
Density Functional Theory and Molecular Docking Investigations of the Chemical and Antibacterial Activities for 1-(4-Hydroxyphenyl)-3-phenylprop-2-en-1-one


Abstract
:1. Introduction
2. Results and Discussion
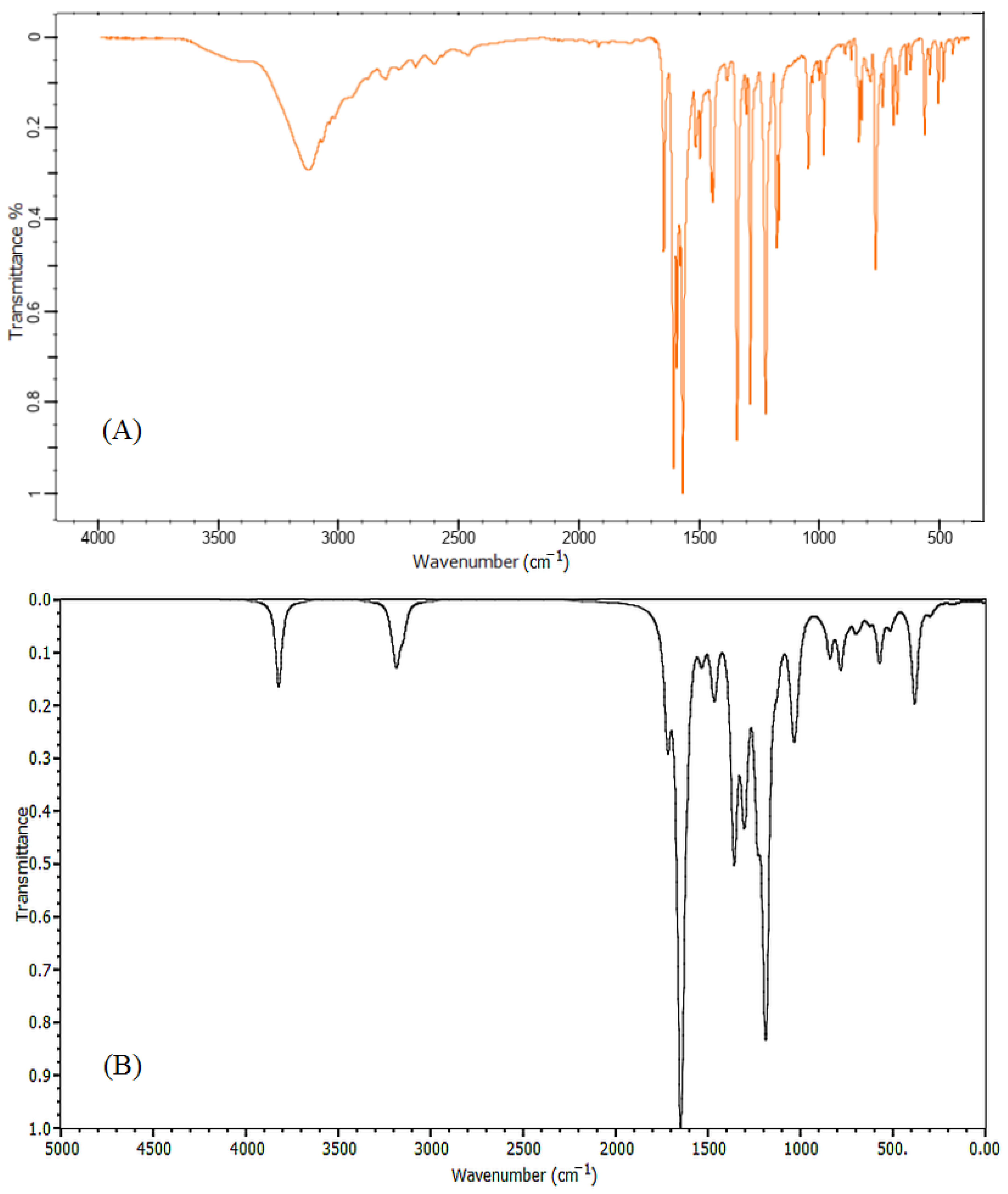
2.1. Vibrational Spectra Analysis
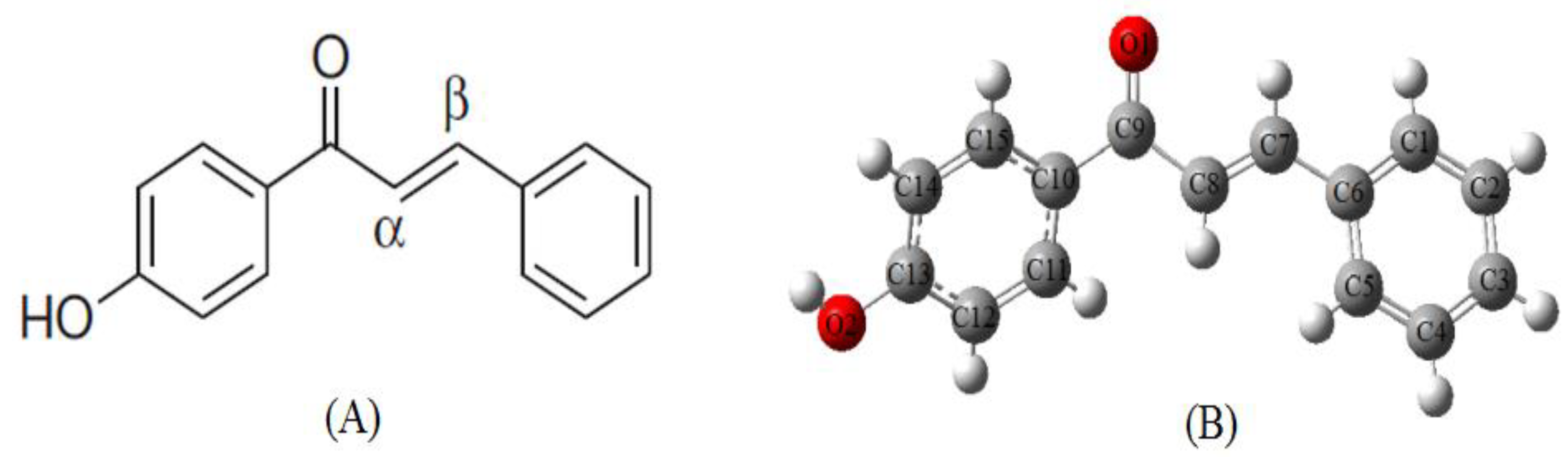
2.2. Molecular Geometry
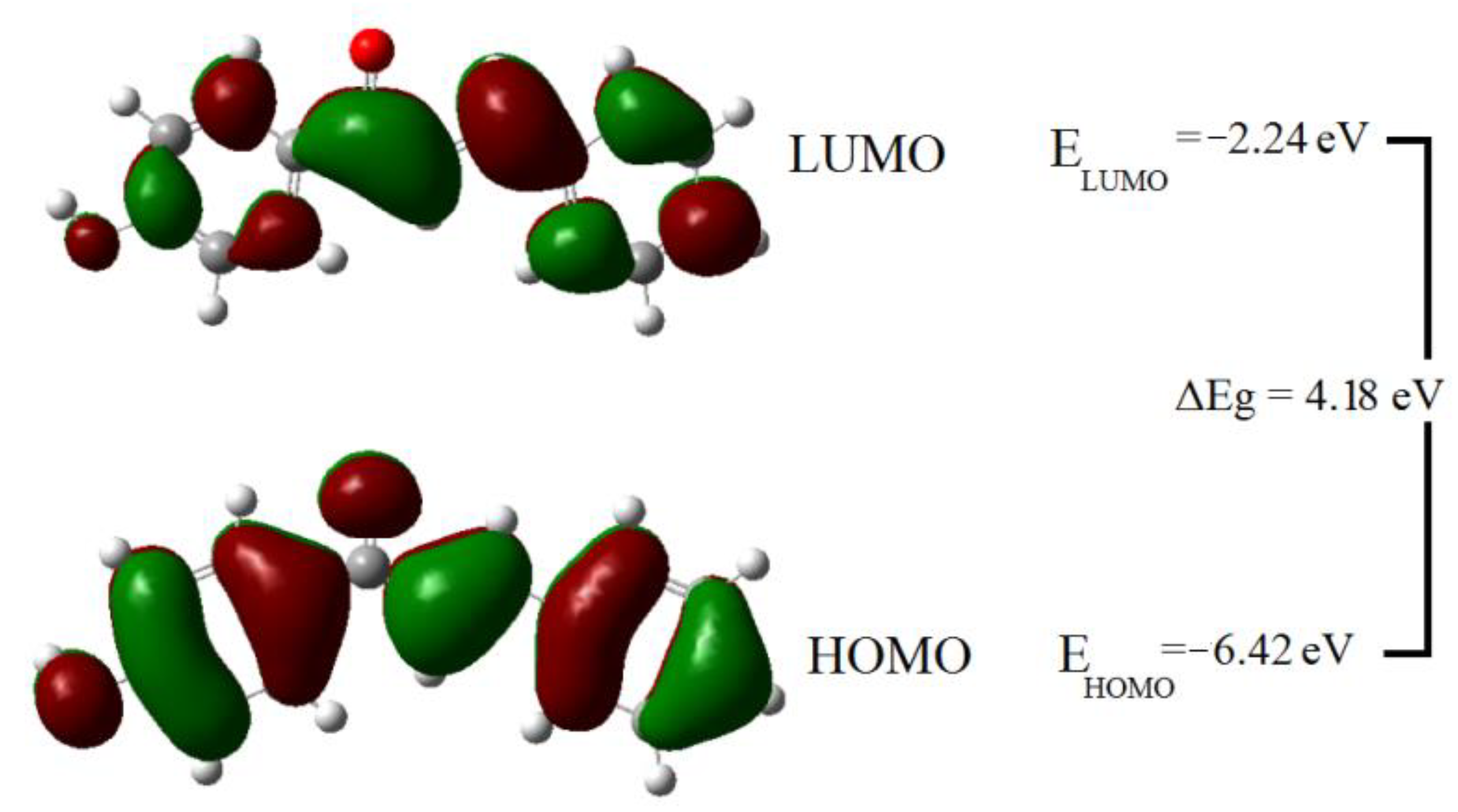
2.3. HOMO and LUMO Analysis
2.4. Analysis of the Conceptual DFT Indices
2.5. Molecular Electrostatic Potential (MEP)
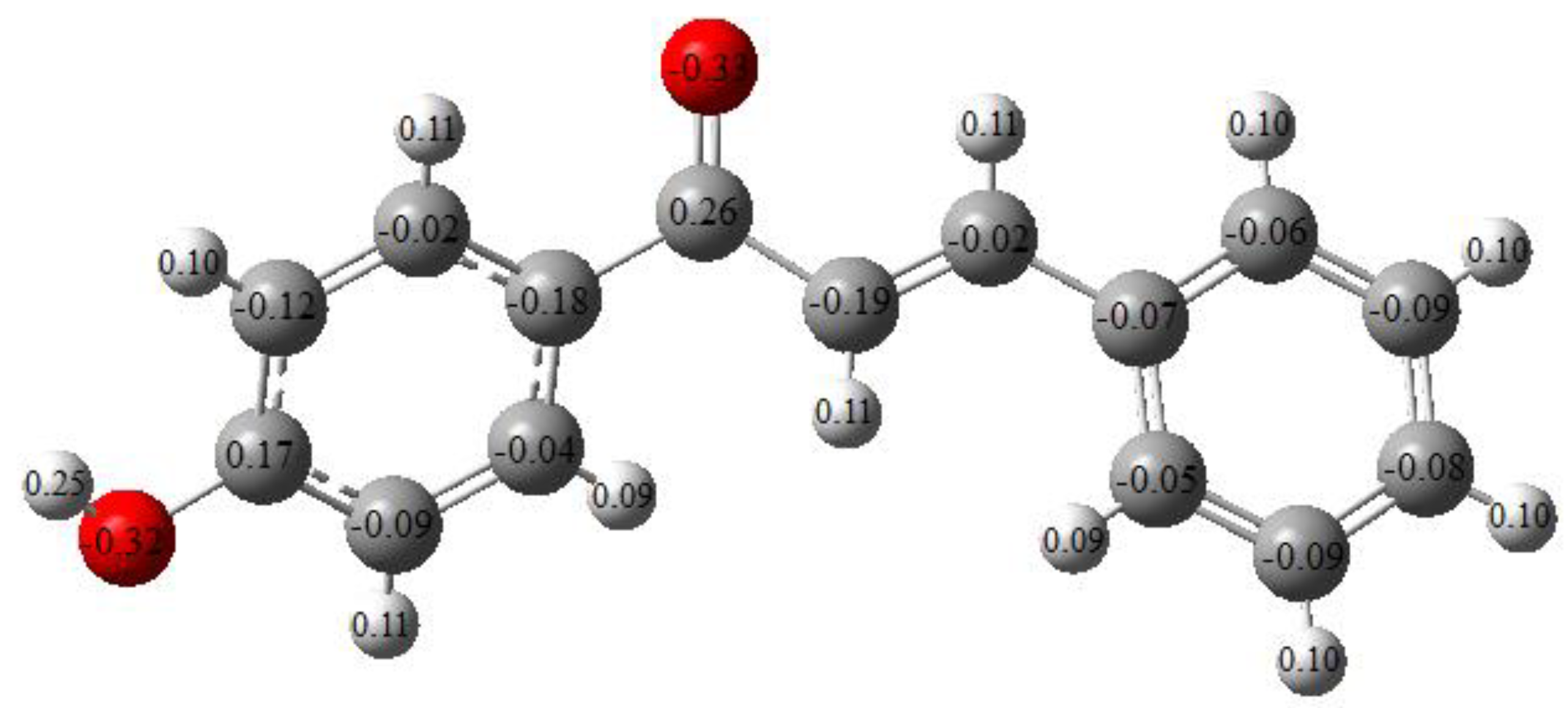
2.6. Mulliken Charge Analysis
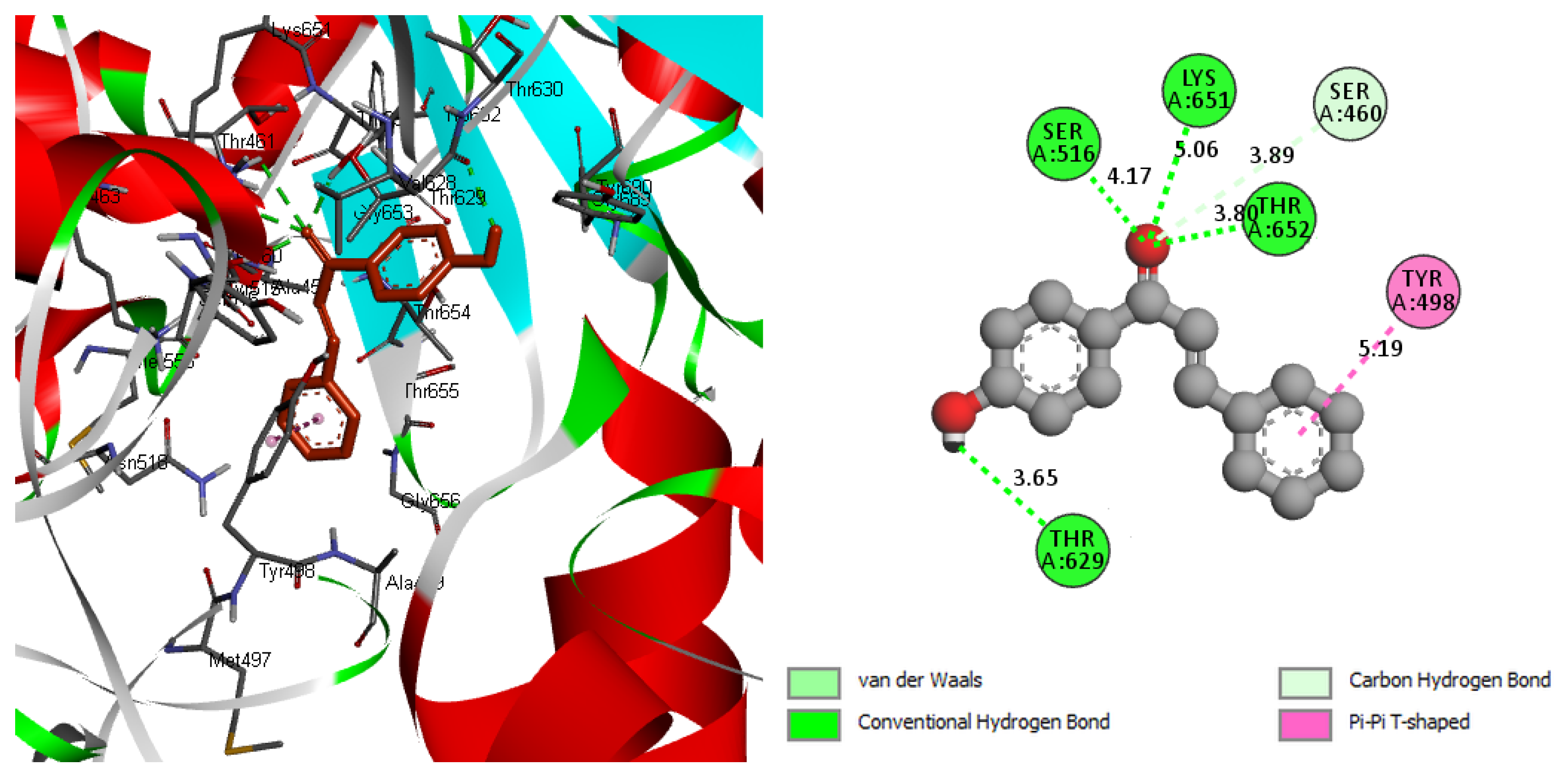
2.7. Molecular Docking
3. Materials and Methods
3.1. Experimental Details
3.2. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
Sample Availability
References
- Rammohan, A.; Reddy, J.S.; Sravya, G.; Rao, C.N.; Zyryanov, G.V. Chalcone Synthesis, Properties and Medicinal Applications: A Review. Environ. Chem. Lett. 2020, 18, 433–458. [Google Scholar] [CrossRef]
- Owaba, A.D.C.; Miediegha, O.; Oladiran, R.R. Chalcones as Sythons for Heterocyclic Compounds- A Review. Int. J. Curr. Res. 2020, 12, 13672–13681. [Google Scholar]
- Banjarnahor, S.D.S.; Artanti, N. Antioxidant Properties of Flavonoids. Med. J. Indones. 2015, 23, 239–244. [Google Scholar] [CrossRef] [Green Version]
- Zhou, B. Diverse Molecular Targets for Chalcones with Varied Bioactivities. Med. Chem. 2015, 5, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Martínez, Y.; Osorio, M.; San Martín, D.; Carvajal, M.; Vergara, A.; Sanchez, E.; Raimondi, M.; Zacchino, S.; Mascayano, C.; Torrent, C.; et al. Antimicrobial, Anti-Inflammatory and Antioxidant Activities of Polyoxygenated Chalcones. J. Braz. Chem. Soc. 2019, 30, 286–304. [Google Scholar] [CrossRef]
