
Electrophilically Activated Nitroalkanes in Synthesis of 3,4-Dihydroquinozalines

 , , ,
, , ,
Abstract
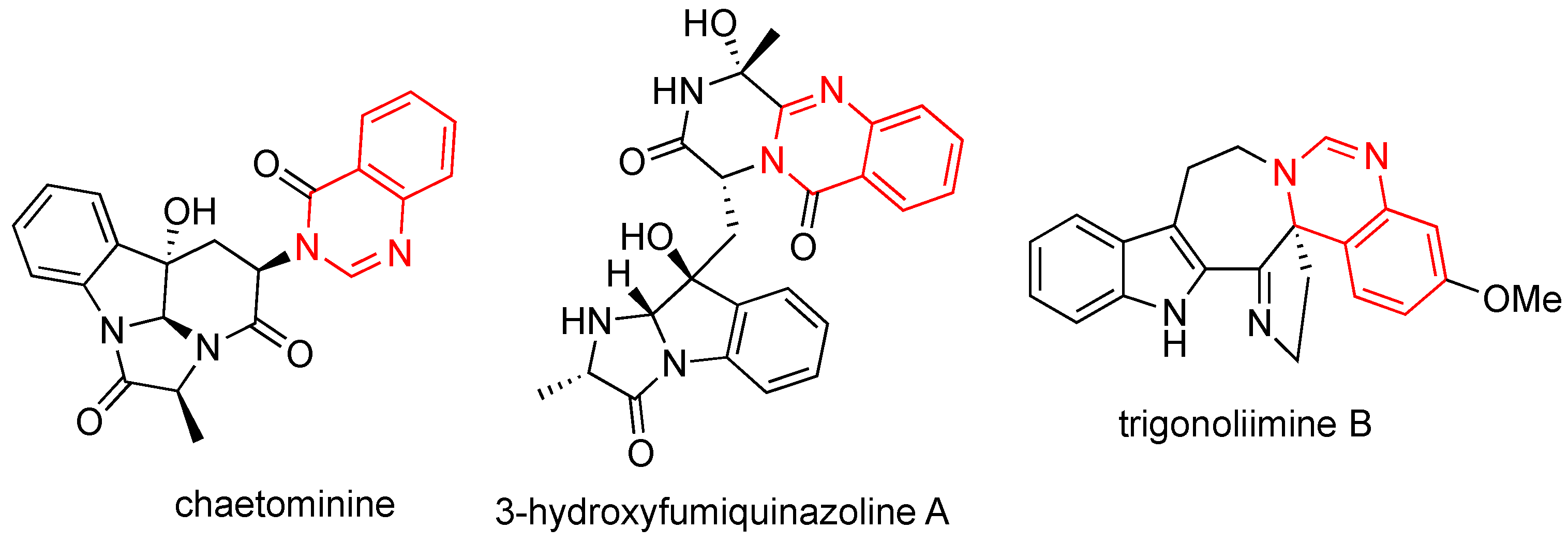
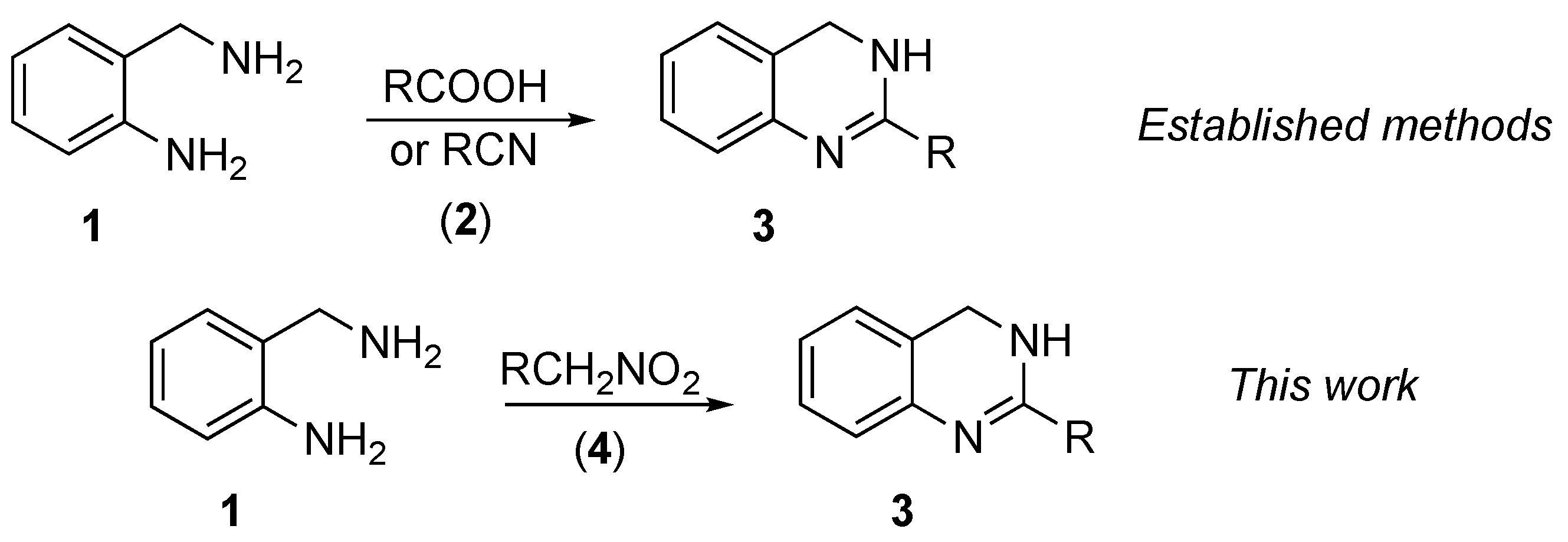
:1. Introduction
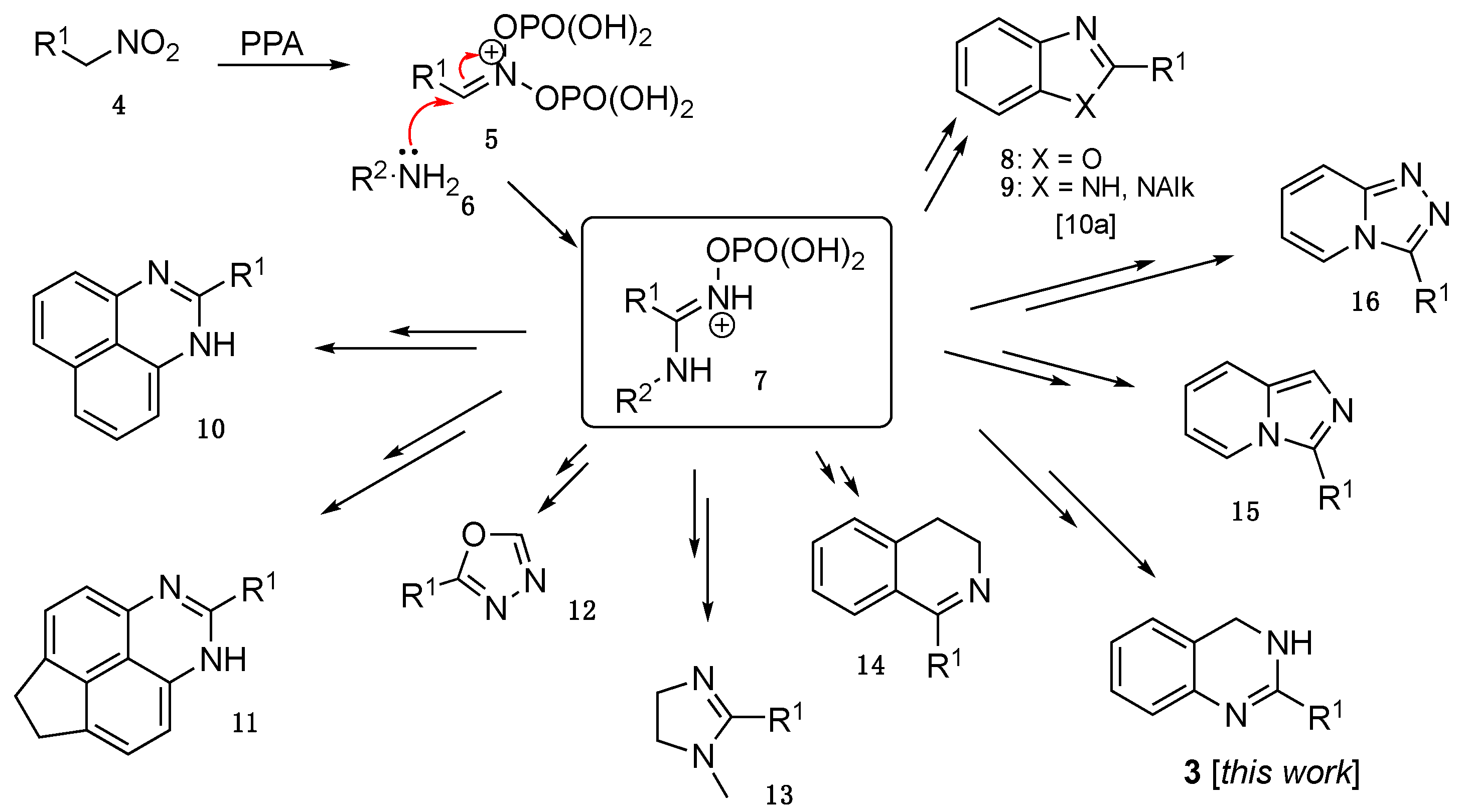
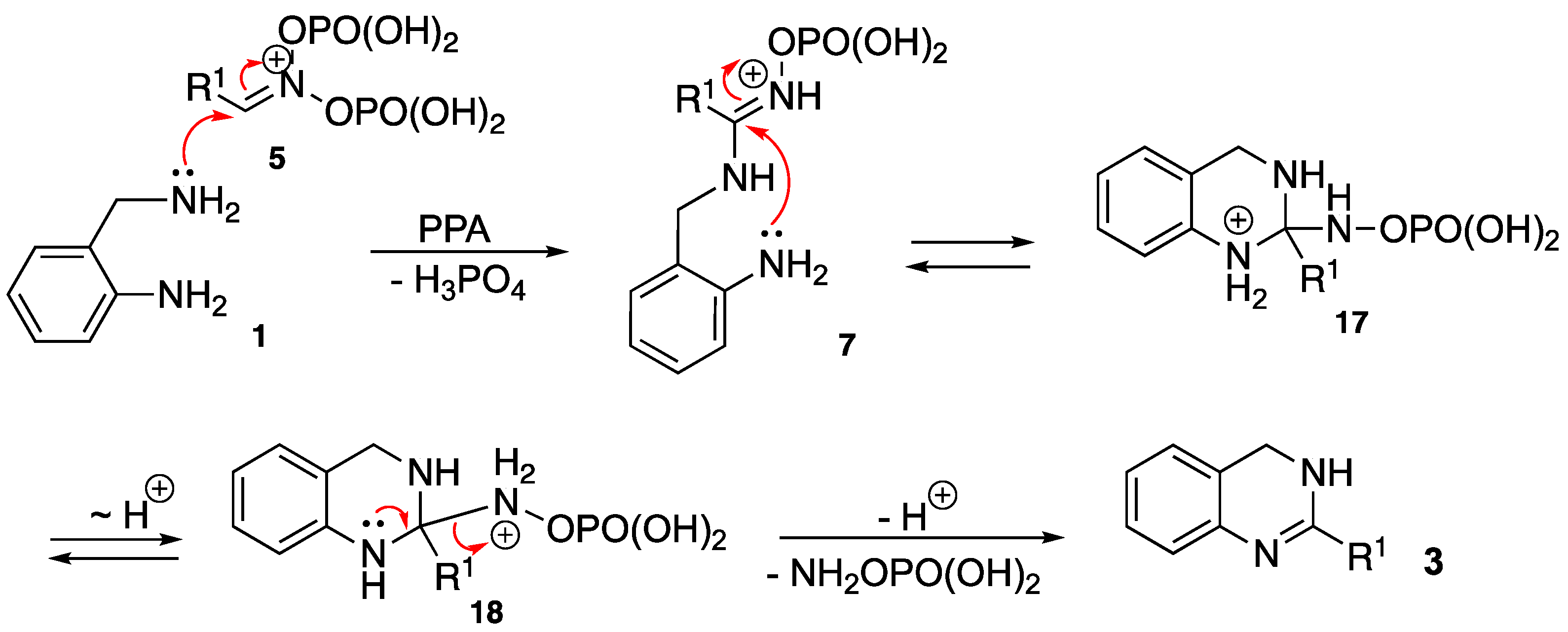
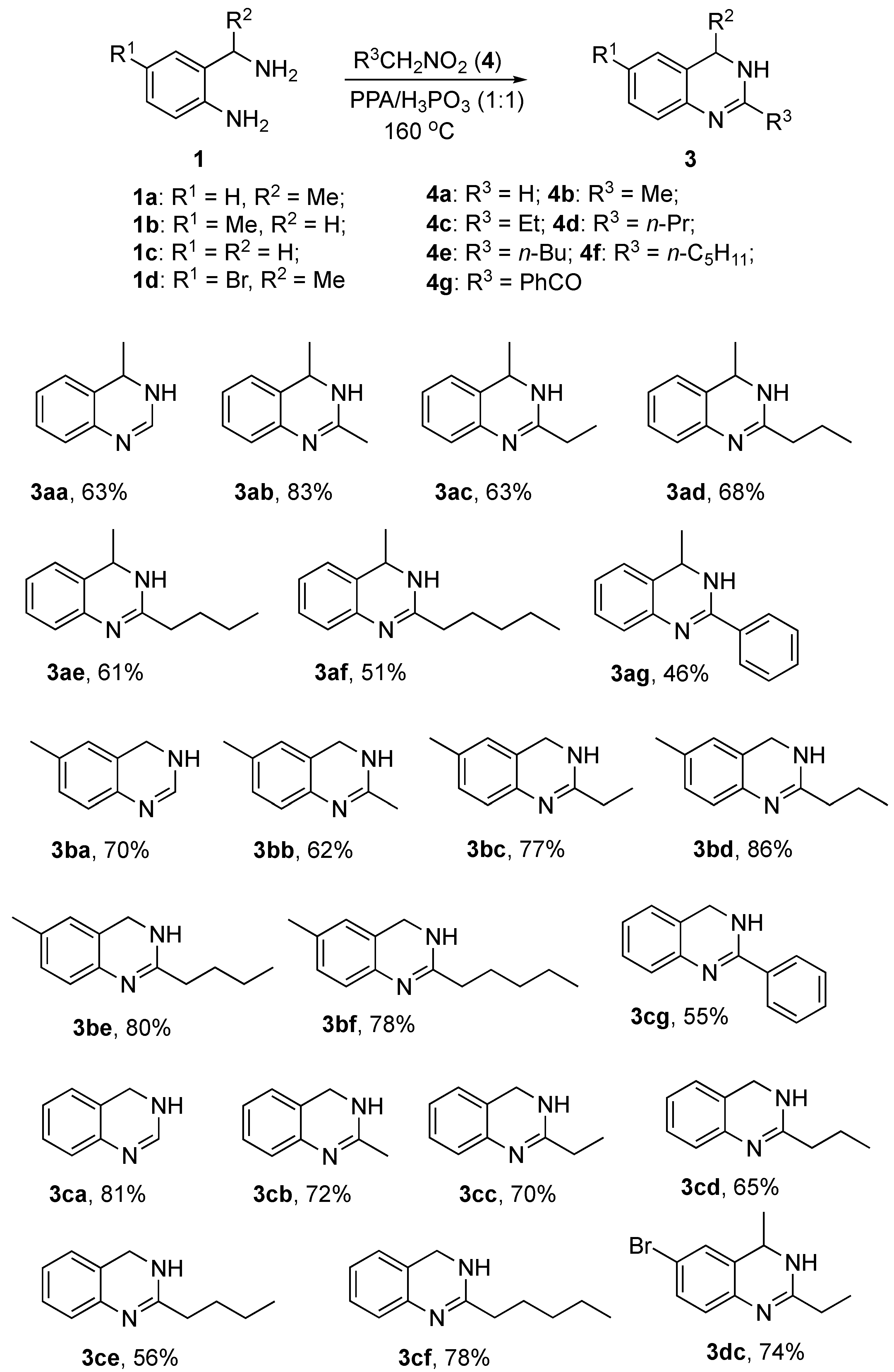
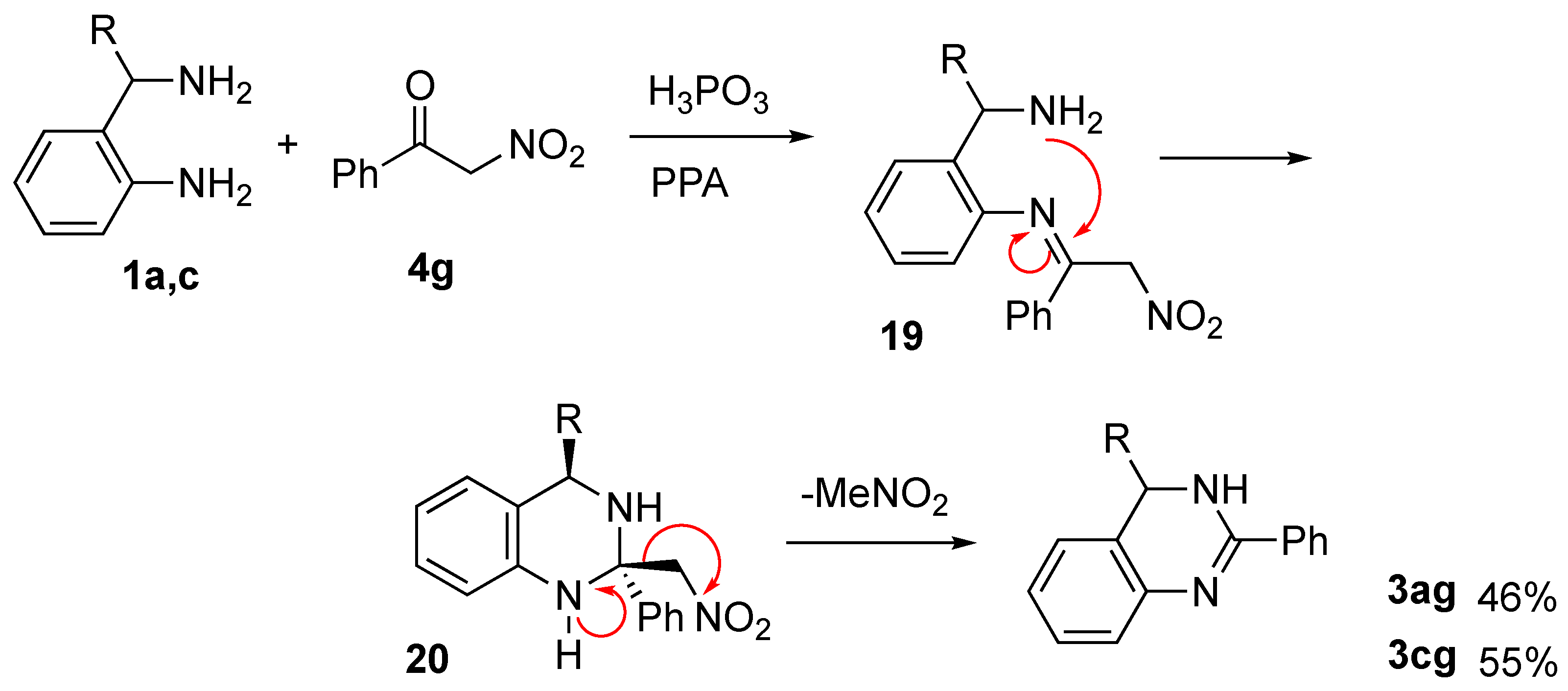
2. Results and Discussion
3. Materials and Methods
- 2-(1-Aminoethyl)aniline (1a):
- Method A:
- Method B:
- 2-(Aminomethyl)-4-methylaniline (1b):
- 2-(Aminomethyl)aniline (1c):
- Method A:
- Method B:
- 2-(1-Aminoethyl)-4-bromoaniline (1d):
- Preparation of 3,4-dihydroquinazolines (3aa–3cf) via annulation of 2-aminobenzylamines and 1-nitroalkanes (General procedure):
- 4-Methyl-3,4-dihydroquinazoline (3aa):
- 2-Methyl-4-methyl-3,4-dihydroquinazoline (3ab):
- 2-Ethyl-4-methyl-3,4-dihydroquinazoline (3ac):
- 4-Methyl-2-propyl-3,4-dihydroquinazoline (3ad):
- 2-Butyl-4-methyl-3,4-dihydroquinazoline (3ae):
- 4-Methyl-2-pentyl-3,4-dihydroquinazoline (3af):
- 4-Methyl-2-phenyl-3,4-dihydroquinazoline (3ag):
- 6-Methyl-3,4-dihydroquinazoline (3ba):
- 2,6-Dimethyl-3,4-dihydroquinazoline (3bb):
- 2-Ethyl-6-methyl-3,4-dihydroquinazoline (3bc):
- 6-Methyl-2-propyl-3,4-dihydroquinazoline (3bd):
- 2-Butyl-6-methyl-3,4-dihydroquinazoline (3be):
- 6-Methyl-2-pentyl-3,4-dihydroquinazoline (3bf):
- 3,4-Dihydroquinazoline (3ca):
- 2-Methyl-3,4-dihydroquinazoline (3cb):
- 2-Ethyl-3,4-dihydroquinazoline (3cc):
- 2-Propyl-3,4-dihydroquinazoline (3cd):
- 2-Butyl-3,4-dihydroquinazoline (3ce):
- 2-Pentyl-3,4-dihydroquinazoline (3cf):
- 2-Phenyl-3,4-dihydroquinazoline (3cg):
- 6-Bromo-2-ethyl-4-methyl-3,4-dihydroquinazoline (3dc):
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Jiao, R.H.; Xu, S.; Liu, J.-Y.; Ge, H.M.; Ding, H.; Xu, C.; Zhu, A.H.L.; Tan, R.X. Chaetominine, a Cytotoxic Alkaloid Produced by Endophytic Chaetomium sp. IFB-E015. Org. Lett. 2006, 8, 5709–5712. [Google Scholar] [CrossRef]
