
Concerning the Role of σ-Hole in Non-Covalent Interactions: Insights from the Study of the Complexes of ArBeO with Simple Ligands


Abstract
:1. Introduction
2. Methods of Bonding Analysis
3. Computational Details
4. Results and Discussion
4.1. The Investigated LAr and L-ArBeO: Predicted Data and Their Accuracy
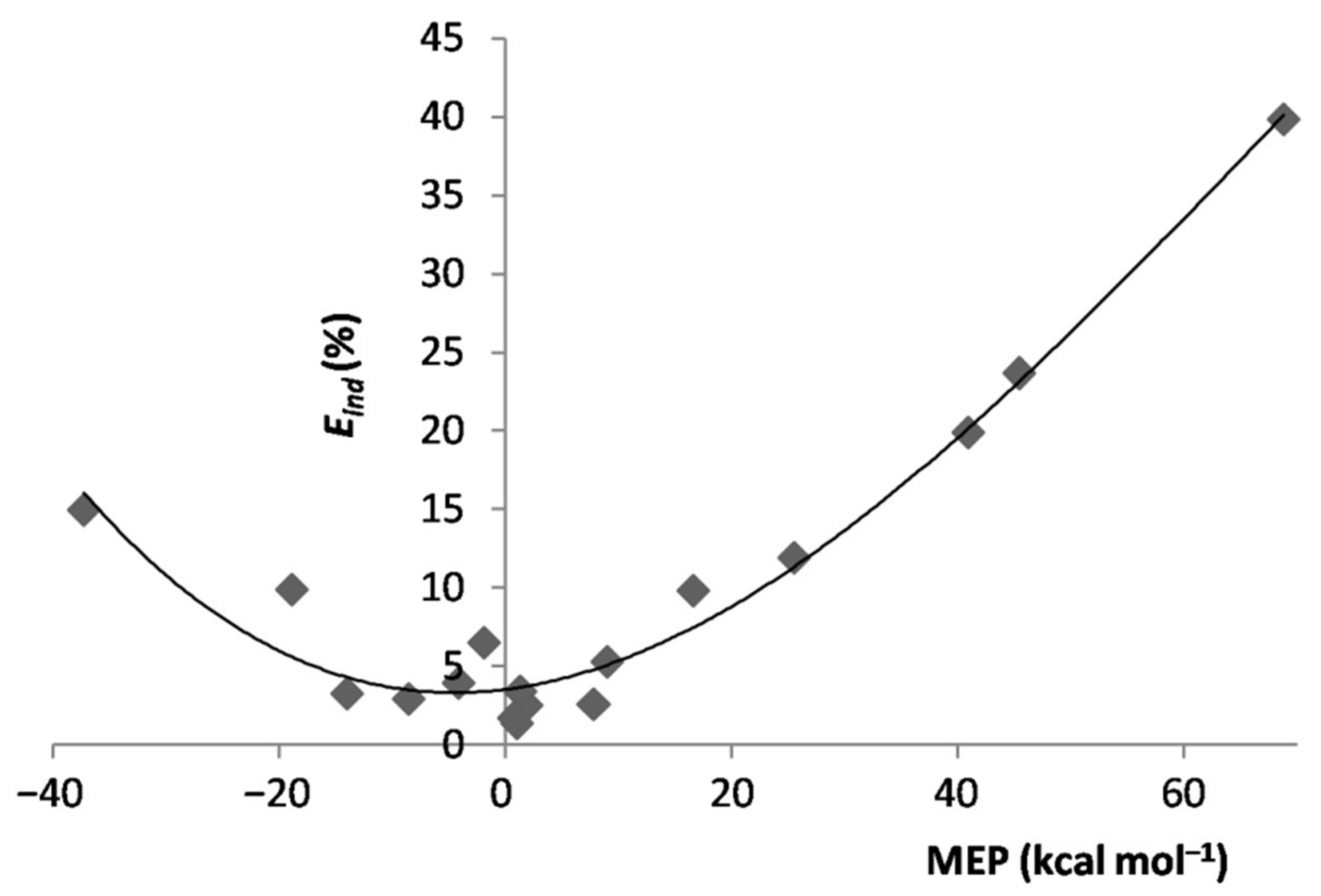
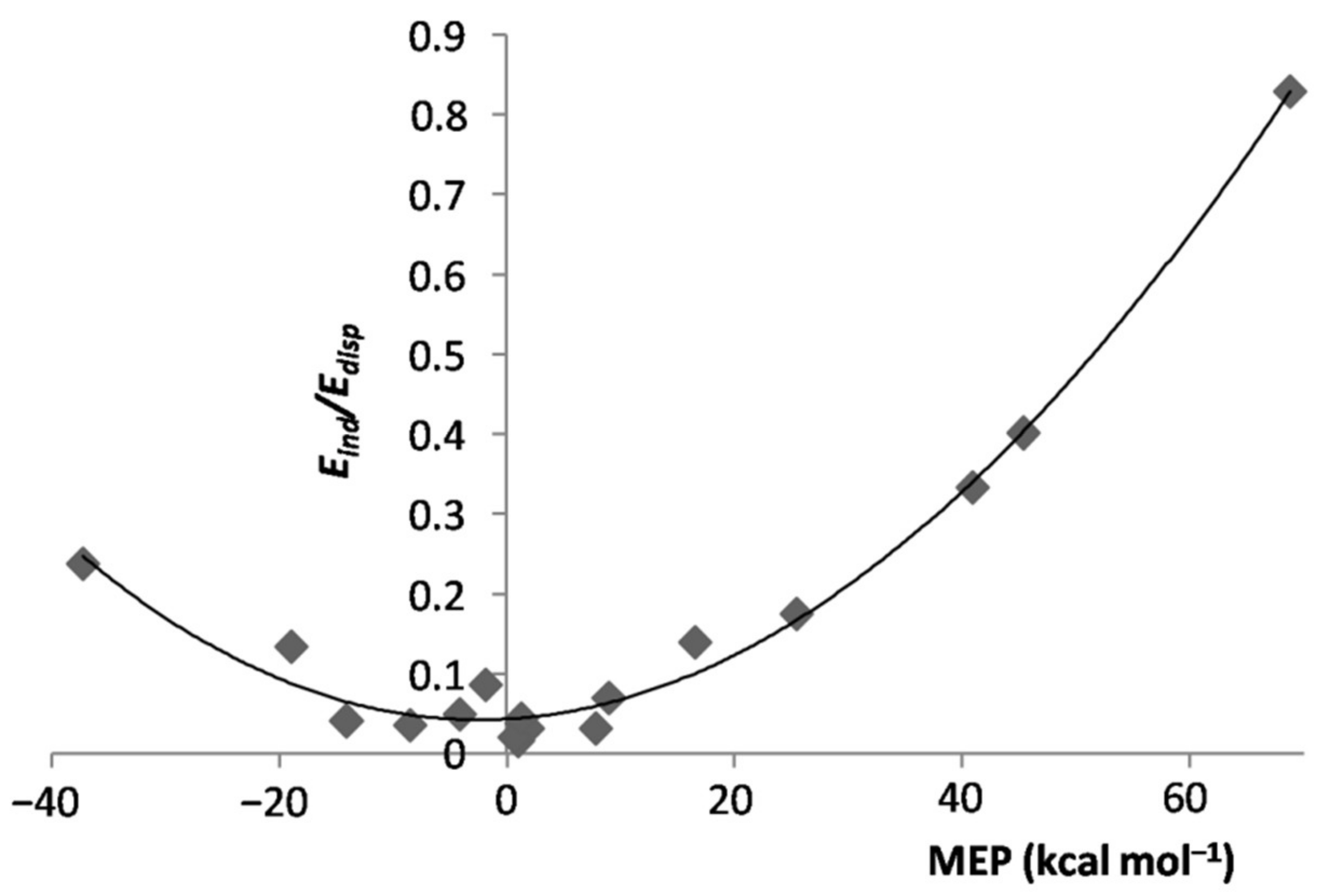
4.2. SAPT Analysis of the LAr: The Role of the MEP of L
4.3. From the LAr to the L-ArBeO: The Role of the σ-Hole of ArBeO
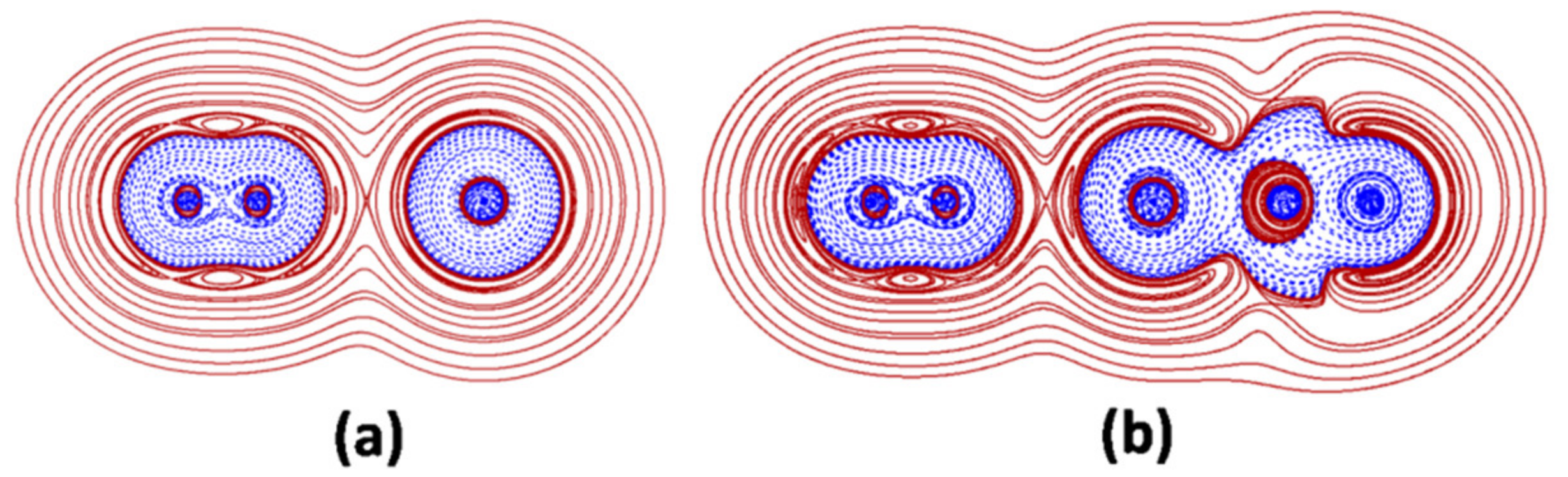
4.4. Bonding Analysi of the LAr and the L-ArBeO
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hobza, P.; Müller-Dethlefs, K. Noncovalent Interactions. Theory and Experiment; RSC Theoretical and Computational Chemistry Series; Royal Society of Chemistry: Cambridge, UK, 2010. [Google Scholar]
- Noncovalent Interactions in the Synthesis and Design of New Compounds; Maharramov, A.M.; Mahmudov, K.T.; Kopylovich, M.N.; Pombeiro, A.J.L. (Eds.) John Wiley and Sons: Hoboken, NJ, USA, 2016. [Google Scholar]
- He, H.; Tan, W.; Guo, J.; Yi, M.; Shy, A.N.; Xu, B. Enzymatic Noncovalent Synthesis. Chem. Rev. 2020, 120, 9994–10078. [Google Scholar] [CrossRef]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the Hydrogen Bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Scheiner, S. Forty Years of Progress in the Study of the Hydrogen Bond. Struct. Chem. 2019, 30, 1119–1128. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Clark, T. σ-Holes. WIREs Comput. Mol. Sci. 2013, 3, 13–20. [Google Scholar]
- Politzer, P.; Murray, J.S.; Clark, T. HalogenBonding and other σ-HoleInteractions: A Perspective. Phys. Chem. ChemPhys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Kolar, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [Green Version]