- Yazdan, S.K. Chemical and Biological Potentials of Chalcones: A Review. Org. Med. Chem. Int. J. 2015, 1, 20–28. [Google Scholar] [CrossRef]
- Silva, M.C.; Duarte, V.S.; Custodio, J.M.F.; Queiroz, J.E.; de Aquino, G.L.B.; Oliver, A.G.; Napolitano, H.B. Comparative Conformational Study of a New Terpenoid-like Chalcone. J. Mol. Struct. 2021, 1228, 129743. [Google Scholar] [CrossRef]
- Ibnaouf, K.H.; Hussein, R.K.; Elkhair, H.M.; Elzupir, A.O. Experimental and Theoretical Study of the Structure, Frontier Molecular Orbital, Tautomerism and Spectral Analysis of 3-(p-Substituted Phenyl)-5-Phenyl-1H-Pyrazole. J. Mol. Liq. 2019, 287, 110675. [Google Scholar] [CrossRef]
- Bonvicini, F.; Gentilomi, G.; Bressan, F.; Gobbi, S.; Rampa, A.; Bisi, A.; Belluti, F. Functionalization of the Chalcone Scaffold for the Discovery of Novel Lead Compounds Targeting Fungal Infections. Molecules 2019, 24, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Anazi, M.; Al-Najjar, B.; Khairuddean, M. Structure-Based Drug Design Studies Toward the Discovery of Novel Chalcone Derivatives as Potential Epidermal Growth Factor Receptor (EGFR) Inhibitors. Molecules 2018, 23, 3203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharib, A.; Pesyan, N.N.; Vojdanifard, L.; Jahangir, M.; Roshani, M.; Moghadasi, S.; Akhavan, H.R. Catalytic Synthesis of 1,3-Diaryl-2-Propene-1-Ones by Using Heteropolyacids as heterogeneous recyclable green catalysts. Bulg. Chem. Commun. 2014, 46, 479–485. [Google Scholar]