- Li, X.-J.; Zhang, Q.; Zhang, A.-L.; Gao, J.-M. Metabolites from Aspergillus fumigatus, an Endophytic Fungus Associated with Melia azedarach, and their Antifungal, Antifeedant, and Toxic Activities. J. Agric. Food Chem. 2012, 60, 3424–3431. [Google Scholar] [CrossRef]
- Tan, C.-J.; Di, Y.-T.; Wang, Y.-H.; Zhang, Y.; Si, Y.-K.; Zhang, Q.; Gao, S.; Hu, X.-J.; Fang, X.; Li, S.-F.; et al. Three New Indole Alkaloids from Trigonostemon lii. Org. Lett. 2010, 12, 2370–2373. [Google Scholar] [CrossRef] [PubMed]
- Buyck, T.; Wang, Q.; Zhu, J. A Concise Total Synthesis of (±)-Trigonoliimine B. Org. Lett. 2012, 14, 1338–1341. [Google Scholar] [CrossRef]
- Feng, P.; Fan, Y.; Xue, F.; Liu, W.; Li, S.; Shi, Y. An Approach to the Hexacyclic Skeleton of Trigonoliimines. Org. Lett. 2011, 13, 5827–5829. [Google Scholar] [CrossRef]
- Han, S.; Morrison, K.C.; Hergenrother, P.J.; Movassaghi, M. Total Synthesis, Stereochemical Assignment, and Biological Activity of All Known (−)-Trigonoliimines. J. Org. Chem. 2013, 79, 473–486. [Google Scholar] [CrossRef]
- Han, S.; Movassaghi, M. Concise Total Synthesis and Stereochemical Revision of all (−)-Trigonoliimines. J. Am. Chem. Soc. 2011, 133, 10768–10771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, B.; Hao, X.-Y.; Zhang, J.-X.; Liu, S.; Hao, X.-J. Rapid Total Synthesis of (±)Trigonoliimine A via a Strecker/Houben–Hoesch Sequence. Org. Lett. 2013, 15, 528–530. [Google Scholar] [CrossRef] [PubMed]
- Beck, H.P.; Kohn, T.; Rubenstein, S.; Hedberg, C.; Schwandner, R.; Hasslinger, K.; Dai, K.; Li, C.; Liang, L.; Wesche, H.; et al. Discovery of potent LPA2 (EDG4) antagonists as potential anticancer agents. Bioorg. Med. Chem. Lett. 2008, 18, 1037–1041. [Google Scholar] [CrossRef] [PubMed]
- Bergman, J.; Brynolf, A. Synthesis of Chrysogine, a Metabolite of Penicillium chrysogenum and some related 2-substituted 4-(3H)-Quinazolinones. Tetrahedron 1990, 46, 1295–1310. [Google Scholar] [CrossRef]
- Matharu, D.S.; Flaherty, D.P.; Simpson, D.S.; Schroeder, C.E.; Chung, D.; Yan, D.; Noah, J.W.; Jonsson, C.B.; White, E.L.; Aubé, J.; et al. Optimization of Potent and Selective Quinazolinediones: Inhibitors of Respiratory Syncytial Virus That Block RNA-Dependent RNA-Polymerase Complex Activity. J. Med. Chem. 2014, 57, 10314–10328. [Google Scholar] [CrossRef] [Green Version]