- Brammer, L. Halogen Bonding, Chalcogen Bonding, Pnictogen Bonding, Tetrel Bonding: Origins, Current Status and Discussion. Faraday Discuss. 2017, 203, 485–507. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Li, Q.; Scheiner, S. Comparative Strengths of Tetrel, Pnicogen, Chalcogen, and Halogen Bonds and ContributingFactors. Molecules 2018, 23, 1681. [Google Scholar] [CrossRef] [Green Version]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Parajuli, R. Does the Recent IUPAC Definition of Hydrogen Bonding Lead to New Intermolecular Interactions? Curr. Sci. 2016, 110, 495–498. [Google Scholar]
- Alkorta, I.; Elguero, J.; Frontera, A. Not only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef] [Green Version]
- Murray, J.S.; Politzer, P. Molecular Electrostatic Potentials and Noncovalent Interactions. WIREs Comput. Mol. Sci. 2017, 7, e1326. [Google Scholar] [CrossRef]
- Bader, R.F.; Carroll, M.T.; Cheeseman, J.R.; Chang, C. Properties of Atoms in Molecules: Atomic Volumes. J. Am. Chem. Soc. 1987, 109, 7968–7979. [Google Scholar] [CrossRef]
- Borocci, S.; Grandinetti, F.; Sanna, N. From LAr to L-ArBeO (L = He, Ne, Ar, HF): Switching on σ-Hole Effects in Non-Covalent Interactions. Chem. Phys. Lett. 2021, 768, 138402. [Google Scholar] [CrossRef]
- Grandinetti, F. Noble Gas. Chemistry: Structure, Bonding, and Gas.-Phase Chemistry; Wiley-VCH: Weinheim, Germany, 2018; Chapter 3; pp. 100–107. [Google Scholar]
- Jeziorski, B.; Moszyński, R.; Ratkiewicz, A.; Rybak, V.; Szalewicz, K.; Williams, H.L. Methods and Techniques in Computational Chemistry: METECC-94; Clementi, E., Ed.; STEF: Cagliari, Italy, 1993; Volume B. [Google Scholar]
