
A Stability-Indicating Ultra Performance Liquid Chromato-Graphic (UPLC) Method for the Determination of a Mycophenolic Acid-Curcumin Conjugate and Its Applications to Chemical Kinetic Studies

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Method Development
2.2. Method Validation
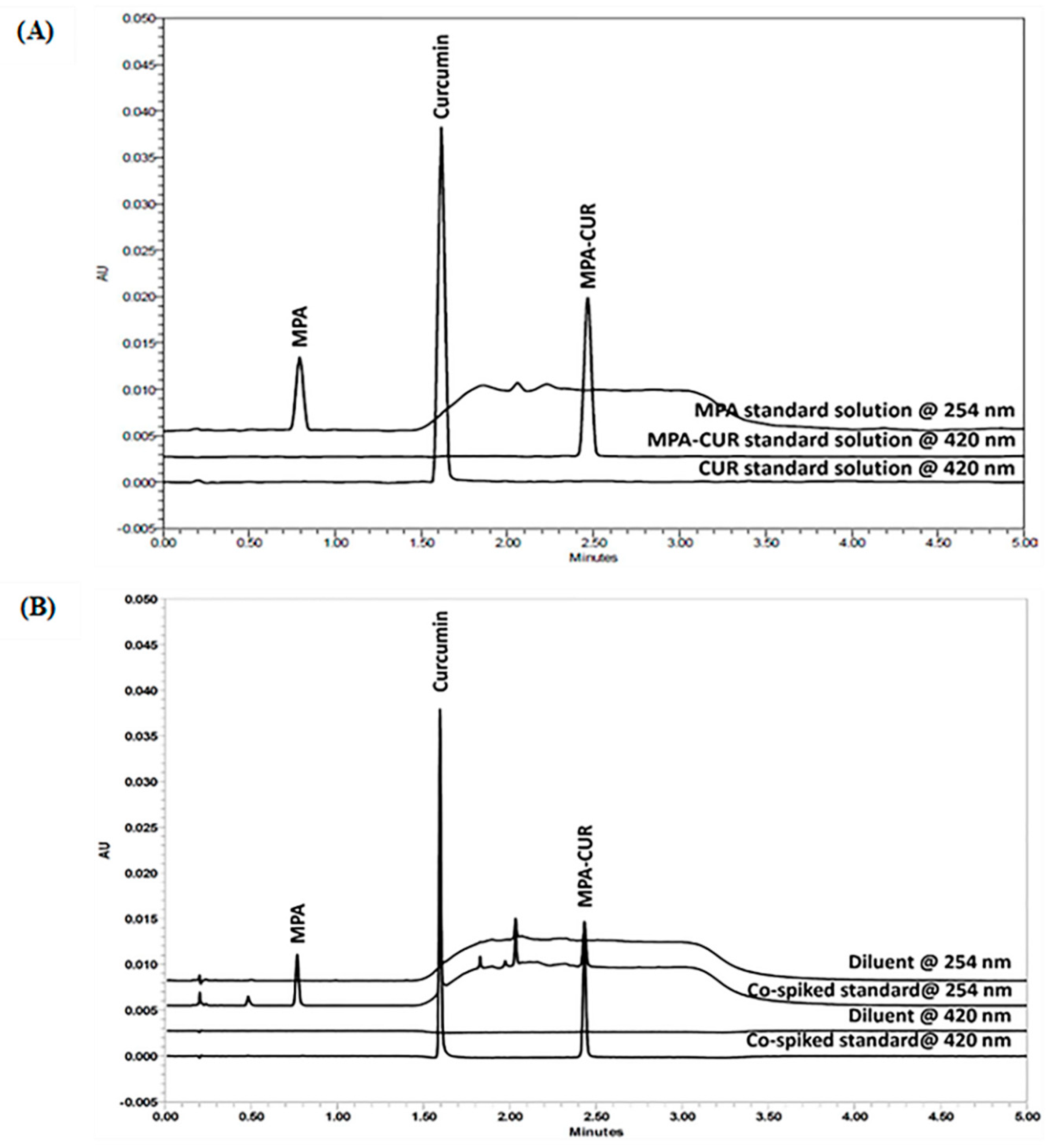
2.2.1. System Suitability
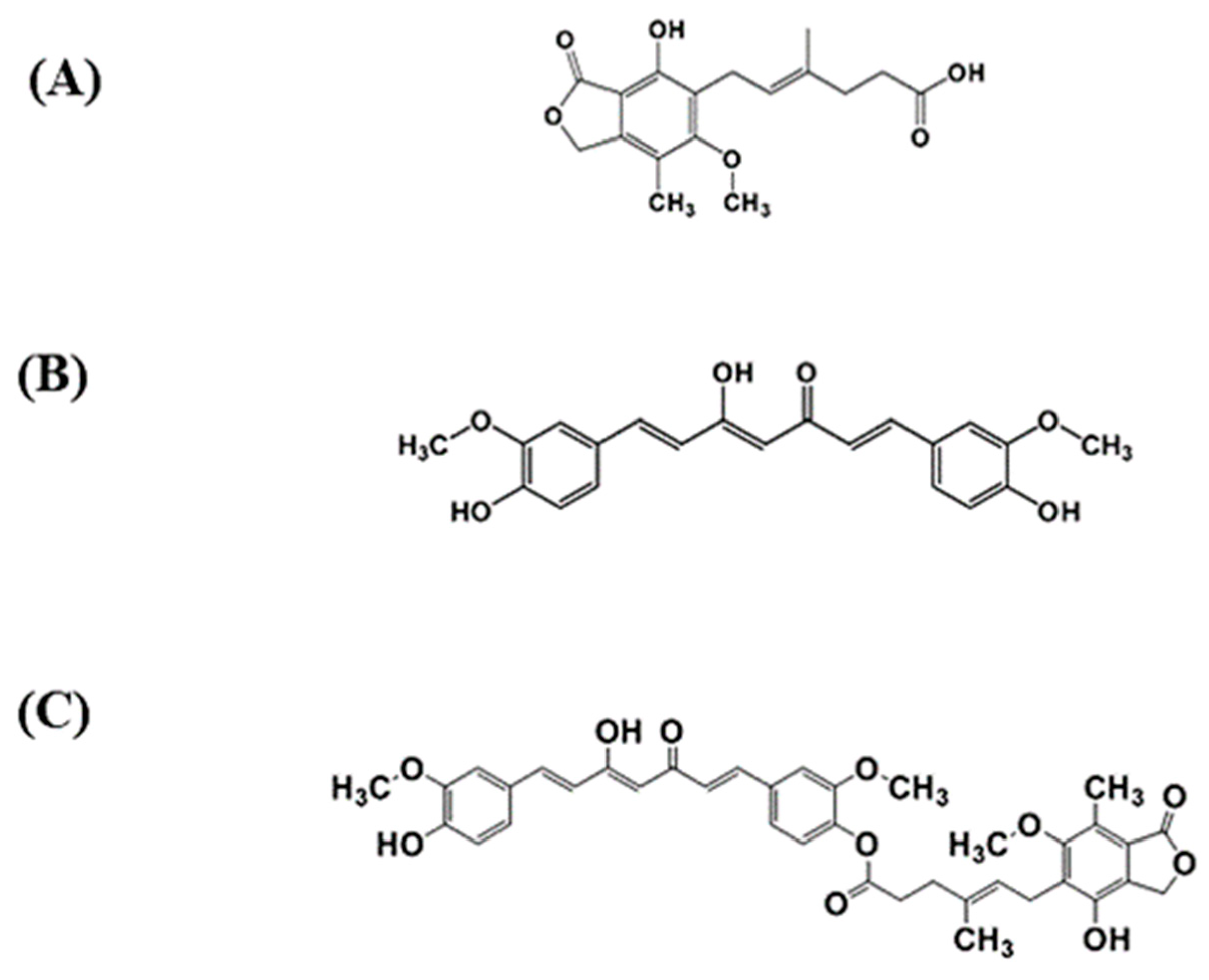
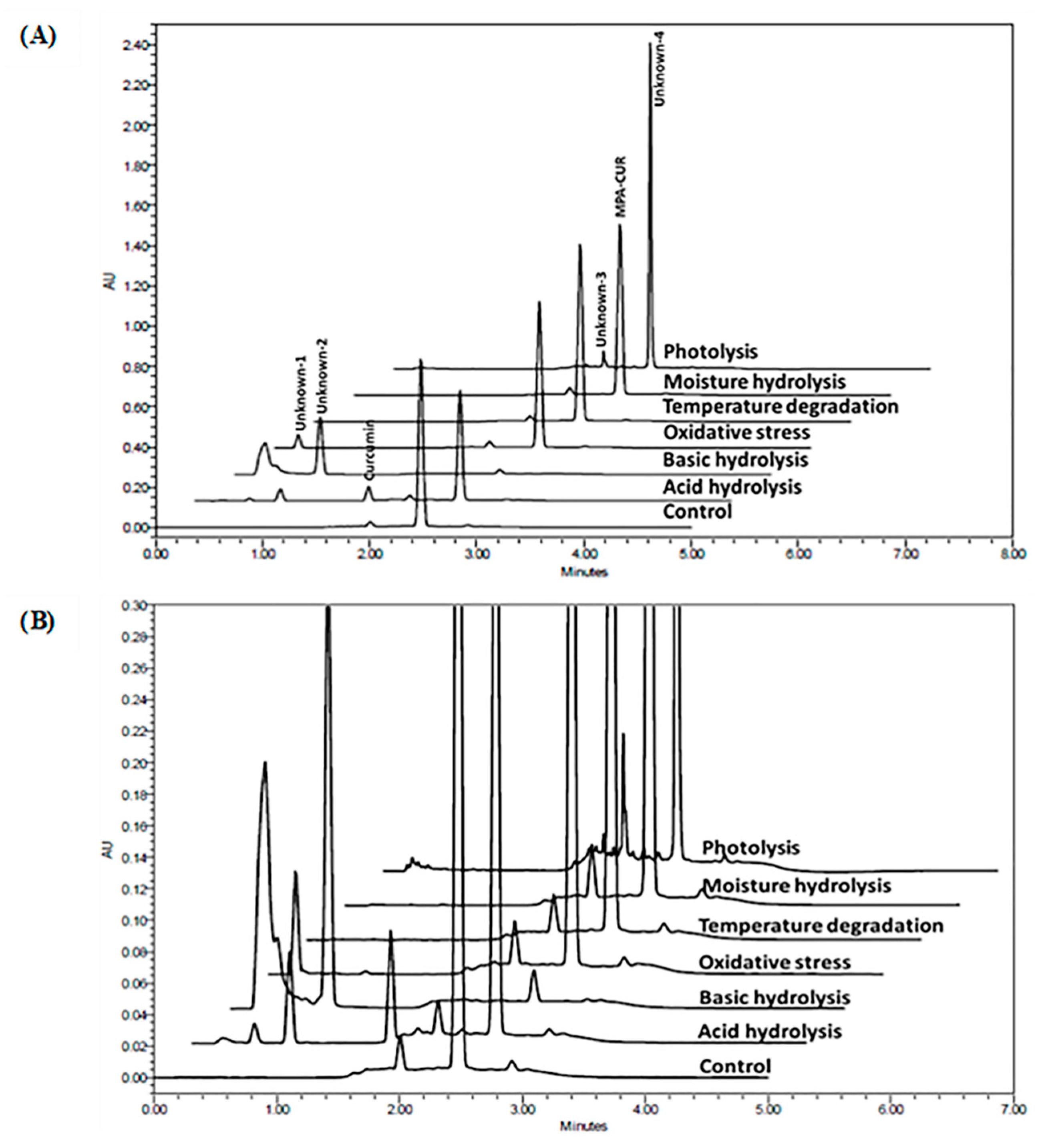
2.2.2. Specificity and Forced Degradation Study
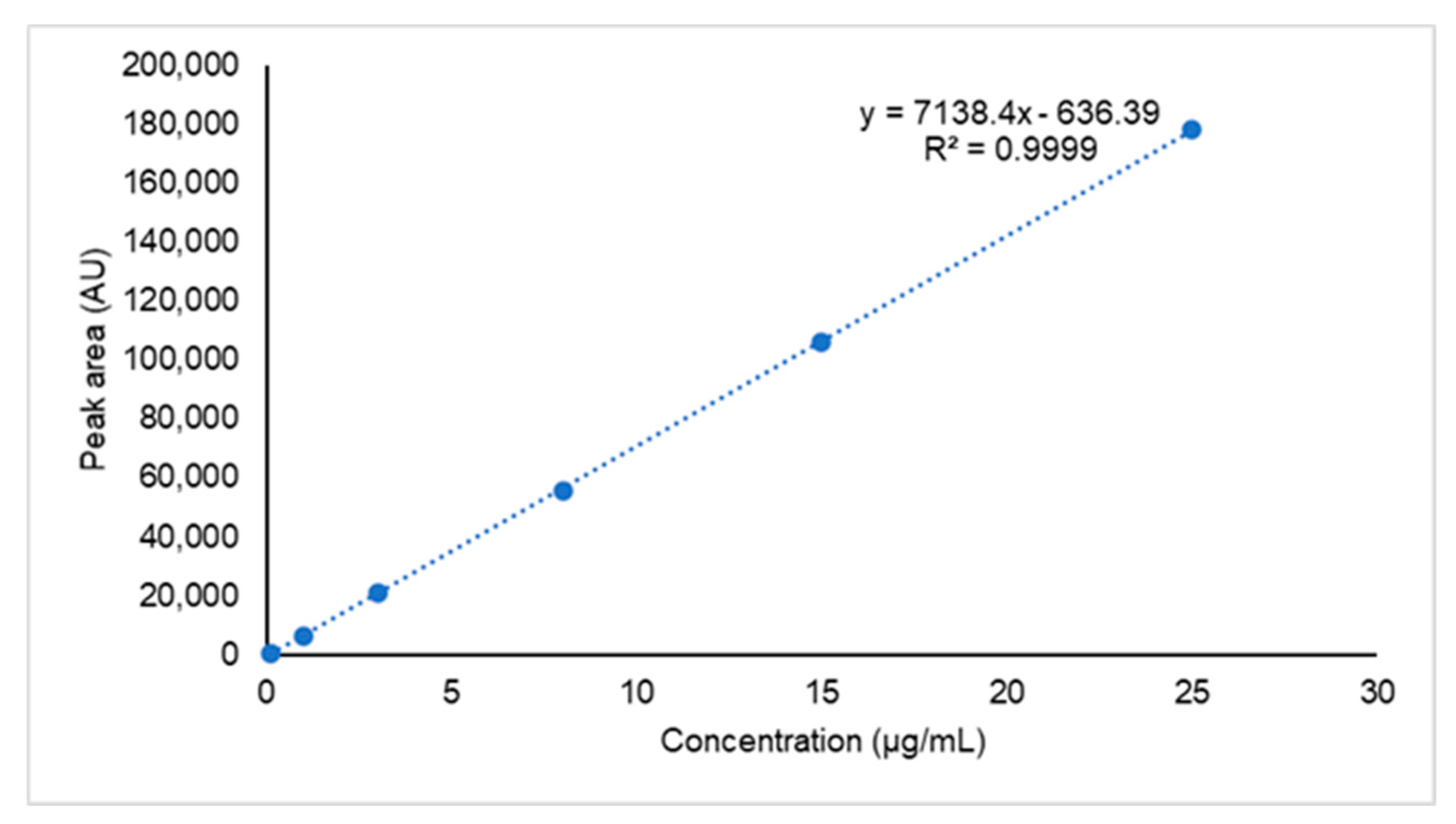
2.2.3. Linearity and Range
2.2.4. Limit of Detection (LOD) and Limit of Quantification (LOQ)
2.2.5. Accuracy and Precision
2.2.6. Robustness
2.2.7. Stability of the MPA-CUR Solutions in the Autosampler
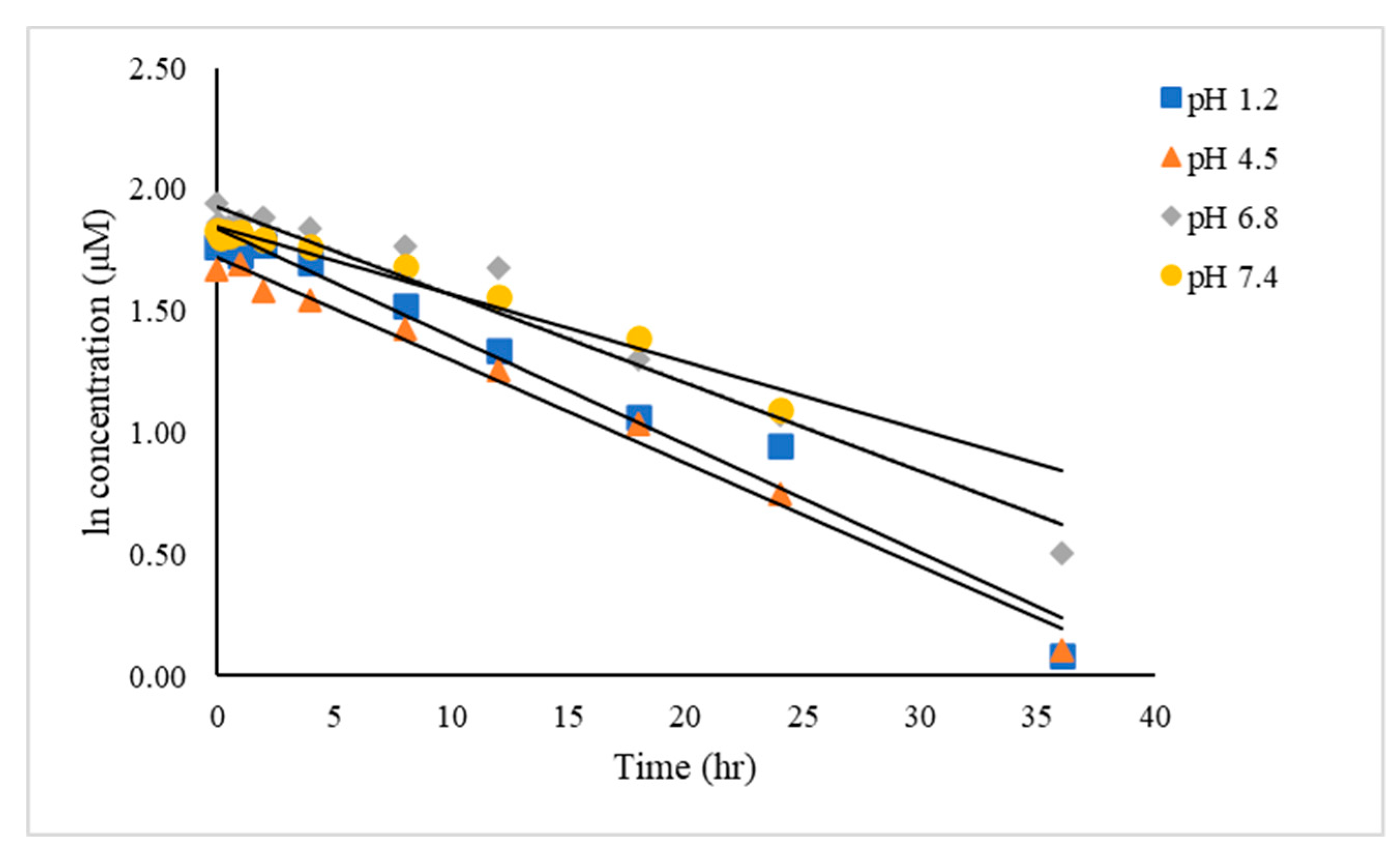
2.3. Application of the MPA-CUR Determination in Chemical Kinetic Studies
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Chromatographic Conditions
3.3. Preparation of the Standard Solution
3.4. System Suitability
3.5. Forced Degradation Studies
3.5.1. A Control Sample
3.5.2. Acid Hydrolysis
3.5.3. Basic Hydrolysis
3.5.4. Moisture Hydrolysis
3.5.5. Oxidative Degradation
3.5.6. Thermal Degradation
3.5.7. Photolysis
3.6. Method Validation
3.6.1. System Suitability
3.6.2. Specificity
3.6.3. Linearity and Range
3.6.4. LOD and LOQ
3.6.5. Accuracy and Precision
3.6.6. Robustness
3.6.7. Stability of the MPA-CUR Solutions in the Autosampler
3.7. Application of the MPA-CUR Determination for Chemical Kinetic Studies
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Aljuffali, I.A.; Chwan-Fwu, L.; Chen, C.; Fang, J. The codrug approach for facilitating drug delivery and bioactivity. Expert Opin. Drug Deliv. 2016, 13, 1311–1325. [Google Scholar] [CrossRef] [PubMed]
- Yuyun, Y.; Ratnatilaka Na Bhuket, P.; Supasena, W.; Suwattananuruk, P.; Praengam, K.; Vajragupta, O.; Muangnoi, C.; Rojsitthisak, P. A novel curcumin-mycophenolic acid conjugate inhibited hyperproliferation of tumor necrosis factor-alpha-induced human keratinocyte cells. Pharmaceutics 2021, 13, 956. [Google Scholar] [CrossRef]
- Karaman, R.; Qtait, A.; Dajani, K.K.; Abu Lafi, S. Design, synthesis, and in vitro kinetics study of atenolol prodrugs for the use in aqueous formulations. Sci. World J. 2014, 2014, 248651. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Yoon, Y.R. Understanding the pharmacokinetics of prodrug and metabolite. Transl Clin. Pharmacol. 2018, 26, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Rasheed, A.; Kumar, C.K.A.; Mishra, A. Synthesis, hydrolysis studies and pharmacodynamic profiles of amide prodrugs of dexibuprofen with amino acids. J. Enzym. Inhib. Med. Chem. 2011, 26, 688–695. [Google Scholar] [CrossRef] [PubMed]
- ICH Steering Committee. Q2(R1): Validation of Analytical Procedures: Text and Methodology, ICH. In Proceedings of the International Conference on Harmonization (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use, Geneva, Switzerland, 2005. [Google Scholar]
- Bakshi, M.; Singh, S. Development of validated stability-indicating assay methods—Critical review. J. Pharm. Biomed. Anal. 2002, 28, 1011–1040. [Google Scholar] [CrossRef]
- Korany, M.A.; Haggag, R.S.; Ragab, M.; Elmallah, O. A validated stability-indicating HPLC method for simultaneous determination of silymarin and curcumin in various dosage forms. Arab. J. Chem. 2017, 10, S1711–S1725. [Google Scholar] [CrossRef] [Green Version]
- Wichitnithad, W.; Nimmannit, U.; Wacharasindhu, S.; Rojsitthisak, P. Synthesis, characterization and biological evaluation of succinate prodrugs of curcuminoids for colon cancer treatment. Molecules 2011, 16, 1888–1900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muangnoi, C.; Jithavech, P.; Ratnatilaka Na Bhuket, P.; Supasena, W.; Wichitnithad, W.; Towiwat, P.; Niwattisaiwong, N.; Haworth, I.S.; Rojsitthisak, P. A curcumin-diglutaric acid conjugated prodrug with improved water solubility and antinociceptive properties compared to curcumin. Biosci. Biotechnol. Biochem. 2018, 82, 1301–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muangnoi, C.; Ratnatilaka Na Bhuket, P.; Jithavech, P.; Supasena, W.; Paraoan, L.; Patumraj, S.; Rojsitthisak, P. Curcumin diethyl disuccinate, a prodrug of curcumin, enhances anti-proliferative effect of curcumin against HepG2 cells via apoptosis induction. Sci. Rep. 2019, 9, 11718. [Google Scholar] [CrossRef] [Green Version]
- Ratnatilaka Na Bhuket, P.; Wichitnithad, W.; Sudtanon, O.; Rojsitthisak, R. A stability-indicating UPLC method for the determination of curcumin diethyl disuccinate, an ester prodrug of curcumin, in raw materials. Heliyon 2020, 6, e04561. [Google Scholar] [CrossRef]
- Kathirvel, S.; Prasad, K.R.; Babu, K.M. Development and validation of HPTLC method for the determination of mycophenolate mofetil in bulk and pharmaceutical formulation. Pharm. Methods 2012, 3, 90–93. [Google Scholar] [CrossRef] [Green Version]
- Protic, A.; Zivanovic, L.; Zecevic, M.; Jocic, B. Development of liquid chromatographic method for simultaneous determination of mycophenolate mofetil and its degradation product mycophenolic acid in dosage form. J. Chromatogr. Sci. 2009, 47, 149–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinay, K.B.; Ravanasiddappa, H.; Raghu, M.; Abdulrahman, S.; Rajendraprasad, N. Spectrophotometric determination of mycophenolate mofetil as its charge-transfer complexes with two pi-acceptors. J. Anal. Methods Chem. 2012, 2012, 875942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syed, M.; Srinivas, N.R. A comprehensive review of the published assays for the quantitation of the immunosuppressant drug mycophenolic acid and its glucuronidated metabolites in biological fluids. Biomed. Chromatogr. 2016, 30, 721–748. [Google Scholar] [CrossRef]
- Chopade, S.S.; Dhaneshwar, S.S. Determination of the mitigating effect of colon-specific bioreversible codrugs of mycophenolic acid and aminosugars in an experimental colitis model in Wistar rats. World J. Gastroenterol. 2018, 24, 1093–1106. [Google Scholar] [CrossRef]
- Atapattu, S.N. Solvation properties of acetone-water mobile phases in reversed-phase liquid chromatography. J. Chromatogr. A 2021, 1650, 462252. [Google Scholar] [CrossRef]
- Yabre, M.; Ferey, L.; Touridomon Some, I.; Gaudin, K. Greening reversed-phase liquid chromatography methods using alternative solvents for pharmaceutical analysis. Molecules 2018, 23, 1065. [Google Scholar] [CrossRef] [Green Version]
- Attimarad, M.V.; Alnajjar, A.O. A conventional HPLC-MS method for the simultaneous determination of ofloxacin and cefixime in plasma: Development and validation. J. Basic Clin. Pharm. 2013, 4, 36–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Naem, O.A.; El-Maraghy, C.M. A validated liquid chromatography-tandem mass spectrometric method for the determination of co-administered ranitidine and metronidazole in plasma of human volunteers. Anal. Methods 2021, 13, 2586–2595. [Google Scholar] [CrossRef]
- Novakova, L.; Matysova, L.; Solich, P. Advantages of application of UPLC in pharmaceutical analysis. Talanta 2006, 68, 908–918. [Google Scholar]
- Gumustas, M.; Kurbanoglu, S.; Uslu, B.; Ozkan, S.A. UPLC versus HPLC on drug analysis: Advantageous, applications and their validation parameters. Chromatographia 2013, 76, 1365–1427. [Google Scholar] [CrossRef]
- Lalitha Devi, M.; Chandrasekhar, K.B. A validated stability-indicating RP-HPLC method for levofloxacin in the presence of degradation products, its process related impurities and identification of oxidative degradant. J. Pharm Biomed. Anal. 2009, 50, 710–717. [Google Scholar] [CrossRef]
- Wang, Y.J.; Pan, M.H.; Chen, A.; Lin, L.; Ho, Y.; Hsieh, C.; Lin, J. Stability of curcumin in buffer solutions and characterization of its degradation products. J. Pharm. Biomed. Anal. 1997, 15, 1867–1876. [Google Scholar] [CrossRef]
- ICH Steering Committee. Q1B: Stability Testing: Photostability Testing of New Drug Substances and Products, ICH. In Proceedings of the International Conference on Harmonization (ICH) of Technical Requirements for Registration of Pharmaceuticals for Human Use, Geneva, Switzerland, 1996. [Google Scholar]
- Rawski, R.I.; Sanecki, P.; Skital, P.; Kijowska, K. Regression analysis in analytical chemistry. Determination and validation of linear and quadratic regression dependencies. South Afr. J. Chem. 2016, 69, 166–173. [Google Scholar] [CrossRef]
- Jurado, J.M.; Alcazar, A.; Muniz-Valencia, R.; Ceballos-Magana, S.; Raposo, F. Some practical considerations for linearity assessment of calibration curves as function of concentration levels according to the fitness-for-purpose approach. Talanta 2017, 172, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.M.; Castel-Branco, M.M.; Falcao, A.C. Linear regression for calibration lines revisited: Weighting schemes for bioanalytical methods. J. Chromatogr. B Life Sci. 2002, 774, 215–222. [Google Scholar]
- Johnson, E.L.; Reynolds, D.; Wright, D.; Pachla, L. Biological sample preparation and data reduction concepts in pharmaceutical analysis. J. Chromatogr. Sci. 1988, 26, 372–379. [Google Scholar] [CrossRef]
- Wichitnithad, W.; Sudtanon, O.; Srisunak, P.; Cheewatanakornkool, K.; Nantaphol, S.; Rojsitthisak, P. Development of a sensitive headspace gas chromatography-mass spectrometry method for the simultaneous determination of nitrosamines in losartan active pharmaceutical ingredients. ACS Omega 2021, 6, 11048–11058. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, S.S.; Chhajed, S.; Attar, S.; Kshirsagar, S. An approach to select linear regression model in bioanalytical method validation. J. Anal. Sci. Technol. 2019, 10, 1. [Google Scholar] [CrossRef]
- Tiwari, G.; Tiwari, R. Bioanalytical method validation: An updated review. Pharm. Methods 2010, 1, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Reis, N.F.; de Assis, J.C.; Fialho, S.L.; Pianetti, G.A.; Fernandes, C. Stability-indicating UHPLC method for determination of nevirapine in its bulk form and tablets: Identification of impurities and degradation kinetic study. J. