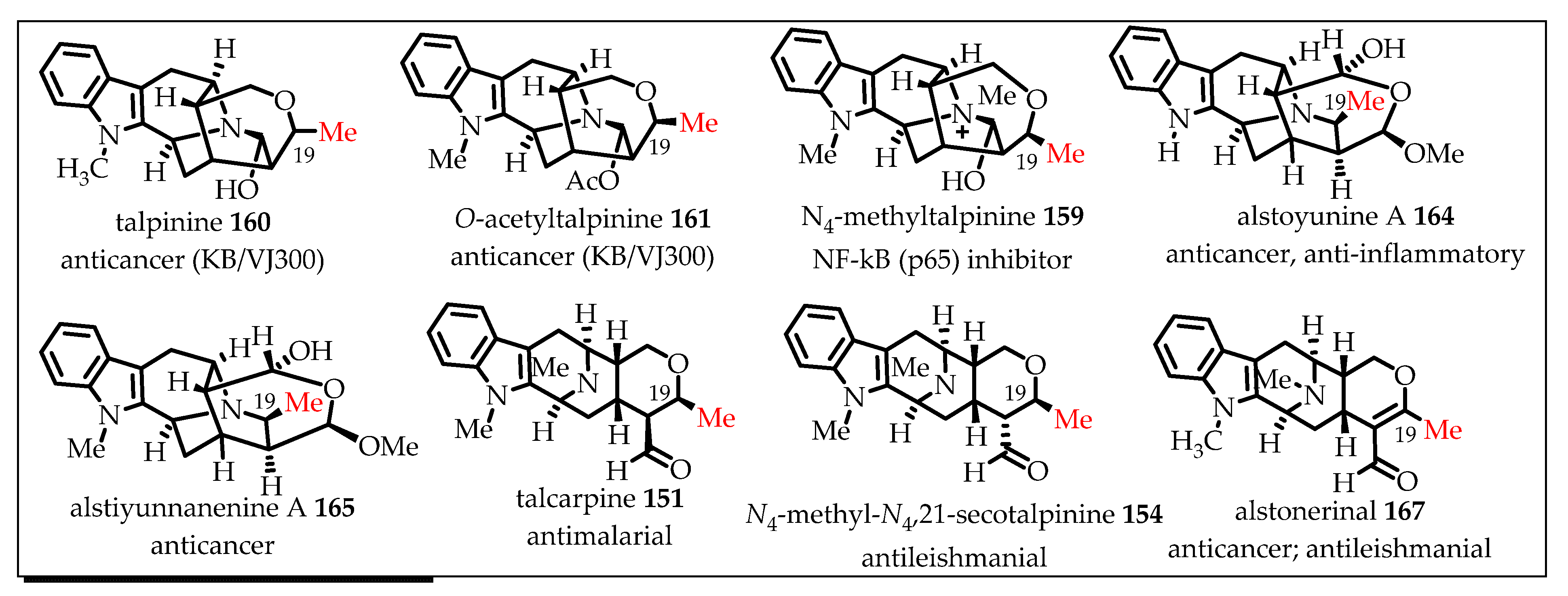
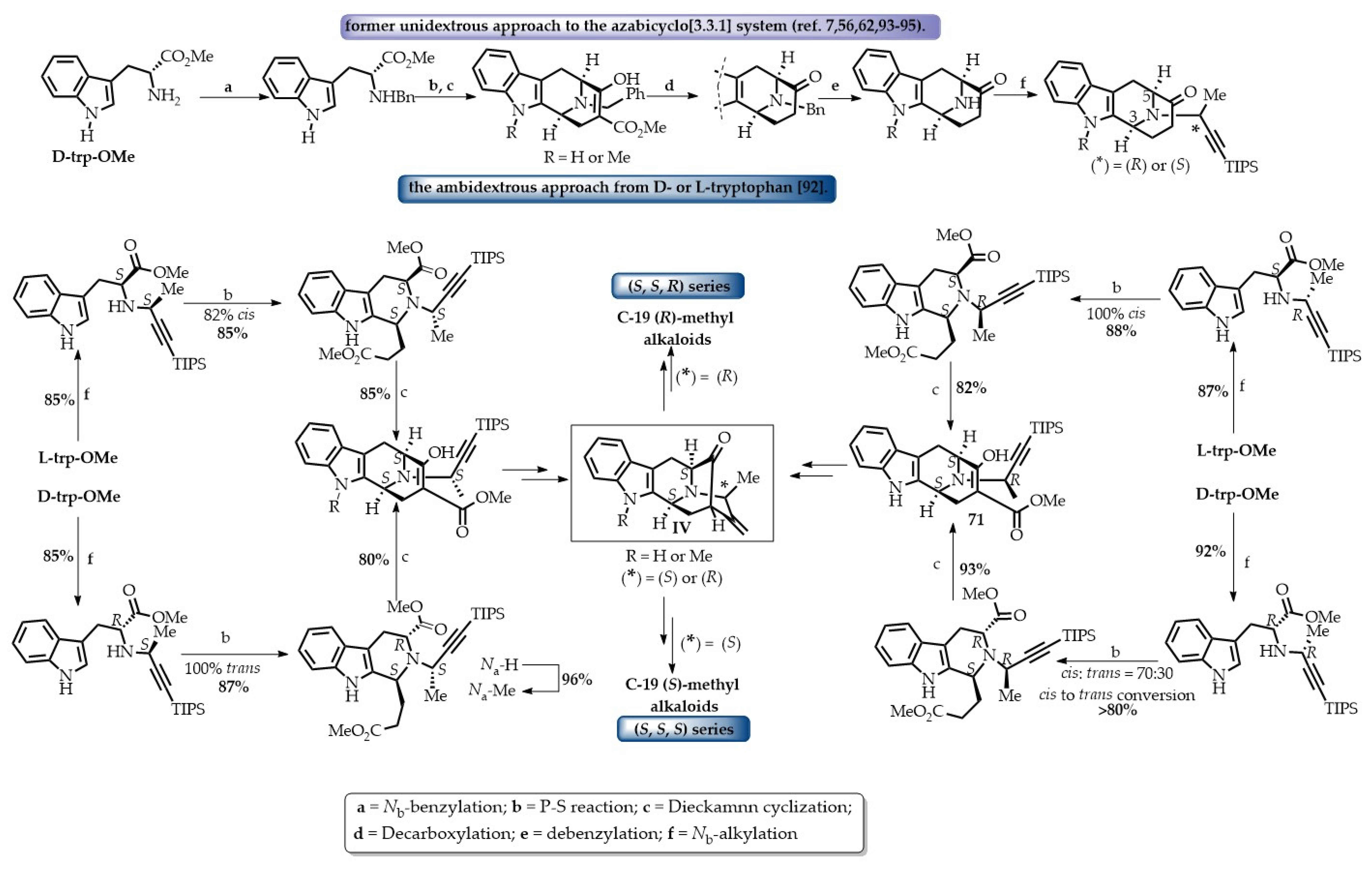
7. C-19 Methyl-Substituted Sarpagine/Macroline/Ajmaline Alkaloids
Enantiospecific Total Synthesis of C-19 Methyl-Substituted Sarpagine/Macroline Alkaloids via the General Strategy Developed in Milwaukee
8. Unnatural (Enantiomers or Synthetic Derivatives) Alkaloids in Drug Discovery
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef]
- Thomford, N.E.; Senthebane, D.A.; Rowe, A.; Munro, D.; Seele, P.; Maroyi, A.; Dzobo, K. Natural products for drug discovery in the 21st century: Innovations for novel drug discovery. Int. J. Mol. Sci. 2018, 19, 1578. [Google Scholar] [CrossRef] [Green Version]
- Harvey, A.L.; Edrada-Ebel, R.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, A.T.; George, G.; Yadav, N.; Jeswani, A.; Auti, P.S. Pharmaceutical Application of Bio-actives from Alstonia Genus: Current Findings and Future Directions. In Bioactive Natural Products for Pharmaceutical Applications; Pal, D., Nayak, A.K., Eds.; Springer International Publishing: New York, NY, USA, 2021; pp. 463–533. [Google Scholar]
- Yeap, J.S.; Saad, H.M.; Tan, C.H.; Sim, K.S.; Lim, S.H.; Low, Y.Y.; Kam, T.S. Macroline-sarpagine bisindole alkaloids with antiproliferative activity from Alstonia penangiana. J. Nat. Prod. 2019, 82, 3121–3132. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.T.; Tiruveedhula, V.V.; Cook, J.M. Synthesis of bisindole alkaloids from the Apocynaceae which contain a macroline or sarpagine unit: A review. Molecules 2016, 21, 1525. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.T.; Deschamps, J.R.; Imler, G.H.; Cook, J.M. Total Synthesis of Sarpagine-Related Bioactive Indole Alkaloids. Chemistry 2018, 24, 2354–2359. [Google Scholar] [CrossRef]
- Tan, M.C.S.; Carranza, M.S.S.; Linis, V.C.; Malabed, R.S.; Oyong, G.G. Antioxidant, cytotoxicity, and antiophidian potential of Alstonia macrophylla bark. ACS Omega 2019, 4, 9488–9496. [Google Scholar] [CrossRef] [Green Version]
- Fielding, B.C.; da Silva Maia Bezerra Filho, C.; Ismail, N.S.M.; de Sousa, D.P. Alkaloids: Therapeutic potential against human coronaviruses. Molecules 2020, 25, 5496. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.E.; Abdul-Hameed, Z.H.; Alotaibi, M.O.; Bawakid, N.O.; Sobahi, T.R.; Abdel-Lateff, A.; Alarif, W.M. Chemical diversity and bioactivities of monoterpene indole alkaloids (MIAs) from six Apocynaceae genera. Molecules 2021, 26, 488. [Google Scholar] [CrossRef]
- Lu, J.J.; Bao, J.L.; Chen, X.P.; Huang, M.; Wang, Y.T. Alkaloids isolated from natural herbs as the anticancer agents. Evid. Based Complement. Altern. Med. 2012, 2012, 485042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvert, M.B.; Sperry, J. Bioinspired total synthesis and structural revision of yuremamine, an alkaloid from the entheogenic plant Mimosa tenuiflora. Chem. Commun. 2015, 51, 6202–6205. [Google Scholar] [CrossRef]
- Namjoshi, O.A.; Cook, J.M. Chapter Two—Sarpagine and Related Alkaloids. In The Alkaloids: Chemistry and Biology; Knӧlker, H.-J., Ed.; Academic Press: San Diego, CA, USA, 2016; Volume 76, pp. 63–169. [Google Scholar]
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S.; Chang, R.S.L.; et al. Methods for drug discovery: Development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef]
- Zeng, J.; Zhang, D.-B.; Zhou, P.-P.; Zhang, Q.-L.; Zhao, L.; Chen, J.-J.; Gao, K. Rauvomines A and B, two monoterpenoid indole alkaloids from Rauvolfia vomitoria. Org. Lett. 2017, 19, 3998–4001. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-Z.; Chen, Q.; Yang, G.-F. A review on recent developments of indole-containing antiviral agents. Eur. Med. Chem. 2015, 89, 421–441. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Kogure, N.; Kitajima, M.; Takayama, H. Total syntheses of pleiocarpamine, normavacurine, and C-mavacurine. Organic Lett. 2019, 21, 3342–3345. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.H. Alkaloids from Alstonia Macrophylla. Ph.D. Thesis, University of Malaya, Kuala Lampur, Malaysia, 2013. [Google Scholar]