- Jeziorski, B.; Moszyński, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Borocci, S.; Giordani, M.; Grandinetti, F. Bonding Motifs of Noble-Gas Compounds as Described by the Local Electron Energy Density. J. Phys. Chem. A 2015, 119, 6528–6541. [Google Scholar] [CrossRef] [PubMed]
- Borocci, S.; Grandinetti, F.; Sanna, N.; Antoniotti, P.; Nunzi, F. Non-Covalent Complexes of the Noble-Gas Atoms: Analyzing the Transition from Physical to Chemical Interactions. J. Comput. Chem. 2019, 40, 2318–2328. [Google Scholar] [CrossRef]
- Borocci, S.; Grandinetti, F.; Sanna, N.; Nunzi, F. Classifying the Chemical Bonds Involving the Noble-Gas Atoms. New J. Chem. 2020, 44, 14536–14550. [Google Scholar] [CrossRef]
- Murray, J.S.; Shields, Z.P.I.; Seybold, P.G.; Politzer, P. Intuitive and Counterintuitive Noncovalent Interactions of Aromatic πRegions with the Hydrogen and the Nitrogen of HCN. J. Comput. Sci. 2015, 10, 209–216. [Google Scholar] [CrossRef]
- Wang, C.; Danovich, D.; Shaik, S.; Wu, W.; Mo, Y. Attraction between Electrophilic Caps: A Counterintuitive Case of Noncovalent Interactions. J. Comput. Chem. 2019, 40, 1015–1022. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. Chemical Bonds without Bonding Electron Density-Does the Difference Electron-Density Analysis Suffice for a Description of the Chemical Bond? Angew. Chem. Int. Ed. Engl. 1984, 23, 627–628. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. A Description of the Chemical Bond in Terms of Local Properties of Electron Density and Energy. Croat. Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Johnson, E.R.; Keinan, S.; Mori-Sanchez, P.; Contreras-Garcia, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narth, C.; Maroun, Z.; Boto, R.A.; Chaudret, R.; Bonnet, M.L.; Piquemal, J.-P.; Contreras-García, J. A Complete NCI Perspective: From New Bonds to Reactivity. In Applications of Topological Methods in Molecular Chemistry; Springer: Cham, Switzerland, 2016; pp. 491–527. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Borocci, S.; Grandinetti, F.; Sanna, N.; Antoniotti, P.; Nunzi, F. Complexes of Helium with Neutral Molecules: Progress toward a Quantitative Scale of Bonding Character. J. Comput. Chem. 2020, 41, 1000–1011. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618. [Google Scholar] [CrossRef] [Green Version]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Head-Gordon, M. A Fifth-Order Perturbation Comparison of Electron Correlation Theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-To-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Welcome to the Website of CFOUR. Available online: http://www.cfour.de (accessed on 20 July 2021).
- Bukowski, R.; Cencek, W.; Jankowski, P.; Jeziorska, M.; Jeziorski, B.; Kucharski, S.A.; Lotrich, V.F.; Metz, M.P.; Misquitta, A.J.; Moszyński, R.; et al. SAPT2016: An Ab Initio Program for Symmetry-Adapted Perturbation Theory Calculations of Intermolecular Interactions Energies. Sequential and Parallel Versions; University of Delaware: Newark, DE, USA; University of Warsaw: Warsaw, Poland, 2016. [Google Scholar]
- Tao, F.-M.; Pan, Y.-K. Ab Initio Potential Energy Curves and Binding Energies of Ar2 and Mg2. Mol. Phys. 1994, 81, 507–518. [Google Scholar] [CrossRef]
- Tao, F.-M.; Klemperer, W. Accurate ab Initio Potential Energy Surfaces of Ar–HF, Ar–H2O, and Ar–NH3. J. Chem. Phys. 1994, 101, 1129–1145. [Google Scholar] [CrossRef]
- Saleh, G.; Gatti, C.; Lo Presti, L. Energetics of Non-Covalent Interactions from Electron and Energy Density Distributions. Comput. Theor. Chem. 2015, 1053, 53–59. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Zou, W. Molden2AIM. Available online: https://github.com/zorkzou/Molden2AIM (accessed on 01 February 2021).