- Guo, Y.; Chen, Y.; Ma, H. Inclusion Mechanism and Heat Stability of the Complex of 4′-Hydroxychalcone and Hydroxypropyl-β-Cyclodextrin. Trop. J. Pharm. Res. 2015, 13, 1971. [Google Scholar] [CrossRef] [Green Version]
- Chimenti, F.; Fioravanti, R.; Bolasco, A.; Chimenti, P.; Secci, D.; Rossi, F.; Yáñez, M.; Orallo, F.; Ortuso, F.; Alcaro, S.; et al. A New Series of Flavones, Thioflavones, and Flavanones as Selective Monoamine Oxidase-B Inhibitors. Bioorg. Med. Chem. 2010, 18, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Elarfi, M.J.; Al-Difar, H.A. Synthesis of Some Heterocyclic Compounds Derived From Chalcones. IIb 2012, 2, 103–107. [Google Scholar]
- Kumar, A.; Kumar, R.; Gupta, A.; Tandon, P.; D’silva, E.D. Molecular Structure, Nonlinear Optical Studies and Spectroscopic Analysis of Chalcone Derivative (2E)-3-[4-(Methylsulfanyl) Phenyl]-1-(3-Bromophenyl) Prop-2-En-1-One by DFT Calculations. J. Mol. Struct. 2017, 1150, 166–178. [Google Scholar] [CrossRef]
- Singh, R.N.; Rawat, P.; Sahu, S. Synthesis, Characterization and Computational Study on Ethyl 4-(3-Furan-2yl-Acryloyl)-3,5-Dimethyl-1H-Pyrrole-2-Carboxylate. J. Mol. Struct. 2014, 1076, 437–445. [Google Scholar] [CrossRef]
- Elzupir, A.O.; Ali, M.K.M.; Hussein, R.K.; Ibrahem, M.A.; Al-Muhanna, M.K.; Ibnaouf, K.H. Molecular Structure, Frontier Molecular Orbital and Spectral Analysis of Dimethylamino Chalcones Efficient Lasing Dyes. J. Mol. Struct. 2019, 1178, 285–289. [Google Scholar] [CrossRef]
- Alwani Zainuri, D.; Arshad, S.; Che Khalib, N.; Fikri Zaini, M.; Razak, I.A. Molecular Structure Investigation On Organic Chalcone Derivative of (E)-3-(4-Bromothiophen-2-1-(3-Nitrophenyl)Prop-2-En-1-One: A Combined Experimental and Theoretical Study. J. Phys. Conf. Ser. 2018, 1083, 12046. [Google Scholar] [CrossRef] [Green Version]