- Keawpradub, N.; Houghton, P.J. Indole alkaloids from Alstonia macrophylla. Phytochemistry 1997, 46, 757–762. [Google Scholar] [CrossRef]
- Keawpradub, N.; Kirby, G.C.; Steele, J.C.; Houghton, P.J. Antiplasmodial activity of extracts and alkaloids of three Alstonia species from Thailand. Planta Med. 1999, 65, 690–694. [Google Scholar] [CrossRef]
- Elderfield, R.C.; Gilman, R.E. Alkaloids of Alstonia muelleriana. Phytochemistry 1972, 11, 339–343. [Google Scholar] [CrossRef] [Green Version]
- Burke, D.E.; Cook, G.A.; Cook, J.M.; Haller, K.G.; Lazar, H.A.; Le Quesne, P.W. Further alkaloids of Alstonia muelleriana. Phytochemistry 1973, 12, 1467–1474. [Google Scholar] [CrossRef] [Green Version]
- Hoard, L.G. The Crystal Structures of Altstonisidine, C42H48N4O4, and Anhydrous Cholesterol, C27H46O. Ph.D. Thesis, University of Michigan, Ann Arbor, MI, USA, 1977. [Google Scholar]
- Hamaker, L.K.; Cook, J.M. The Synthesis of Macroline Related Sarpagine Alkaloids. In Alkaloids: Chemical and Biological Perspectives; Pelletier, S.W., Ed.; Elsevier Publications: Amsterdam, The Netherlands, 1995; Volume 9. [Google Scholar]
- Kitajima, M.; Takayama, H. Chapter Four-Monoterpenoid Bisindole Alkaloids. In The Alkaloids: Chemistry and Biology; Knölker, H.-J., Ed.; Academic Press: San Diego, CA, USA, 2016; Volume 76, pp. 259–310. [Google Scholar]
- Lim, S.-H.; Tan, S.-J.; Low, Y.-Y.; Kam, T.-S. Lumutinines A–D, linearly fused macroline–macroline and macroline–sarpagine bisindoles from alstonia macrophylla. J. Nat. Prod. 2011, 74, 2556–2562. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.-H.; Low, Y.-Y.; Subramaniam, G.; Abdullah, Z.; Thomas, N.F.; Kam, T.-S. Lumusidines A− D and villalstonidine F, macroline–macroline and macroline–pleiocarpamine bisindole alkaloids from Alstonia macrophylla. Phytochemistry 2013, 87, 148–156. [Google Scholar] [CrossRef]
- Tan, S.-J.; Lim, K.-H.; Subramaniam, G.; Kam, T.-S. Macroline–sarpagine and macroline–pleiocarpamine bisindole alkaloids from Alstonia angustifolia. Phytochemistry 2013, 85, 194–202. [Google Scholar] [CrossRef]
- Sharp, T.M. 265. The alkaloids of Alstonia barks. Part II. A. macrophylla, wall., A. somersetensis, FM Bailey, A. verticillosa, F. Muell., A. villosa, blum. J. Chem. Soc. 1934, 1227–1232. [Google Scholar] [CrossRef]
- Kishi, T.; Hesse, M.; Vetter, W.; Gemenden, C.; Taylor, W.; Schmid, H. Macralstonin. Helv. Chim. Acta 1966, 49, 946–964. [Google Scholar] [CrossRef]
- Hart, N.; Johns, S.; Lamberton, J. Tertiary alkaloids of Alstonia spectabilis and Alstonia glabriflora (Apocynaceae). Aust. J. Chem. 1972, 25, 2739–2741. [Google Scholar] [CrossRef] [Green Version]
- Keawprdub, N.; Houghton, P.; Eno-Amooquaye, E.; Burke, P. Activity of extracts and alkaloids of Thai Alstonia species against human lung cancer cell lines. Planta Med. 1997, 63, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.-H.; Low, Y.-Y.; Tan, S.-J.; Lim, K.-H.; Thomas, N.F.; Kam, T.-S. Perhentidines A–C: Macroline–macroline bisindoles from Alstonia and the absolute configuration of perhentinine and macralstonine. J. Nat. Prod. 2012, 75, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Changwichit, K.; Khorana, N.; Suwanborirux, K.; Waranuch, N.; Limpeanchob, N.; Wisuitiprot, W.; Suphrom, N.; Ingkaninan, K. Bisindole alkaloids and secoiridoids from Alstonia macrophylla Wall. ex G. Don. Fitoterapia 2011, 82, 798–804. [Google Scholar] [CrossRef]
- Cook, J.; Le Quesne, P. Macralstonine from Alstonia muelleriana. Phytochemistry 1971, 10, 437–439. [Google Scholar] [CrossRef] [Green Version]
- Ghedira, K.; Zeches-Hanrot, M.; Richard, B.; Massiot, G.; Le Men-Olivier, L.; Sevenet, T.; Goh, S. Alkaloids of Alstonia angustifolia. Phytochemistry 1988, 27, 3955–3962. [Google Scholar] [CrossRef]
- Cook, J.M.; Le Quesne, P.; Elderfield, R. Alstonerine, a new indole alkaloid from Alstonia muelleriana. J. Chem. Soc. Chem. Commun. 1969, 1306–1307. [Google Scholar] [CrossRef]
- Hesse, M.; Hürzeler, H.; Gemenden, C.; Joshi, B.; Taylor, W.; Schmid, H. Die Struktur des Alstonia-Alkaloides Villalstonin Vorläufige Mitteilung. Helv. Chim. Acta 1965, 48, 689–704. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Terrazas, C.; Muñoz Acuña, U.; Ninh, T.N.; Chai, H.; Carcache de Blanco, E.J.; Soejarto, D.D.; Satoskar, A.R.; Kinghorn, A.D. Bioactive indole alkaloids isolated from Alstonia angustifolia. Phytochem. Lett. 2014, 10, 54–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordman, C.; Kumra, S. The structure of villalstonine1. J. Am. Chem. Soc. 1965, 87, 2059–2060. [Google Scholar] [CrossRef]
- Buckingham, J.; Baggaley, K.H.; Roberts, A.D.; Szabo, L.F. Dictionary of Alkaloids with CD-ROM; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Keawpradub, N.; Eno-Amooquaye, E.; Burke, P.; Houghton, P. Cytotoxic activity of indole alkaloids from Alstonia macrophylla. Planta Med. 1999, 65, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.; Allen, D.; Cai, Y.; Phillipson, J.; Said, I.; Kirby, G.; Warhurst, D. In vitro antiamoebic and antiplasmodial activities of alkaloids isolated from Alstonia angustifolia roots. Phytother. Res. 1992, 6, 121–124. [Google Scholar] [CrossRef]
- Gan, T.; Cook, J.M. General approach for the synthesis of macroline/sarpagine related indole alkaloids via the asymmetric Pictet-Spengler reaction: The enantiospecific synthesis of (−)-anhydromacrosalhine-methine. Tetrahedron Lett. 1996, 37, 5033–5036. [Google Scholar] [CrossRef]
- Bi, Y.; Zhang, L.-H.; Hamaker, L.K.; Cook, J.M. Enantiospecific synthesis of (-)-alstonerine and (+)-macroline as well as a partial synthesis of (+)-villalstonine. J. Am. Chem. Soc. 1994, 116, 9027–9041. [Google Scholar] [CrossRef]
- Mayerl, F.; Hesse, M. Macrocarpamin, ein neues Bisindolalkaloid aus Alstonia macrophylla WALL. 167. Mittelung über organische Naturstoffe. Helv. Chim. Acta 1978, 61, 337–351. [Google Scholar] [CrossRef]
- Khan, Z.M.; Hesse, M.; Schmid, H. Die Struktur des quartären Alkaloides Macrosalhin. Helv. Chim. Acta 1967, 50, 1002–1010. [Google Scholar] [CrossRef]
- Kump, W.; Schmid, H. Über die alkaloide von Pleiocarpa mutica BENTH. Helv. Chim. Acta 1961, 44, 1503–1516. [Google Scholar] [CrossRef]
- Kam, T.-S.; Subramaniam, G.; Chen, W. Alkaloids from Kopsia dasyrachis. Phytochemistry 1999, 51, 159–169. [Google Scholar] [CrossRef]
- Bartlett, M.; Sklar, R.; Smith, A.; Taylor, W. The alkaloids of Hunteria eburnea Pichon. III. 1 The tertiary bases. J. Org. Chem. 1963, 28, 2197–2199. [Google Scholar] [CrossRef]
- Burke, D.E.; Cook, J.M.; Le Quesne, P. Biomimetic synthesis of the bisindole alkaloids villalstonine and alstonisidine. J. Am. Chem. Soc. 1973, 95, 546–552. [Google Scholar] [CrossRef]
- Kam, T.-S.; Tan, S.-J.; Ng, S.-W.; Komiyama, K. Bipleiophylline, an unprecedented cytotoxic bisindole alkaloid constituted from the bridging of two indole moieties by an aromatic spacer unit. Org. Lett. 2008, 10, 3749–3752. [Google Scholar] [CrossRef]
- Jiménez, J.-M.; Zulaica, E.; Bennasar, M.-L.; Bosch, J. A new synthetic entry to the alkaloids of the mavacurine group. First total synthesis of (±)-2, 7-dihydropleiocarpamine. J. Chem. Soc. Chem. Commun. 1993, 732–733. [Google Scholar] [CrossRef]
- Jarret, M.; Tap, A.; Kouklovsky, C.; Poupon, E.; Evanno, L.; Vincent, G. Bioinspired oxidative cyclization of the geissoschizine skeleton for the total synthesis of (-)-17-nor-excelsinidine. Angew. Chem. Int. Ed. 2018, 57, 12294–12298. [Google Scholar] [CrossRef] [PubMed]
- Le Men, J.; Taylor, W.I. A uniform numbering system for indole alkaloids. Experientia 1965, 21, 508–510. [Google Scholar] [CrossRef]
- Rahman, M.T.; Cook, J.M. The C-19 Methyl Substituted Sarpagine-Macroline-Ajmaline Alkaloids: Diversity, Occurrence, Bioactivity, and Synthesis. In Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Ruppert, M.; Woll, J.; Giritch, A.; Genady, E.; Ma, X.; Stöckigt, J. Functional expression of an ajmaline pathway-specific esterase from Rauvolfia in a novel plant-virus expression system. Planta 2005, 222, 888–898. [Google Scholar] [CrossRef]
- Mattern-Dogru, E.; Ma, X.; Hartmann, J.; Decker, H.; Stöckigt, J. Potential activ-site residues in polyneuridine aldehyde esterase, a central enzyme of indole alkaloid biosynthesis, by modelling and site-directed mutagenesis. Eur. J. Biochem. 2002, 269, 2889–2896. [Google Scholar] [CrossRef] [PubMed]
- Pfitzner, A.; Stöckigt, J. Biogenetic link between sarpagine and ajmaline type alkaloids. Tetrahedron Lett. 1983, 24, 5197–5200. [Google Scholar] [CrossRef]
- Wu, F.; Kerčmar, P.; Zhang, C.; Stöckigt, J. Sarpagan-Ajmalan-Type Indoles: Biosynthesis, structural biology, and chemo-enzymatic significance. In The Alkaloids: Chemistry and Biology; Knӧlker, H.-J., Ed.; Academic Press: San Diego, CA, USA, 2016; Volume 76, pp. 1–61. [Google Scholar]
- Esmond, R.W.; Le Quesne, P.W. Biomimetic synthesis of macroline. J. Am. Chem. Soc. 1980, 102, 7116–7117. [Google Scholar] [CrossRef]
- Rahman, M.T.; Deschamps, J.R.; Imler, G.H.; Schwabacher, A.W.; Cook, J.M. Total synthesis of macrocarpines D and E via an enolate-driven copper-mediated cross-coupling process: Replacement of catalytic palladium with copper iodide. Org. Lett. 2016, 18, 4174–4177. [Google Scholar] [CrossRef] [Green Version]
- Edwankar, C.R.; Edwankar, R.V.; Namjoshi, O.A.; Rallapalli, S.K.; Yang, J.; Cook, J.M. Recent progress in the total synthesis of indole alkaloids. Curr. Opin. Drug Discov. Dev. 2009, 12, 752–771. [Google Scholar]
- Edwankar, C.R. Part. I: The First Regio-and Atropdiastereoselective Total Synthesis of the Dimeric Indole Alkaloid (+)-Dispegatrine, as Well as The First Total Synthesis of the Sarpagine Alkaloids (+)-Spegatrine, Lochvinerine,(+)-Lochneram and an Improved Total Synthesis of (+)-10-Methoxyvellosimine,(+)-Lochnerine and (+)-Sarpagine. Part. II: Studies Directed toward the Total Synthesis of The Carbon-19 Methyl Substituted Sarpagine-Macroline Alkaloids (+)-Macro-salhine Chloride as Well as Macrocarpine A, B and C. Ph.D. Thesis, The University of Wisconsin-Milwaukee, Milwaukee, WI, USA, 2011. [Google Scholar]
- Gan, T.; Cook, J.M. Partial synthesis of the antiamoebic bisindole alkaloid (−)-macrocarpamine. Tetrahedron Lett. 1996, 37, 5037–5038. [Google Scholar] [CrossRef]
- Liao, X.; Zhou, H.; Wearing, X.Z.; Ma, J.; Cook, J.M. The first regiospecific, enantiospecific total synthesis of 6-oxoalstophylline and an improved total synthesis of alstonerine and alstophylline as well as the bisindole alkaloid macralstonine. Org. Lett. 2005, 7, 3501–3504. [Google Scholar] [CrossRef]
- Kam, T.-S.; Choo, Y.-M.; Komiyama, K. Unusual spirocyclic macroline alkaloids, nitrogenous derivatives, and a cytotoxic bisindole from Alstonia. Tetrahedron 2004, 60, 3957–3966. [Google Scholar] [CrossRef]
- Kishi, T.; Hesse, M.; Gemenden, C.; Taylor, W.; Schmid, H. Alstophyllin, ein neues indolalkaloid aus Alstonia macrophylla WALL. Helv. Chim. Acta 1965, 48, 1349–1362. [Google Scholar] [CrossRef]
- Liu, X.; Deschamp, J.R.; Cook, J.M. Regiospecific, enantiospecific total synthesis of the alkoxy-substituted indole bases, 16-e pi-N a-methylgardneral, 11-methoxyaffinisine, and 11-methoxymacroline as well as the indole alkaloids alstophylline and macralstonine. Org. Lett. 2002, 4, 3339–3342. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.S.; Hamaker, L.K.; La Loggia, A.J.; Cook, J.M. Entry into 6-methoxy-D (+)-tryptophans. Stereospecific synthesis of 1-benzenesulfonyl-6-methoxy-D (+)-tryptophan ethyl ester. Synth. Commun. 1992, 22, 2077–2102. [Google Scholar] [CrossRef]
- Ma, C.; Liu, X.; Li, X.; Flippen-Anderson, J.; Yu, S.; Cook, J.M. Efficient asymmetric synthesis of biologically important tryptophan analogues via a palladium-mediated heteroannulation reaction. J. Org. Chem. 2001, 66, 4525–4542. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, J.; Nagashima, H.; Hori, K. A new preparative method for 1, 3-dicarbonyl compounds by the regioselective oxidation of alpha, beta-unsaturated carbonyl compounds, catalyzed by PdCl2 using hydroperoxides as the reoxidant of Pd0. Chem. Lett. 1980, 9, 257–260. [Google Scholar] [CrossRef]
- Takayama, H.; Phisalaphong, C.; Kitajima, M.; Aimi, N.; Sakai, S.-I. An efficient synthetic pathway to the macroline-type indole alkaloids, talcarpine and alstonerine from ajmaline. Tetrahedron 1991, 47, 1383–1392. [Google Scholar] [CrossRef]
- Zhang, L.-H.; Bi, Y.-Z.; Yu, F.-X.; Menzia, G.; Cook, J.M. Stereospecificity in the Pictet-Spengler reaction. Enantiospecific synthesis of (6S, 10S)-5# 75)-5-methyl-9-oxo-12-benzyl-6, 7, 8, 9, 10, 11-hexahydro-6, 10-imino-5H-cyclooct [b] indole, a template for preparation of macroline/sarpagine alkaloids. Heterocycles 1992, 34, 517–547. [Google Scholar]
- Zhang, L.; Cook, J. General approach to the synthesis of macroline-related alkaloids. Stereospecific total synthesis of (-)-alstonerine. J. Am. Chem. Soc. 1990, 112, 4088–4090. [Google Scholar] [CrossRef]
- Liao, X.; Zhou, H.; Yu, J.; Cook, J.M. An improved total synthesis of (+)-macroline and alstonerine as well as the formal total synthesis of (−)-talcarpine and (−)-anhydromacrosalhine− methine. J. Org. Chem. 2006, 71, 8884–8890. [Google Scholar] [CrossRef]
- Gorman, M.; Sweeny, J. Perivine. Tetrahedron Lett. 1964, 5, 3105–3111. [Google Scholar] [CrossRef]
- Neukomm, G.; Kletzhäundler, E.; Hesse, M. Die absolute konfiguration von macrolin, einem abbauprodukt des alkaloides villalstonin 179. Mitteilung über organische naturstoffe. Helv. Chim. Acta 1981, 64, 90–96. [Google Scholar] [CrossRef]
- Bi, Y.; Cook, J.M. General approach for the synthesis of macroline/sarpagine alkaloids. The total synthesis of (+)-macroline. Tetrahedron Lett. 1993, 34, 4501–4504. [Google Scholar] [CrossRef]
- Hesse, M.; Bodmer, F.; Gemenden, C.; Joshi, B.; Taylor, W.; Schmid, H. Die struktur des Alstonia-alkaloides villalstonin. Helv. Chim. Acta 1966, 49, 1173–1182. [Google Scholar] [CrossRef]
- Tran, Y.S.; Kwon, O. An application of the phosphine-catalyzed [4 + 2] annulation in indole alkaloid synthesis: Formal syntheses of (±)-alstonerine and (±)-macroline. Org. Lett. 2005, 7, 4289–4291. [Google Scholar] [CrossRef]
- Kadam, V.D.; Rao, B.S.S.; Mahesh, S.; Chakraborty, M.; Vemulapalli, S.P.B.; Dayaka, S.N.; Sudhakar, G. Stereoselective access to the core structure of macroline-type indole alkaloids: Total synthesis of macroline and alstomicine. Org. Lett. 2018, 20, 4782–4786. [Google Scholar] [CrossRef]
- Zhao, S.; Liao, X.; Wang, T.; Flippen-Anderson, J.; Cook, J.M. The enantiospecific, stereospecific total synthesis of the ring-A oxygenated sarpagine indole alkaloids (+)-majvinine,(+)-10-methoxyaffinisine, and (+)-N a-methylsarpagine, as well as the total synthesis of the Alstonia bisindole alkaloid macralstonidine. J. Org. Chem. 2003, 68, 6279–6295. [Google Scholar] [CrossRef]
- Zhao, S.; Liao, X.; Cook, J.M. Enantiospecific, stereospecific total synthesis of (+)-majvinine,(+)-10-methoxyaffinisine, and (+)-N a-methylsarpagine as well as the total synthesis of the alstonia bisindole macralstonidine. Org. Lett. 2002, 4, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Burke, D.E.; Cook, J.M.; Le Quesne, P. Biomimetic synthesis and structure of the bisindole alkaloid alstonisidine. J. Chem. Soc. Chem. Commun. 1972, 697. [Google Scholar] [CrossRef]
- Lin, M.; Yang, B.-Q.; Yu, D.-Q. Studies on the quaternary alkaloids of Rauvolfia verticillata (lour.) Baill var. Hainanensis Tsiang. Acta Pharmacol. Sin. 1986, 21, 114–118. [Google Scholar]
- Edwankar, C.R.; Edwankar, R.V.; Deschamps, J.R.; Cook, J.M. Nature-inspired stereospecific total synthesis of p-(+)-dispegatrine and four other monomeric sarpagine indole alkaloids. Angew. Chem. Int. Ed. 2012, 51, 11762–11765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwankar, C.R.; Edwankar, R.V.; Namjoshi, O.A.; Liao, X.; Cook, J.M. Stereospecific approach to the synthesis of ring-A oxygenated sarpagine indole alkaloids. Total synthesis of the dimeric indole alkaloid P-(+)-dispegatrine and six other monomeric indole alkaloids. J. Org. Chem. 2013, 78, 6471–6487. [Google Scholar] [CrossRef] [Green Version]
- Burke, D.E.; DeMarkey, C.A.; Le Quesne, P.; Cook, J.M. Biomimetic synthesis of the bis-indole alkaloid macralstonine. J. Chem. Soc. Chem. Commun. 1972, 1346–1347. [Google Scholar] [CrossRef]
- Zhang, L.-H.; Cook, J.M. Pictet-Spengler reactions in aprotic media: Nb-benzyl promoted retention of optical activity in the synthesis of an indolo substituted azabicyclo[3.3.1]nonane, a key template for the synthesis of macroline alkaloids. Heterocycles 1988, 27, 2795–2802. [Google Scholar] [CrossRef]
- Liao, X. The First Total Synthesis of The Indole Alkaloids, Macralstonidine, 6-Oxoalstophylline, 10-Methoxyvellosimine, Lochnerine, Sarpagine and an Improved Total Synthesis of Macralstonine and Macroline, as Well as The Formal Total Synthesis of Dispegatrine. Ph.D. Thesis, University of Wisconsin-Milwaukee, Milwaukee, WI, USA, 2007. [Google Scholar]
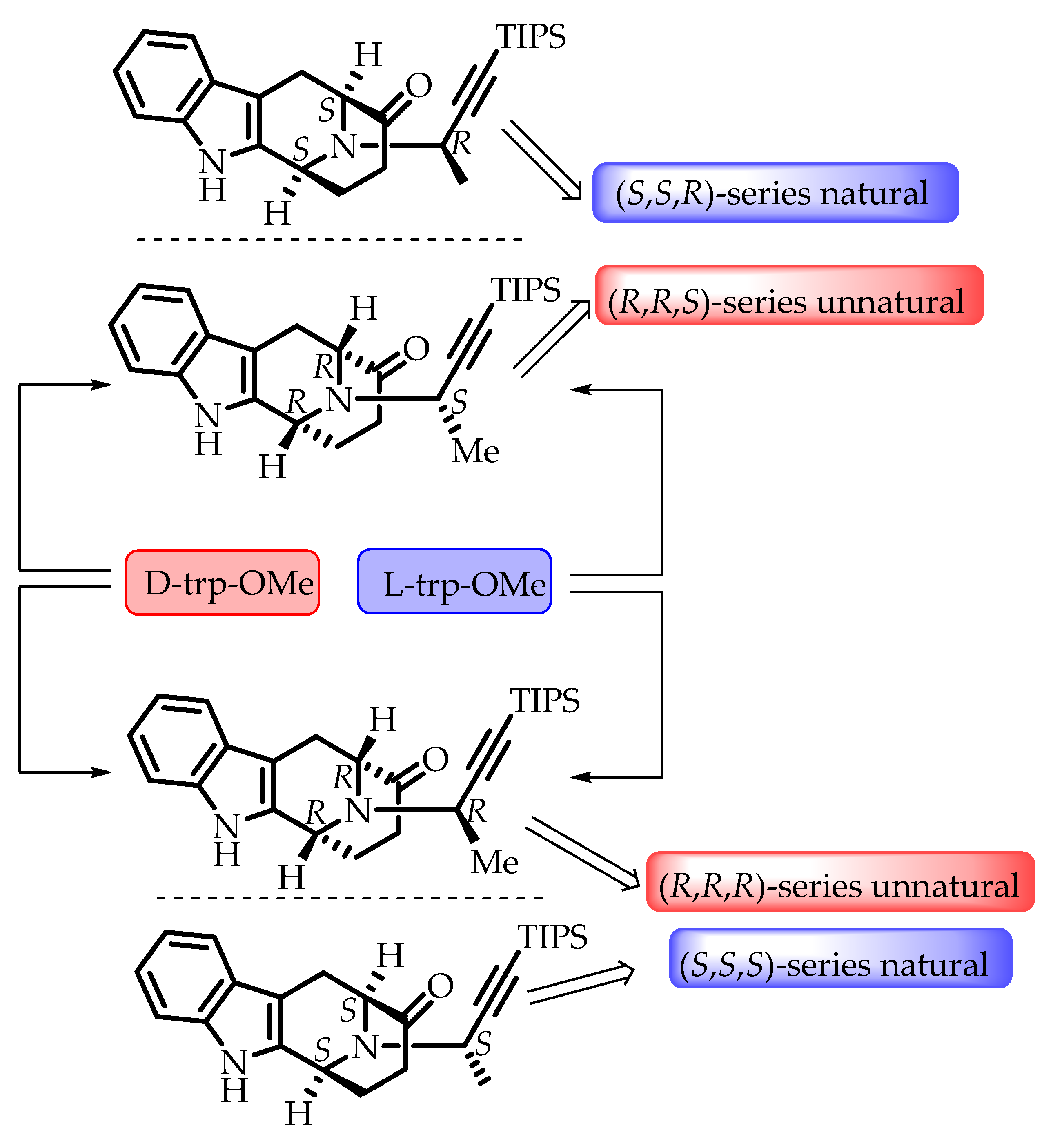
- Rahman, M.T.; Cook, J.M. The ambidextrous Pictet–Spengler reaction: Access to the (+)- or (–)-enantiomers of the bioactive C-19 methyl-substituted sarpagine/macroline/ajmaline alkaloids from either d- or l-tryptophan. Synthesis 2019, 51, 1980–1988. [Google Scholar] [CrossRef]
- Edwankar, R.V.; Edwankar, C.R.; Deschamps, J.R.; Cook, J.M. General strategy for synthesis of C-19 methyl-substituted sarpagine/macroline/ajmaline indole alkaloids including total synthesis of 19 (S), 20 (R)-dihydroperaksine, 19 (S), 20 (R)-dihydroperaksine-17-al, and peraksine. J. Org. Chem. 2014, 79, 10030–10048. [Google Scholar] [CrossRef] [PubMed]
- Edwankar, R.V.; Edwankar, C.R.; Deschamps, J.; Cook, J.M. Regiospecific, enantiospecific total synthesis of C-19 methyl substituted sarpagine alkaloids dihydroperaksine-17-al and dihydroperaksine. Org. Lett. 2011, 13, 5216–5219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, M.T.; Cook, J.M. Unprecedented stereocontrol in the synthesis of 1, 2, 3-trisubstituted tetrahydro-β-carbolines via a new asymmetric Pictet—Spengler reaction towards sarpagine-type indole alkaloids. Eur. J. Org. Chem. 2018, 2018, 3224–3229. [Google Scholar] [CrossRef]
- Sheludko, Y.; Gerasimenko, I.; Kolshorn, H.; Stöckigt, J. New alkaloids of the sarpagine group from Rauvolfia serpentina hairy root culture. J. Nat. Prod. 2002, 65, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
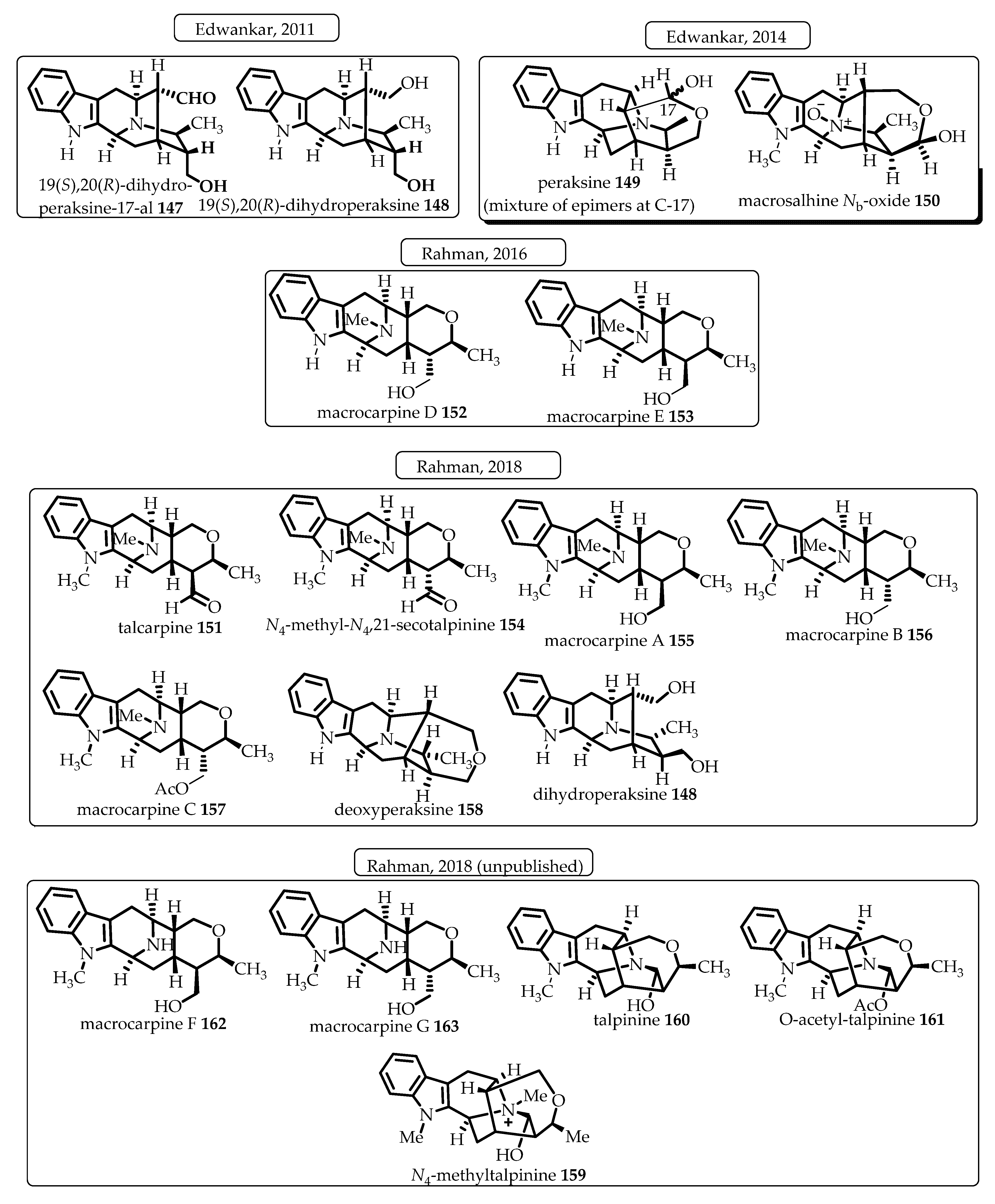
- Rahman, M.T. Shorter and Improved Access to the Key Tetracyclic Core of Sarpagine-Macroline-Ajmaline Indole Alkaloids: The Total Synthesis of Alkaloids Macrocarpines Ag, Talcarpine, N (4)-methyl-n (4), 21-secotalpinine, Deoxyperaksine, Dihydroperaksine, Talpinine, O-acetyltalpinine, and N (4)-methyltalpinine. Ph.D. Thesis, University of Wisconsin-Milwaukee, Milwaukee, WI, USA, 2018. [Google Scholar]
- Brossi, A.; Pei, X.-F. Chapter 3 Biological Activity of Unnatural Alkaloid Enantiomers. In The Alkaloids: Chemistry and Biology; Cordell, G.A., Ed.; Academic Press: San Diego CA, USA, 1998; Volume 50, pp. 109–139. [Google Scholar]
- Liotta, D.C.; Painter, G.R. Discovery and development of the anti-human immunodeficiency virus drug, emtricitabine (Emtriva, FTC). Acc. Chem. Res. 2016, 49, 2091–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, P.L.; Rower, J.E. Zidovudine and Lamivudine for HIV Infection. Clin. Med. Rev. Ther. 2010, 2, a2004. [Google Scholar]