- Lee, T.J.; Taylor, P.R. A Diagnostic for Determining the Quality of Single-Reference Electron Correlation Methods. Int. J. Quantum Chem. 1989, 36, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Keil, M.; Danielson, L.J.; Dunlop, P.J. On Obtaining Interatomic Potentials from Multiproperty Fits to Experimental Data. J. Chem. Phys. 1991, 94, 296–309. [Google Scholar] [CrossRef]
- Barrow, D.A.; Aziz, R.A. The Neon-Argon Potential Revisited. J. Chem. Phys. 1988, 89, 6189–6194. [Google Scholar] [CrossRef]
- Aziz, R.A. A Highly Accurate Interatomic Potential for Argon. J. Chem. Phys. 1993, 99, 4518–4525. [Google Scholar] [CrossRef]
- Dham, A.K.; Meath, W.J.; Jechow, J.W.; McCourt, F.R.W. New Exchange-Coulomb N2-Ar Potential-Energy Surface and its Comparison with other Recent N2-Ar Potential-Energy Surfaces. J. Chem. Phys. 2006, 124, 034308. [Google Scholar] [CrossRef]
- Sumiyoshi, Y.; Endo, Y. Three-Dimensional Potential Energy Surface of Ar-CO. J. Chem. Phys. 2015, 142, 024314. [Google Scholar] [CrossRef]
- Chan, K.W.; Power, T.D.; Jai-nhuknan, J.; Cybulski, S.M. An ab Initio Study of He-F2, Ne-F2, and Ar-F2 van der Waals Complexes. J. Chem. Phys. 1999, 110, 860–869. [Google Scholar] [CrossRef]
- Rohrbacher, A.; Janda, K.C.; Beneventi, L.; Casavecchia, P.; Volpi, G.G. Differential Scattering Cross Sections for HeCl2, NeCl2, and ArCl2: Multiproperty Fits of Potential Energy Surfaces. J. Phys. Chem. A 1997, 101, 6528–6537. [Google Scholar] [CrossRef]
- Prosmiti, R.; Villareal, P.; Delgado-Barrio, G. Structure and Bonding of ArClF: Intermolecular Potential Surface. Isr. J. Chem. 2003, 43, 279–286. [Google Scholar] [CrossRef]
- Hutson, J.M. Vibrational Dependence of the Anisotropic Intermolecular Potential of Ar-HF. J. Chem. Phys. 1992, 96, 6752–6767. [Google Scholar] [CrossRef]
- Jouypazadeh, H.; Solimannejad, M.; Farrokhpour, H. New Potential Energy Surface and Rovibrational Spectra of Ar···HCl. Comput. Theor. Chem. 2016, 1083, 64–71. [Google Scholar] [CrossRef]
- Loreau, J.; Liévin, J.; Scribano, Y.; van der Avoird, A. Potential Energy Surface and Bound States of the NH3-Ar and ND3-Ar Complexes. J. Chem. Phys. 2014, 141, 224303. [Google Scholar] [CrossRef] [Green Version]
- Chałasiński, G.; Szczęśniak, M.M. Origins of Structure and Energetics of van der Waals Clusters from ab Initio Calculations. Chem. Rev. 1994, 94, 1723–1765. [Google Scholar] [CrossRef]
- Nunzi, F.; Pannacci, G.; Tarantelli, F.; Belpassi, L.; Cappelletti, D.; Falcinelli, S.; Pirani, F. Leading Interaction Components in the Structure and Reactivity of Noble Gases Compounds. Molecules 2020, 25, 2367. [Google Scholar] [CrossRef] [PubMed]
- Maroulis, G. Accurate Electric Multipole Moment, Static Polarizability and Hyperpolarizability Derivatives for N2. J. Chem. Phys. 2003, 118, 2673–2687. [Google Scholar] [CrossRef]
- Maroulis, G. Electric Polarizability and Hyperpolarizability of Carbon Monoxide. J. Phys. Chem. 1996, 100, 13466–13473. [Google Scholar] [CrossRef]
- Maroulis, G. On the Bond-Length Dependence of the Static Electric Polarizability and Hyperpolarizability of F2. Chem. Phys. Lett. 2007, 442, 265–269. [Google Scholar] [CrossRef]
- Maroulis, G. Accurate Dipole Polarizability for Cl2 (X1Σg+). Mol. Phys. 1992, 77, 1085–1094. [Google Scholar] [CrossRef]
- Miller, K.J.; Savchik, J.A. A New Empirical Method to Calculate Average Molecular Polarizabilities. J. Am. Chem. Soc. 1979, 101, 7206–7213. [Google Scholar] [CrossRef]
- Sadlej, A.J. Electric Properties of Diatomic Interhalogens. A Study of the Electron Correlation and Relativistic Contributions. J. Chem. Phys. 1992, 96, 2048–2053. [Google Scholar] [CrossRef]
- Maroulis, G. Electric Multipole Moment, Dipole and Quadrupole (Hyper)Polarizability Derivatives for HF (X1Σ+). J. Mol. Struct. (THEOCHEM) 2003, 633, 177–197. [Google Scholar] [CrossRef]
- Maroulis, G. A Systematic Study of Basis Set, Electron Correlation, and Geometry Effects on the Electric Multipole Moments, Polarizability, and Hyperpolarizability of HCl. J. Chem. Phys. 1998, 108, 5432–5448. [Google Scholar] [CrossRef]
- Zeiss, G.D.; Meath, W.J. Dispersion Energy Constants C6 (A,B), Dipole Oscillator Strength Sums and Refractivities for Li, N, O, H2, N2, O2, NH3, H2O, NO and N2O. Mol. Phys. 1977, 33, 1155–1176. [Google Scholar] [CrossRef]
- Clark, T. Polarization, Donor-Acceptor Interactions, and Covalent Contributions in Weak Interactions: A Clarification. J. Mol. Model. 2017, 23, 297. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Politzer, P. Interaction and Polarization Energy Relationships in σ-Hole and π-Hole Bonding. Crystals 2020, 10, 76. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lit. | DoP(Ar)b | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HeAr | −0.0119 | −0.0040 | −0.1049 | 0.0638 | −0.0570 | −0.0588 c | 9.85 | 3.35 | 86.80 | 0.026 |
| He-ArBeO | −0.0204 | −0.0432 | −0.1686 | 0.1218 | −0.1104 | 8.80 | 18.61 | 72.59 | ||
| NeAr | −0.0433 | −0.0037 | −0.2306 | 0.1534 | −0.1242 | −0.1342 d | 15.61 | 1.34 | 83.05 | 0.039 |
| Ne-ArBeO | −0.0507 | −0.0797 | −0.3371 | 0.2561 | −0.2114 | 10.85 | 17.04 | 72.11 | ||
| ArAr | −0.1332 | −0.0179 | −0.5641 | 0.4311 | −0.2841 | −0.2846 e | 18.62 | 2.50 | 78.88 | 0.099 |
| Ar-ArBeO | −0.1745 | −0.2428 | −0.7345 | 0.6422 | −0.5096 | 15.15 | 21.08 | 63.77 | ||
| N2−Ar | −0.1074 | −0.0189 | −0.5284 | 0.4039 | −0.2508 | −0.2221 f | 16.41 | 2.88 | 80.71 | −0.14 |
| N2-ArBeO | −0.9782 | −0.3132 | −0.9032 | 1.0386 | −1.1560 | 44.57 | 14.27 | 41.16 | ||
| N2-Ar (Tg) | −0.1635 | −0.0223 | −0.6912 | 0.5682 | −0.3088 | −0.2913 f | 18.64 | 2.54 | 78.82 | 0.36 |
| N2-ArBeO (T) | 0.2296 | −0.1944 | −0.7061 | 0.5104 | −0.1605 | 20.32 | 17.20 | 62.48 | ||
| OC-Ar | −0.1063 | −0.0191 | −0.4698 | 0.3506 | −0.2446 | −0.2078 h | 17.86 | 3.21 | 78.93 | −0.29 |
| OC-ArBeO | −1.5489 | −0.3531 | −0.9869 | 1.3982 | −1.4907 | 53.61 | 12.22 | 34.16 | ||
| CO-Ar | −0.1224 | −0.0290 | −0.5916 | 0.4618 | −0.2812 | −0.2391 h | 16.47 | 3.91 | 79.62 | −0.034 |
| CO-ArBeO | −0.9018 | −0.3204 | −0.8840 | 0.8791 | −1.2271 | 42.81 | 15.21 | 41.97 | ||
| F2-Ar | −0.2343 | −0.1155 | −0.8264 | 0.8015 | −0.3747 | −0.3510 i | 19.92 | 9.82 | 70.26 | 0.69 |
| F2-ArBeO | 0.4667 | −0.2444 | −0.6561 | 0.3806 | −0.0532 | 34.14 | 17.86 | 48.00 | ||
| F2-Ar (T) | −0.1406 | −0.0133 | −0.6521 | 0.4774 | −0.3286 | −0.3146 i | 17.44 | 1.65 | 80.91 | 0.013 |
| F2-ArBeO (T) | −0.3956 | −0.2249 | −0.9411 | 0.8385 | −0.7231 | 25.33 | 14.40 | 60.27 | ||
| Cl2-Ar | −0.4113 | −0.2373 | −1.3498 | 1.3329 | −0.6655 | −0.6487 j | 20.58 | 11.87 | 67.54 | 1.28 |
| Cl2-ArBeO | 1.0857 | −0.3846 | −0.8325 | 0.3932 | 0.2618 | 47.15 | 16.70 | 36.15 | ||
| Cl2-Ar (T) | −0.3778 | −0.0603 | −1.3592 | 1.1461 | −0.6512 | −0.6314 j | 21.02 | 3.35 | 75.63 | 0.20 |
| Cl2-ArBeO (T) | −0.9942 | −0.6121 | −1.7521 | 1.7508 | −1.6076 | 29.60 | 18.22 | 52.17 | ||
| FCl-Ar | −0.5476 | −0.5337 | −1.6006 | 1.8462 | −0.8357 | −0.8101 k | 20.42 | 19.90 | 59.68 | 1.89 |
| ClF-Ar | −0.2064 | −0.0716 | −0.8296 | 0.6875 | −0.4201 | −0.3687 k | 18.63 | 6.47 | 74.90 | 0.021 |
| ClF-ArBeO | −1.2759 | −0.4951 | −1.1938 | 1.2087 | −1.7561 | 43.04 | 16.70 | 40.27 | ||
| FH-Ar | −0.2437 | −0.8019 | −0.9663 | 1.4098 | −0.6020 | −0.6037 l | 12.11 | 39.86 | 48.03 | 3.80 |
| HF-Ar | −0.1203 | −0.0705 | −0.5234 | 0.4182 | −0.2960 | −0.3067 l | 16.84 | 9.87 | 73.29 | −0.54 |
| HF-ArBeO | −3.5413 | −0.4587 | −1.1374 | 1.6639 | −3.4735 | 68.93 | 8.92 | 22.14 | ||
| ClH-Ar | −0.3268 | −0.4413 | −1.0998 | 1.3561 | −0.5118 | −0.5050 m | 17.50 | 23.63 | 58.87 | 2.45 |
| HCl-Ar | −0.2153 | −0.0611 | −0.8788 | 0.7188 | −0.4364 | −0.4288 m | 18.64 | 5.29 | 76.07 | 0.52 |
| HCl-ArBeO | −0.3947 | −0.3613 | −0.9692 | 0.7563 | −0.9689 | 22.88 | 20.94 | 56.18 | ||
| H3N-Ar | −0.2052 | −0.1364 | −0.5726 | 0.5809 | −0.3333 | −0.2966 n | 22.45 | 14.92 | 62.63 | −0.98 |
| H3N-ArBeO | −6.8566 | −1.0230 | −1.6735 | 4.1151 | −5.4380 | 71.77 | 10.71 | 17.52 |
| Bond | Ωs | N(Ωs) | ρs(ave) | Hs(ave/max/min) | |
|---|---|---|---|---|---|
| He-Ar | HeAr | 0.0212 | 0.024 | 0.0011 | 0.00040/0.00042/0.00038 |
| He-ArBeO | 0.0234 | 0.043 | 0.0018 | 0.00079/0.00082/0.00076 | |
| Ne-Ar | NeAr | 0.0316 | 0.063 | 0.0020 | 0.00059/0.00061/0.00057 |
| Ne-ArBeO | 0.0372 | 0.11 | 0.0030 | 0.00084/0.00103/0.00090 | |
| Ar-Ar | ArAr | 0.0948 | 0.27 | 0.0029 | 0.00074/0.00078/0.00069 |
| Ar-ArBeO | 0.0942 | 0.35 | 0.0038 | 0.00113/0.00122/0.00105 | |
| N-Ar | N2-Ar | 0.0788 | 0.22 | 0.0028 | 0.00076/0.00080/0.00071 |
| N2-ArBeO | 0.0904 | 0.46 | 0.0051 | 0.00162/0.00172/0.00150 | |
| N2-Ar (T a) | 0.1570 | 0.46 | 0.0029 | 0.00075/0.00082/0.00067 | |
| N2-ArBeO (T) | 0.1344 | 0.40 | 0.0030 | 0.00086/0.00093/0.00078 | |
| C-Ar | OC-Ar | 0.0914 | 0.21 | 0.0023 | 0.00060/0.00063/0.00056 |
| OC-ArBeO | 0.1203 | 0.64 | 0.0053 | 0.00150/0.00167/0.00139 | |
| O-Ar | CO-Ar | 0.0736 | 0.24 | 0.0032 | 0.00086/0.00091/0.00080 |
| CO-ArBeO | 0.0785 | 0.40 | 0.0051 | 0.00159/0.00168/0.00148 | |
| F-Ar | F2-Ar | 0.0966 | 0.46 | 0.0047 | 0.00154/0.00176/0.00137 |
| F2-ArBeO | 0.0686 | 0.24 | 0.0035 | 0.00124/0.00134/0.00113 | |
| F2-Ar (T) | 0.2052 | 0.59 | 0.0029 | 0.00068/0.00076/0.00058 | |
| F2-ArBeO (T) | 0.2254 | 0.95 | 0.0042 | 0.00111/0.00126/0.00096 | |
| Cl-Ar | Cl2-Ar | 0.1687 | 0.90 | 0.0053 | 0.00153/0.00165/0.00139 |
| Cl2-ArBeO | 0.1139 | 0.32 | 0.0028 | 0.00095/0.00101/0.00087 | |
| Cl2-Ar (T) | 0.4444 | 1.56 | 0.0035 | 0.00088/0.00095/0.00076 | |
| Cl2-ArBeO (T) | 0.4482 | 2.08 | 0.0046 | 0.00127/0.00142/0.00111 | |
| Cl-Ar | FCl-Ar | 0.2167 | 1.44 | 0.0067 | 0.00184/0.00199/0.00165 |
| F-Ar | ClF-Ar | 0.0866 | 0.38 | 0.0043 | 0.00121/0.00132/0.00111 |
| ClF-ArBeO | 0.0920 | 0.61 | 0.0066 | 0.00212/0.00233/0.00193 | |
| H-Ar | FH-Ar | 0.0777 | 0.65 | 0.0084 | 0.00148/0.00158/0.00134 |
| F-Ar | HF-Ar | 0.0700 | 0.23 | 0.0032 | 0.00092/0.00097/0.00086 |
| HF-ArBeO | 0.0936 | 0.73 | 0.0078 | 0.00259/0.00286/0.00234 | |
| H-Ar | ClH-Ar | 0.0922 | 0.64 | 0.0069 | 0.00129/0.00140/0.00123 |
| Cl-Ar | HCl-Ar | 0.1268 | 0.46 | 0.0037 | 0.00112/0.00120/0.00102 |
| HCl-ArBeO | 0.1170 | 0.47 | 0.0041 | 0.00135/0.00145/0.00124 | |
| N-Ar | H3N-Ar | 0.1113 | 0.31 | 0.0028 | 0.00055/0.00063/0.00051 |
| H3N-ArBeO | 0.1815 | 1.65 | 0.0091 | 0.00185/0.00209/0.00168 |
| L | MEP point | R/θ | ||||
|---|---|---|---|---|---|---|
| N2 | VS,Min(N): −8.53 | VS,Max(perp): 7.82 | 1.114 | 1.75 c | 2.20 d | 1.52 d |
| CO | VS,Min(C): −14.04 | VS,Min(O): −4.12 | 1.139 | 1.95 e | 2.31 e | 1.77 e |
| F2 | VS,Max(F): 16.57 | VS,Max(perp): 0.76 | 1.401 | 1.25 f | 1.84 f | 0.96 f |
| Cl2 | VS,Max(Cl): 25.50 | VS,Max(perp): 1.26 | 1.999 | 4.59 g | 6.27 g | 3.75 g |
| ClF | VS,Max(Cl): 40.94 | VS,Max(F): −1.86 | 1.639 | 2.68 h | 3.37 i | 2.34 j |
| HF | VS,Max(H): 68.78 | VS,Max(F): −18.91 | 0.922 | 0.83 k | 0.94 k | 0.77 k |
| HCl | VS,Max(H): 45.38 | VS,Max(Cl): 9.00 | 1.275 | 2.58 l | 2.74 l | 2.50 l |
| NH3 | VS,Min(N): −37.25 | 1.012/106.8 | 2.15 m | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borocci, S.; Grandinetti, F.; Sanna, N. Concerning the Role of σ-Hole in Non-Covalent Interactions: Insights from the Study of the Complexes of ArBeO with Simple Ligands. Molecules 2021, 26, 4477. https://doi.org/10.3390/molecules26154477
Borocci S, Grandinetti F, Sanna N. Concerning the Role of σ-Hole in Non-Covalent Interactions: Insights from the Study of the Complexes of ArBeO with Simple Ligands. Molecules. 2021; 26(15):4477. https://doi.org/10.3390/molecules26154477
Chicago/Turabian StyleBorocci, Stefano, Felice Grandinetti, and Nico Sanna. 2021. "Concerning the Role of σ-Hole in Non-Covalent Interactions: Insights from the Study of the Complexes of ArBeO with Simple Ligands" Molecules 26, no. 15: 4477. https://doi.org/10.3390/molecules26154477
APA StyleBorocci, S., Grandinetti, F., & Sanna, N. (2021). Concerning the Role of σ-Hole in Non-Covalent Interactions: Insights from the Study of the Complexes of ArBeO with Simple Ligands. Molecules, 26(15), 4477. https://doi.org/10.3390/molecules26154477