- McAulay, K.; Hoyt, E.A.; Thomas, M.; Schimpl, M.; Bodnarchuk, M.S.; Lewis, H.J.; Barratt, D.; Bhavsar, D.; Robinson, D.M.; Deery, M.J.; et al. Alkynyl Benzoxazines and Dihydroquinazolines as Cysteine Targeting Covalent Warheads and their Application in Identification of Selective Irreversible Kinase Inhibitors. J. Am. Chem. Soc. 2020, 142, 10358–10372. [Google Scholar] [CrossRef]
- Van Veghel, D.; Cleynhens, J.; Pearce, L.V.; DeAndrea-Lazarus, I.; Blumberg, P.M.; Van Laere, K.; Verbruggen, A.; Bormans, G. New Transient Receptor Potential Vanilloid Subfamily Member 1 Positron Emission Tomography Radioligands: Synthesis, Radiolabeling, and Preclinical Evaluation. ACS Chem. Neurosci. 2013, 4, 624–634. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; An, S.; Zhu, Y.; Zhang, J.; Kang, Y.; Liu, P.; Wang, Y.; Li, J. Copper-catalyzed intermolecular cyclization of nitriles and 2-aminobenzylamine for 3,4-dihydroquinazolines and quinazolines synthesis via cascade coupling and aerobic oxidation. RSC Adv. 2014, 4, 49888–49891. [Google Scholar] [CrossRef]
- Amini-Rentsch, L.; Vanoli, E.; Richard-Bildstein, S.; Marti, R.; Vilé, G. A Novel and Efficient Continuous-Flow Route To Prepare Trifluoromethylated N-Fused Heterocycles for Drug Discovery and Pharmaceutical Manufacturing. Ind. Eng. Chem. Res. 2019, 58, 10164–10171. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Shi, J.; Luo, N.; Ding, M.; Bao, X. Synthesis, Crystal Structure, and Agricultural Antimicrobial Evaluation of Novel Quinazoline Thioether Derivatives Incorporating the 1,2,4-Triazolo[4,3-a]pyridine Moiety. J. Agric. Food Chem. 2019, 67, 11598–11606. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, G.-S.; Yao, Z.-J. Short and Efficient Total Synthesis of Luotonin A and 22-Hydroxyacuminatine Using a Common Cascade Strategy. J. Org. Chem. 2007, 72, 6270–6272. [Google Scholar] [CrossRef]