- Barker, T.J.; Duncan, K.K.; Otrubova, K.; Boger, D.L. Potent vinblastine C20′ ureas displaying additionally improved activity against a vinblastine-resistant cancer cell cine. ACS Med. Chem. Lett. 2013, 4, 985–988. [Google Scholar] [CrossRef]
- Wender, P.A.; DeChristopher, B.A.; Schrier, A.J. Efficient synthetic access to a new family of highly potent bryostatin analogues via a Prins-driven macrocyclization strategy. J. Am. Chem. Soc. 2008, 130, 6658–6659. [Google Scholar] [CrossRef] [Green Version]
- Carney, D.W.; Lukesh, J.C.; Brody, D.M.; Brütsch, M.M.; Boger, D.L. Ultrapotent vinblastines in which added molecular complexity further disrupts the target tubulin dimer–dimer interface. Proc. Natl. Acad. Sci. USA 2016, 113, 9691–9698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, H.; Colby, D.A.; Seto, S.; Va, P.; Tam, A.; Kakei, H.; Rayl, T.J.; Hwang, I.; Boger, D.L. Total synthesis of vinblastine, vincristine, related natural products, and key structural analogues. J. Am. Chem. Soc. 2009, 131, 4904–4916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotoh, H.; Duncan, K.K.; Robertson, W.M.; Boger, D.L. 10′-fluorovinblastine and 10′-fluorovincristine: Synthesis of a key series of modified Vinca alkaloids. ACS Med. Chem. Lett. 2011, 2, 948–952. [Google Scholar] [CrossRef] [PubMed]


























| Types | Indole Alkaloids | |
|---|---|---|
 |  |  |
 |  | |
 |  | |
 |  | |
 |  | |
| Bisindole Alkaloids Reviewed Herein | ||
|---|---|---|
| Macroline-macroline | ||
 |  |  |
 |  |  |
 |  |  |
 |  |  |
 |  |  |
| Macroline-sarpagine | ||
 |  |  |
 |  |  |
 |  |  |
 |  | |
| Macroline-ajmaline | ||
 |  | |
| Macroline-pleiocarpamine | ||
 |  |  |
 |  |  |
 | ||
| Bisindoles | Bioactivity | References |
|---|---|---|
| (+)-Alstomacroline 1 | Antimalarial, with IC50 values of 1.12 ± 0.35 and 10.0 ± 0.4 μM against the K1 strain and T9-96 strain of P. falciparum, respectively. | [20] |
| (+)-Alstomacrophylline 2 | Antimalarial, with an IC50 value of 1.10 ± 0.30 μM against the K1 strain of P. falciparum. | [20] |
| Angustilongines E, F, G, H, J, and K (6–11) | Anticancer, cytotoxic against various human cancer cell lines including KB, vincristine-resistant KB, HCCT 116, PC-3, MDA-MB-231, LNCaP, MCF7, HT-29, and A549 cells with IC50 values ranging from 0.02 to 9.0 μM. | [5] |
| (−)-Lumusidine A, B, and C (14–16) | Anticancer, moderately cytotoxic in KB/VJ300 cells with IC50 values of 0.16, 0.70, and 1.19 μg/mL (μM), respectively. The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (−)-Lumusidine D 17 | Anticancer, cytotoxic in KB/VJ300 cells with an IC50 value of 5.03 μg/mL (μM). The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| Lumutinine A, B, C, D, and E (18–22) | Anticancer, moderately cytotoxic in KB/VJ300 cells with IC50 0.21, 0.10, 4.61, 3.93, and 2.74 μg/mL (μM) values, respectively. The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (+)-Macralstonine 24 | Anticancer, strongly cytotoxic in KB/VJ300 cells with an IC50 1.71 μg/mL (μM) value. The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| Antimalarial, active against the K1 strain of P. falciparum with an IC50 8.92 ± 2.95 μM value. | [20] | |
| (−)-Anhydromacralstonine 27 | Anticancer, moderately cytotoxic in KB/VJ300 cells with an IC50 value of 0.44 μg/mL (μM). The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (+)-O-Acetyl macralstonine 25 | Antimalarial, with IC50 values 0.53 ± 0.09 and 12.4 ± 1.6 (μM) against the K1 strain and T9-96 strain of P. falciparum, respectively. | [20] |
| (+)-O-Methyl macralstonine 26 | Antimalarial, active against the K1 strain of P. falciparum with an IC50 0.85 ± 0.20 μM value. | [20] |
| O-Acetyl-E-seco-macralstonine 53 | Anticancer, strongly cytotoxic in KB/VJ300 cells with an IC50 value of 0.27 μg/mL (μM). The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (−)-Perhentidine A 36 and (-)-perhentidine B 37 | Anticancer, strongly cytotoxic in KB/VJ300 cells with IC50 values of 2.29 and 0.84 μg/mL (μM), respectively. The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (−)-O-Acetylperhentidine A 54 and (-)-O-Acetylperhentidine B 55 | Anticancer, strongly cytotoxic in KB/VJ300 cells with IC50 0.36 and 0.28 μg/mL (μM) values, respectively. The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (−)-Perhentinine 39 and O-Acetylperhentinine 56 | Anticancer, cytotoxic in KB/VJ300 cells with IC50 values of 0.52 and 0.30 μg/mL (μM), respectively. The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (+)-Macralstonidine 23 | Anticancer, moderately cytotoxic in KB/VJ300 cells with an IC50 value of 0.13 μg/mL (μM). The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (+)-Macrocarpamine 21 | Anticancer, strongly cytotoxic in KB/VJ300 cells with an IC50 value of 0.53 μg/mL (μM). The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| Strong antimalarial activity against the K1 strain of P. falciparum with an IC50 value of 0.36 ± 0.06 μM. Active against the T9-96 strain of P. falciparum with an IC50 >39 μM value. | [20] | |
| Strong antiprotozoal activity in vitro against E. histolytica and P. falciparum with ED50 8.12 (95% C.I.) μM and ED50 9.36 (95% C.I.) μM values, respectively. | [43] | |
| (+)-Villalstonine 43 | Anticancer, cytotoxic in KB/VJ300 cells with an IC50 value of 0.42 μg/mL (μM). The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| Anticancer, cytotoxic against the HT-29 cell line with an ED50 8.0 μM value (paclitaxel was used as the positive control). | [39] | |
| Antimalarial, with IC50 values of 0.27 ± 0.06 and 0.94 ± 0.07 μM against the K1 strain and T9-96 strain of P. falciparum, respectively. | [20] | |
| Antiamoebic activity against E. histolytica with an ED50 of 2.04 μM. | [43] | |
| Villalstonine N(4)-oxide 44 | Antileishmanial activity against promastigotes of L. mexicana with an IC50 value of 80.3 μM (amphotericin B was used as the positive control). | [39] |
| Antimalarial, active against the K1 strain of P. falciparum with an IC50 10.7 ± 1.9 (μM) value. | [20] | |
| (+)-Villalstonidine B 48 and (+)-villalstonidine F 52 | Anticancer, strongly cytotoxic in KB/VJ300 cells with IC50 values of 0.35 and 5.64 μg/mL (μM), respectively. The assay with 0.12 μM added vincristine did not influence KB/VJ300 cell growth. | [18] |
| (+)-Villalstodinine D 50 | Antileishmanial, active against promastigotes of L. mexicana with an IC50 value of 120.4 μM (amphotericin B was used as the positive control). | [39] |
| (+)-Villalstonidine E 51 | Anticancer, cytotoxic against HT-29 cell lines with an ED50 6.5 μM value (paclitaxel was used as the positive control). | [39] |
| Antileishmanial against promastigotes of L. mexicana with an IC50 78 μM value (amphotericin B was used as the positive control). | [39] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, K.P.; Rahman, M.T.; Cook, J.M. Bisindole Alkaloids from the Alstonia Species: Recent Isolation, Bioactivity, Biosynthesis, and Synthesis. Molecules 2021, 26, 3459. https://doi.org/10.3390/molecules26113459
Pandey KP, Rahman MT, Cook JM. Bisindole Alkaloids from the Alstonia Species: Recent Isolation, Bioactivity, Biosynthesis, and Synthesis. Molecules. 2021; 26(11):3459. https://doi.org/10.3390/molecules26113459
Chicago/Turabian StylePandey, Kamal P., Md Toufiqur Rahman, and James M. Cook. 2021. "Bisindole Alkaloids from the Alstonia Species: Recent Isolation, Bioactivity, Biosynthesis, and Synthesis" Molecules 26, no. 11: 3459. https://doi.org/10.3390/molecules26113459
APA StylePandey, K. P., Rahman, M. T., & Cook, J. M. (2021). Bisindole Alkaloids from the Alstonia Species: Recent Isolation, Bioactivity, Biosynthesis, and Synthesis. Molecules, 26(11), 3459. https://doi.org/10.3390/molecules26113459