- Hassan, A.T.; Hussein, R.K.; Abou-krisha, M.; Mohamed, A. Density Functional Theory Investigation of Some Pyridine Dicarboxylic Acids Derivatives as Corrosion Inhibitors. Int. J. Electrochem. Sci. 2020, 15, 4274–4286. [Google Scholar] [CrossRef]
- Zaini, M.F.; Arshad, S.; Thanigaimani, K.; Khalib, N.C.; Zainuri, D.A.; Abdullah, M.; Razak, I.A. New Halogenated Chalcones: Synthesis, Crystal Structure, Spectroscopic and Theoretical Analyses for Third-Order Nonlinear Optical Properties. J. Mol. Struct. 2019, 1195, 606–619. [Google Scholar] [CrossRef]
- Chidan Kumar, C.S.; Govindarasu, K.; Fun, H.-K.; Kavitha, E.; Chandraju, S.; Quah, C.K. Synthesis, Molecular Structure, Spectroscopic Characterization and Quantum Chemical Calculation Studies of (2E)-1-(5-Chlorothiophen-2-Yl)-3-(2,3,4-Trimethoxyphenyl)Prop-2-En-1-One. J. Mol. Struct. 2015, 1085, 63–77. [Google Scholar] [CrossRef]
- Espinoza-Hicks, J.C.; Camacho-Dávila, A.A.; Flores-Holguín, N.R.; Nevárez-Moorillón, G.V.; Glossman-Mitnik, D.; Rodríguez-Valdez, L.M. Experimental and Quantum Chemical Studies of a Novel Synthetic Prenylated Chalcone. Chem. Cent. J. 2013, 7, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yalcin, G.; Burmaoglu, S.; Yildiz, I.; Algul, O. Molecular Docking Studies on Fluoro-Substituted Chalcones as Potential DprE1 Enzyme Inhibitors. J. Mol. Struct. 2018, 1164, 50–56. [Google Scholar] [CrossRef]
- Hussein, R.K.; Elkhair, H.M. Molecular Docking Identification for the Efficacy of Some Zinc Complexes with Chloroquine and Hydroxychloroquine against Main Protease of COVID-19. J. Mol. Struct. 2021, 1231, 129979. [Google Scholar] [CrossRef]
- Nayak, P.S.; Narayana, B.; Sarojini, B.K.; Fernades, J.; Bharath, B.R.; Madhu, L.N. Synthesis, Molecular Docking and Biological Evaluation of Novel Bis-Pyrazole Derivatives for Analgesic, Anti-Inflammatory and Antimicrobial Activities. Med. Chem. Res. 2015, 24, 4191–4206. [Google Scholar] [CrossRef]
- Gerhards, M.; Jansen, A.; Unterberg, C.; Kleinermanns, K. OH Stretching Vibrations of the Phenol (H2O)1+ Cation. Chem. Phys. Lett. 2001, 344, 113–119. [Google Scholar] [CrossRef]
- Coates, J. Interpretation of Infrared Spectra, A Practical Approach. In Encyclopedia of Analytical Chemistry; Meyers, R.A., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 2006; p. a5606. [Google Scholar]
- Galabov, B.; Simov, D. The Stretching Vibration of Carbonyl Groups in Cyclic Ketones. Chem. Phys. Lett. 1970, 5, 549–551. [Google Scholar] [CrossRef]
- Balan, V.; Mihai, C.-T.; Cojocaru, F.-D.; Uritu, C.-M.; Dodi, G.; Botezat, D.; Gardikiotis, I. Vibrational Spectroscopy Fingerprinting in Medicine: From Molecular to Clinical Practice. Materials 2019, 12, 2884. [Google Scholar] [CrossRef] [Green Version]
- Gipson, K.; Stevens, K.; Brown, P.; Ballato, J. Infrared Spectroscopic Characterization of Photoluminescent Polymer Nanocomposites. J. Spectrosc. 2015, 2015, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Parthasarathi, V.; Praveen, V.N.; Thamotharan, S.; Vijayalakshmi, L.; Bhaskar, A. 1-(4-Hydroxyphenyl)-3-Phenylprop-2-En-1-One. Acta Crystallogr. Sect. E Struct. Rep. Online 2002, 58, o86–o87. [Google Scholar] [CrossRef]
- Domingo, L.; Ríos-Gutiérrez, M.; Pérez, P. Applications of the Conceptual Density Functional Theory Indices to Organic Chemistry Reactivity. Molecules 2016, 21, 748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuneda, T.; Song, J.-W.; Suzuki, S.; Hirao, K. On Koopmans’ Theorem in Density Functional Theory. J. Chem. Phys. 2010, 133, 174101. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Pérez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Mou, J.; Liu, Y.; Gong, X.; Yang, Y.; An, L. An Ab Initio Simulation of the UV/Visible Spectra of Substituted Chalcones. Open Chem. 2010, 8, 928–936. [Google Scholar] [CrossRef]
- Domingo, L.R.; Pérez, P. The Nucleophilicity N Index in Organic Chemistry. Org. Biomol. Chem. 2011, 9, 7168. [Google Scholar] [CrossRef]
- Alam, M.S.; Rahman, S.M.M.; Lee, D.-U. Synthesis, Biological Evaluation, Quantitative-SAR and Docking Studies of Novel Chalcone Derivatives as Antibacterial and Antioxidant Agents. Chem. Pap. 2015, 69, 1118–1129. [Google Scholar] [CrossRef]
- Chen, D.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of Protein-Ligand Binding Affinity by Hydrogen Bond Pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef] [Green Version]
- Singh, N.; Kumar, N.; Rathee, G.; Sood, D.; Singh, A.; Tomar, V.; Dass, S.K.; Chandra, R. Privileged Scaffold Chalcone: Synthesis, Characterization and Its Mechanistic Interaction Studies with BSA Employing Spectroscopic and Chemoinformatics Approaches. ACS Omega 2020, 5, 2267–2279. [Google Scholar] [CrossRef] [Green Version]
- John Wiley & Sons, Inc. SpectraBase; SpectraBase Compound ID=15yGNIg0GtJ SpectraBase Spectrum ID=BpE7xdvOvip. Available online: https://spectrabase.com/spectrum/BpE7xdvOvip (accessed on 20 May 2021).
- Hertwig, R.H.; Koch, W. On the Parameterization of the Local Correlation Functional. What Is Becke-3-LYP? Chem. Phys. Lett. 1997, 268, 345–351. [Google Scholar] [CrossRef]
- Raghavachari, K. Perspective on “Density Functional Thermochemistry. III. The Role of Exact Exchange”. Theor. Chem. Acc. Theory Comput. Model. Theor. Chim. Acta 2000, 103, 361–363. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. Gauss View; Semichem Inc.: Shawnee, KS, USA, 2009. [Google Scholar]
- Reed, P.; Atilano, M.L.; Alves, R.; Hoiczyk, E.; Sher, X.; Reichmann, N.T.; Pereira, P.M.; Roemer, T.; Filipe, S.R.; Pereira-Leal, J.B.; et al. Staphylococcus Aureus Survives with a Minimal Peptidoglycan Synthesis Machine but Sacrifices Virulence and Antibiotic Resistance. PLoS Pathog. 2015, 11, e1004891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beise, F.; Labischinski, H.; Bradaczek, H. On the Relationships between Molecular Conformation, Affinity towards Penicillin-Binding Proteins, and Biological Activity of Penicillin G-Sulfoxide. Z. Naturforschung C 1988, 43, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navratna, V.; Nadig, S.; Sood, V.; Prasad, K.; Arakere, G.; Gopal, B. Molecular Basis for the Role of Staphylococcus Aureus Penicillin Binding Protein 4 in Antimicrobial Resistance. J. Bacteriol. 2010, 192, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiblier, C.; Luczak-Kadlubowska, A.; Holdener, E.; Alborn, D.; Schneider, T.; Wiedemann, I.; Pinho, M.; Sahl, H.-G.; Rohrer, S.; Berger-Bächi, B.; et al. The Staphylococcus Aureus Membrane Protein SA2056 Interacts with Peptidoglycan Synthesis Enzymes. Antibiotics 2013, 2, 11–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twinkle, A.R.; Leenaraj, D.R.; Ratkovic, Z.; Arunsasi, B.S.; Bright, K.C.; Reshma, R. Ferrocenyl Chalcone Derivative (E)-3-(2-Methylpyrimidin-5-Yl)-1-Ferroceynlprop-2-En-1-One: Synthesis, Structural Analysis, Docking Study and Their Antibacterial Evaluation. J. Mol. Struct. 2020, 1210, 128049. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. Discovery Studio Modelling Environment, Release 2017; Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Observed Frequencies | Calculated | Frequencies | Assignment | ||
|---|---|---|---|---|---|
| No | FT-IR | FT Raman | Unscaled | Scaled | |
| 1 | ⎯⎯ | ⎯⎯ | 3825 | 3787 | ν (O–H) |
| 2 | ⎯⎯ | ⎯⎯ | 3212 | 3179 | ν (C–H) |
| 3 | ⎯⎯ | ⎯⎯ | 3203 | 3170 | ν (C–H) |
| 4 | ⎯⎯ | ⎯⎯ | 3197 | 3165 | ν (C–H) |
| 5 | ⎯⎯ | ⎯⎯ | 3193 | 3161 | ν (C–H) R s |
| 7 | 3120 | 3060 | 3184 | 3152 | ν (C–H) |
| 8 | ⎯⎯ | ⎯⎯ | 3177 | 3145 | ν (C–H) R as |
| 9 | ⎯⎯ | ⎯⎯ | 3163 | 3131 | ν (C–H) R as |
| 10 | ⎯⎯ | ⎯⎯ | 3150 | 3118 | ν (C–H) |
| 11 | ⎯⎯ | ⎯⎯ | 3149 | 3117 | ν (C–H) |
| 12 | 1648 | 1645 | 1723 | 1705 | ν (C=O) |
| 13 | 1607 | 1594 | 1654 | 1637 | ν (C=C) |
| 14 | 1594 | ⎯⎯ | 1645 | 1628 | ν (C=C) + ν (C=O) |
| 15 | 1574 | 1552 | 1637 | 1620 | ν (C=C) + ν (C=O) |
| 16 | 1569 | ⎯⎯ | 1623 | 1606 | ν (C–H) R as |
| 17 | 1551 | ⎯⎯ | 1615 | 1599 | ν (C–H) R s |
| 18 | 1333 | 1319 | 1364 | 1350 | γ (C–H)R |
| 19 | ⎯⎯ | ⎯⎯ | 1321 | 1308 | γ (C–H) |
| 20 | 1281 | ⎯⎯ | 1301 | 1287 | ν (C–O) |
| 21 | 1165 | 1198 | 1188 | 1176 | α(C–H) |
| 22 | ⎯⎯ | ⎯⎯ | 1184 | 1172 | α(C–H) |
| 23 | ⎯⎯ | ⎯⎯ | 1130 | 1118 | α(C–H) |
| 24 | ⎯⎯ | ⎯⎯ | 1104 | 1093 | α(C–H) |
| 25 | 978 | 998 | 994 | 984 | twist (C–H) |
| 26 | ⎯⎯ | ⎯⎯ | 980 | 970 | twist (C–H) |
| 27 | ⎯⎯ | ⎯⎯ | 950 | 940 | twist (C–H) |
| 28 | ⎯⎯ | ⎯⎯ | 931 | 921 | twist (C–H) |
| 29 | 772 | ⎯⎯ | 784 | 776 | ω (C–H) |
| 30 | ⎯⎯ | ⎯⎯ | 744 | 737 | ω (C–H) as+ (C=C) |
| 31 | ⎯⎯ | ⎯⎯ | 705 | 698 | ω (C–H) R |
| Bond Length (Å) | Exp. | B3LYB/6-311g(d,p) | Bond Length (Å) | Exp. | B3LYB/6-311g(d,p) |
|---|---|---|---|---|---|
| C9-O1 | 1.24 | 1.23 | C7-C8 | 1.32 | 1.34 |
| C13-O2 | 1.34 | 1.36 | C8-C9 | 1.47 | 1.48 |
| C1-C2 | 1.38 | 1.39 | C9-C10 | 1.46 | 1.49 |
| C2-C3 | 1.36 | 1.39 | C10-C11 | 1.39 | 1.40 |
| C3-C4 | 1.36 | 1.39 | C11-C12 | 1.37 | 1.38 |
| C4-C5 | 1.38 | 1.38 | C12-C13 | 1.39 | 1.39 |
| C6-C1 | 1.38 | 1.40 | C13-C14 | 1.38 | 1.40 |
| C5-C6 | 1.38 | 1.40 | C15-C14 | 1.37 | 1.38 |
| C6-C7 | 1.46 | 1.46 | C10-C15 | 1.39 | 1.40 |
| Angle (◦) | Exp. | B3LYB/6-311g(d,p) | Angle (◦) | Exp. | B3LYB/6-311g(d,p) |
| C13-O2-H | 109.5 | 109.40 | C10-C9-C8 | 118.68 | 119.23 |
| C11-C10-C15 | 117.61 | 117.97 | C12-C11-C10 | 121.63 | 121.30 |
| C11-C10-C9 | 122.0 | 124.56 | C14-C15-C10 | 121.2 | 121.31 |
| C15-C10-C9 | 120.39 | 117.44 | O2-C13-C14 | 122.98 | 122.67 |
| C5-C6-C1 | 117.90 | 118.03 | O2-C13-C12 | 117.64 | 117.54 |
| C5-C6-C7 | 122.47 | 123.45 | C8-C7-C6 | 126.6 | 124.95 |
| C1-C6-C7 | 119.70 | 118.51 | C8-C7-H7 | 116.7 | 115.23 |
| O1-C9-C10 | 121.17 | 119.95 | C7-C8-C9 | 122.4 | 121.9 |
| O1-C9-C8 | 120.15 | 120.83 | C6-C1-C2 | 121 | 120.87 |
| C6-C5-C4 | 120.90 | 120.12 | C3-C2-C1 | 119.9 | 117.35 |
| C14-C13-C12 | 119.40 | 118.55 | C15-C14-C13 | 120.14 | 119.27 |
| C11-C12-C13 | 120 | 119.80 | C4-C3-C2 | 120.2 | 119.86 |
| C7-C8-C9-O1 | −11.40 | −11.37 | C1-C6-C5-C4 | −1.2 | −1.19 |
| C10-C9-C8-C7 | 168.50 | 167.42 | C7-C6-C5-C4 | 177.1 | 175.54 |
| C5-C6-C1-C2 | 1.5 | 1.31 | C6-C5-C4-C3 | 0.7 | 0.68 |
| C5-C6-C7-C8 | −7.8 | −7.2 | C6-C1-C2-C3 | −1.2 | −1.17 |
| C7-C6-C1-C2 | −176.9 | −175.66 | C6-C7-C8-C9 | −177.17 | −176.58 |
| C1-C6-C7-C8 | 170.5 | 169.42 | C9-C10-C11-C12 | −179.4 | −178.32 |
| C12-C11-C10-C9 | −179.4 | −178.01 | C11-C10-C15-C14 | 0.8 | 0.78 |
| C9-C10-C15-C14 | −179.15 | −177.89 | C10-C15-C14-C13 | −1.7 | −1.67 |
| C11-C12-C13-O2 | −179.3 | −177.53 | C12-C13-C14-C15 | 1.2 | 1.17 |
| C15-C14-C13-O2 | −179.4 | −179.11 | C10-C11-C12-C13 | −1.1 | −1.15 |
| C11-C10-C9-O1 | 155.7 | 153.64 | C14-C13-C12-C11 | 0.2 | 0.17 |
| C15-C10-C9-O1 | −24.4 | −23.05 | C15-C10-C9-C8 | 155.73 | 154.22 |
| C11-C10-C9-C8 | −24.2 | −25.21 | C15-C10-C11-C12 | 0.6 | 0.56 |
| Molecular Property | EHOMO (eV) | ELUMO (eV) | η (eV) | S (eV) −1 | χ (eV) | ω (eV) | N (eV) |
|---|---|---|---|---|---|---|---|
| Definition | ⎯⎯ | ⎯⎯ | |||||
| Value | −6.42 | −2.24 | 4.18 | 0.24 | 4.33 | 2.25 | 2.95 |
| Binding Energy (kcal/mol) | Inhibition Constant Ki (µM) | Intermolecular Energy (kcal/mol) | Bonded Residues | |
|---|---|---|---|---|
| (PBP-1b) protein | −7.40 | 3.74 | −8.6 | THR629, THR625 LYS651, SER516 SER460, TYR498 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deghady, A.M.; Hussein, R.K.; Alhamzani, A.G.; Mera, A. Density Functional Theory and Molecular Docking Investigations of the Chemical and Antibacterial Activities for 1-(4-Hydroxyphenyl)-3-phenylprop-2-en-1-one. Molecules 2021, 26, 3631. https://doi.org/10.3390/molecules26123631
Deghady AM, Hussein RK, Alhamzani AG, Mera A. Density Functional Theory and Molecular Docking Investigations of the Chemical and Antibacterial Activities for 1-(4-Hydroxyphenyl)-3-phenylprop-2-en-1-one. Molecules. 2021; 26(12):3631. https://doi.org/10.3390/molecules26123631
Chicago/Turabian StyleDeghady, Ahmed M., Rageh K. Hussein, Abdulrahman G. Alhamzani, and Abeer Mera. 2021. "Density Functional Theory and Molecular Docking Investigations of the Chemical and Antibacterial Activities for 1-(4-Hydroxyphenyl)-3-phenylprop-2-en-1-one" Molecules 26, no. 12: 3631. https://doi.org/10.3390/molecules26123631
APA StyleDeghady, A. M., Hussein, R. K., Alhamzani, A. G., & Mera, A. (2021). Density Functional Theory and Molecular Docking Investigations of the Chemical and Antibacterial Activities for 1-(4-Hydroxyphenyl)-3-phenylprop-2-en-1-one. Molecules, 26(12), 3631. https://doi.org/10.3390/molecules26123631