- Reddy, M.B.; Prasanth, K.; Anandhan, R. Visible-light induced copper(i)-catalyzed oxidative cyclization of o-aminobenzamides with methanol and ethanol via HAT. Org. Biomol. Chem. 2020, 18, 9601–9605. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Hati, S. Synthesis of Quinazolines and Dihydroquinazolines: O-Iodoxybenzoic Acid Mediated Tandem Reaction of o-Aminobenzylamine with Aldehydes. Synthesis 2016, 48, 1389–1398. [Google Scholar] [CrossRef]
- Wang, G.-W.; Miao, C.-B.; Kang, H. Benign and Efficient Synthesis of 2-Substituted 4(3H)-Quinazolinones Mediated by Iron(III) Chloride Hexahydrate in Refluxing Water. Bull. Chem. Soc. Jpn. 2006, 79, 1426–1430. [Google Scholar] [CrossRef]
- Aksenov, N.A.; Aksenov, A.V.; Ovcharov, S.N.; Aksenov, D.A.; Rubin, M. Electrophilically Activated Nitroalkanes in Reactions with Carbon Based Nucleophiles. Front. Chem. 2020, 8, 77. [Google Scholar] [CrossRef] [Green Version]
- Aksenov, A.V.; Aksenov, N.A.; Orazova, N.A.; Aksenov, D.A.; Dmitriev, M.V.; Rubin, M. Direct metal-free synthesis of diarylamines from 2-nitropropane via the twofold C–H functionalization of arenes. RSC Adv. 2015, 5, 84849–84855. [Google Scholar] [CrossRef]
- Aksenov, N.A.; Aksenov, A.V.; Nadein, O.N.; Aksenov, D.A.; Smirnov, A.N.; Rubin, M. One-pot synthesis of benzoxazoles via the metal-free ortho-C–H functionalization of phenols with nitroalkanes. RSC Adv. 2015, 5, 71620–71626. [Google Scholar] [CrossRef]
- Aksenov, A.; Aksenov, N.; Nadein, O.; Aksenova, I. Nitroethane in Polyphosphoric Acid: A New Reagent for Acetamidation and Amination of Aromatic Compounds. Synlett 2010, 2010, 2628–2630. [Google Scholar] [CrossRef]
- Aksenov, A.V.; Aksenov, N.A.; Arutiunov, N.A.; Malyuga, V.V.; Ovcharov, S.N.; Rubin, M. Electrophilically activated nitroalkanes in reaction with aliphatic diamines en route to imidazolines. RSC Adv. 2019, 9, 39458–39465. [Google Scholar] [CrossRef] [Green Version]
- Aksenov, D.A.; Arutiunov, N.A.; Maliuga, V.V.; Aksenov, A.V.; Rubin, M. Synthesis of imidazo[1,5-a]pyridines via cyclocondensation of 2-(aminomethyl)pyridines with electrophilically activated nitroalkanes. Beilstein J. Org. Chem. 2020, 16, 2903–2910. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, N.A.; Malyuga, V.V.; Abakarov, G.M.; Voskressensky, L.G.; Aksenov, A.; Aksenov, D.A. Synthesis of 3,4-dihydroisoquinolines using nitroalkanes in polyphosphoric acid. Russ. Chem. Bull. 2019, 68, 1047–1051. [Google Scholar] [CrossRef]
- Aksenov, A.V.; Smirnov, A.N.; Aksenov, N.; Bijieva, A.S.; Aksenova, I.; Rubin, M. Benzimidazoles and benzoxazoles via the nucleophilic addition of anilines to nitroalkanes. Org. Biomol. Chem. 2015, 13, 4289–4295. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, A.V.; Aksenov, N.A.; Ovcharov, D.S.; Aksenov, D.A.; Griaznov, G.; Voskressensky, L.; Rubin, M. Rational design of an efficient one-pot synthesis of 6H-pyrrolo[2,3,4-gh]perimidines in polyphosphoric acid. RSC Adv. 2016, 6, 82425–82431. [Google Scholar] [CrossRef] [Green Version]
- Aksenov, A.V.; Ovcharov, D.S.; Aksenov, N.A.; Aksenov, D.A.; Nadein, O.N.; Rubin, M. Dual role of polyphosphoric acid-activated nitroalkanes in oxidative peri-annulations: Efficient synthesis of 1,3,6,8-tetraazapyrenes. RSC Adv. 2017, 7, 29927–29932. [Google Scholar] [CrossRef] [Green Version]
- Aksenov, A.V.; Aksenov, N.A.; Ovcharov, D.S.; Shcherbakov, S.V.; Smirnova, A.N.; Aksenova, I.V.; Goncharov, V.I.; Rubin, M.A. Electrophilically activated nitroalkanes in the synthesis of 6,7-dihydro-1H-cyclopenta[g]perimidines. Russ. J. Org. Chem. 2017, 53, 1081–1084. [Google Scholar] [CrossRef]
- Aksenov, N.A.; Arutiunov, N.A.; Kirillov, N.K.; Aksenov, D.A.; Aksenov, A.V.; Rubin, M. Preparation of 1,3,4-oxadiazoles and 1,3,4-thiadiazoles via chemoselective cyclocondensation of electrophilically activated nitroalkanes to (thio)semicarbazides or thiohydrazides. Chem. Heterocycl. Compd. 2020, 56, 1–6. [Google Scholar] [CrossRef]
- Aksenov, A.V.; Khamraev, V.; Aksenov, N.A.; Kirilov, N.K.; Domenyuk, D.A.; Zelensky, V.A.; Rubin, M. Electrophilic activation of nitroalkanes in efficient synthesis of 1,3,4-oxadiazoles. RSC Adv. 2019, 9, 6636–6642. [Google Scholar] [CrossRef] [Green Version]
- Nef, J.U. Ueber die Constitution der Salze der Nitroparaffine. Justus Liebigs Ann. Chem. 1894, 280, 263–291. [Google Scholar] [CrossRef]
- Ballini, R.; Petrini, M. Recent synthetic developments in the nitro to carbonyl conversion (Nef reaction). Tetrahedron 2004, 60, 1017–1047. [Google Scholar] [CrossRef]
- Ballini, R.; Petrini, M. The Nitro to Carbonyl Conversion (Nef Reaction): New Perspectives for a Classical Transformation. Adv. Synth. Catal. 2015, 357, 2371–2402. [Google Scholar] [CrossRef]
- Aksenov, N.A.; Aksenov, A.V.; Kirilov, N.K.; Arutiunov, N.A.; Aksenov, D.A.; Maslivetc, V.; Zhao, Z.; Du, L.; Rubin, M.; Kornienko, A. Nitroalkanes as electrophiles: Synthesis of triazole-fused heterocycles with neuroblastoma differentiation activity. Org. Biomol. Chem. 2020, 18, 6651–6664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhao, Y.; Xuan, L.; Ding, C. SO2F2-Activated Efficient Beckmann Rearrangement of Ketoximes for Accessing Amides and Lactams. Eur. J. Org. Chem. 2019, 2019, 4911–4915. [Google Scholar] [CrossRef]
- Scheinbaum, M.L. Addition of dinitrogen trioxide to nonconjugated dienes. J. Org. Chem. 1970, 35, 2785–2790. [Google Scholar] [CrossRef]
- Pearson, D.E.; Cole, W.E. The Beckmann Rearrangement. V. The Rearrangement Rates of Someortho-Substituted Acetophenone Oximes (1). J. Org. Chem. 1955, 20, 488–493. [Google Scholar] [CrossRef]
- Kano, S.; Tanaka, Y.; Sugino, E.; Hibino, S. Reduction of Some Functional Groups with Titanium(IV) Chloride/Sodium Borohydride. Synthesis 1980, 1980, 695–697. [Google Scholar] [CrossRef]
- Bischler, A. Zur Kenntniss der Phenmiazinderivate. Ber. Dtsch. Chem. Ges. 1893, 26, 1891–1903. [Google Scholar] [CrossRef] [Green Version]
- Aneja, B.; Irfan, M.; Kapil, C.; Jairajpuri, M.A.; Maguire, R.; Kavanagh, K.; Rizvi, M.; Manzoor, N.; Azam, A.; Abid, M. Effect of novel triazole–amino acid hybrids on growth and virulence of Candida species: In vitro and in vivo studies. Org. Biomol. Chem. 2016, 14, 10599–10619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcaide, B.; Almendros, P.; Busto, E.; Herrera, F.; Lázaro-Milla, C.; Luna, A. Photopromoted Entry to Benzothiophenes, Benzoselenophenes, 3H-Indoles, Isocoumarins, Benzosultams, and (Thio)flavones by Gold-Catalyzed Arylative Heterocyclization of Alkynes. Adv. Synth. Catal. 2017, 359, 2640–2652. [Google Scholar] [CrossRef] [Green Version]
- Roman, D.S.; Takahashi, Y.; Charette, A.B. Potassiumtert-Butoxide Promoted Intramolecular Arylation via a Radical Pathway. Org. Lett. 2011, 13, 3242–3245. [Google Scholar] [CrossRef]
- Kurosu, M.; Lin, M.-H.; Kishi, Y. Fe/Cr- and Co/Cr-Mediated Catalytic Asymmetric 2-Haloallylations of Aldehydes. J. Am. Chem. Soc. 2004, 126, 12248–12249. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.-L.; Han, Y.; Yuan, C.-Z.; Wang, Y.; Xiahou, Z.-K.; Liao, J.; Gao, R.-T.; Mao, B.-B.; Zhao, B.-L.; Li, Z.-F.; et al. Synthesis and antiproliferative activity of 4-substituted-piperazine-1-carbodithioate derivatives of 2,4-diaminoquinazoline. Eur. J. Med. Chem. 2013, 64, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, T.; Kim, D.I.; Cho, E.J. Base-Promoted Synthesis of 2-Aryl Quinazolines from 2-Aminobenzylamines in Water. J. Org. Chem. 2018, 83, 7423–7430. [Google Scholar] [CrossRef] [PubMed]
- Wiley, R.H.; Wakefield, B.J. Infrared Spectra of the Nitrile N-Oxides: Some New Furoxans. J. Org. Chem. 1960, 25, 546–551. [Google Scholar] [CrossRef]
- Grosso, J.A.; Nichols, D.E.; Nichols, M.B.; Yim, G.K.W. Synthesis and adrenergic blocking effects of 2-(alkylamino)-3,4-dihydroquinazolines. J. Med. Chem. 1980, 23, 1261–1264. [Google Scholar] [CrossRef] [PubMed]
- Francesconi, L.C.; Yang, Y.Y.; Kung, M.-P.; Zhang, X.X.; Billings, J.J.; Guo, Y.-Z.; Kung, H.F. Technetium-99 m N,N′-Bis(2-mercapto-2-methylpropyl)-2-aminobenzylamine: Technetium-99 m Complexes of a Novel Bis(aminoethanethiol) Ligand. J. Med. Chem. 1994, 37, 3282–3288. [Google Scholar] [CrossRef] [PubMed]
- Kempter, G.; Ziegner, H.J. Heterocycles from aminoketones. XVII. Quinazolines from 1,4-diamines. Z. Chem. 1971, 11, 12–13. [Google Scholar] [CrossRef]
- Richards, J.E.; Hooper, A.J.J.; Bayfield, O.W.; Cockett, M.C.R.; Dear, G.J.; Holmes, A.J.; John, R.O.; Mewis, R.E.; Pridmore, N.; Roberts, A.D.; et al. Using hyperpolarised NMR and DFT to rationalise the unexpected hydrogenation of quinazoline to 3,4-dihydroquinazoline. Chem. Commun. 2018, 54, 10375–10378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ried, W.; Stahlhofen, P. Über heterocyclische Siebenringsysteme, IV. Mitteil.: Synthesen und Eigenschaften von 4.5-Benzo-[hept-1.2.6-oxdiazinen]. Chem. Ber. 1954, 87, 1814–1824. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Medium | t, °C | Time, h | Yield, % 1 | |
|---|---|---|---|---|
| 1 | H3PO4 86%, 2 g | 110 | 4 | 0 |
| 2 | H3PO4 86%, 2 g | 120 | 4 | 0 |
| 3 | H3PO4 86%, 1 g PPA 87%, 1 g | 120 | 4 | 0 |
| 4 | PPA 87%, 2 g | 120 | 4 | 0 |
| 5 | PPA 87%, 1 g; H3PO3, 1 g | 130 | 4 | 9 |
| 6 | PPA 87%, 1 g; H3PO3, 1 g | 150 | 2 | 40 |
| 7 | PPA 87%, 1 g; H3PO3, 1 g | 160 | 2 | 63 2 |
| 8 | H3PO3, 2 g | 160 | 2 | 51 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aksenov, A.V.; Grishin, I.Y.; Aksenov, N.A.; Malyuga, V.V.; Aksenov, D.A.; Nobi, M.A.; Rubin, M. Electrophilically Activated Nitroalkanes in Synthesis of 3,4-Dihydroquinozalines. Molecules 2021, 26, 4274. https://doi.org/10.3390/molecules26144274
Aksenov AV, Grishin IY, Aksenov NA, Malyuga VV, Aksenov DA, Nobi MA, Rubin M. Electrophilically Activated Nitroalkanes in Synthesis of 3,4-Dihydroquinozalines. Molecules. 2021; 26(14):4274. https://doi.org/10.3390/molecules26144274
Chicago/Turabian StyleAksenov, Alexander V., Igor Yu. Grishin, Nicolai A. Aksenov, Vladimir V. Malyuga, Dmitrii A. Aksenov, Mezvah A. Nobi, and Michael Rubin. 2021. "Electrophilically Activated Nitroalkanes in Synthesis of 3,4-Dihydroquinozalines" Molecules 26, no. 14: 4274. https://doi.org/10.3390/molecules26144274
APA StyleAksenov, A. V., Grishin, I. Y., Aksenov, N. A., Malyuga, V. V., Aksenov, D. A., Nobi, M. A., & Rubin, M. (2021). Electrophilically Activated Nitroalkanes in Synthesis of 3,4-Dihydroquinozalines. Molecules, 26(14), 4274. https://doi.org/10.3390/molecules26144274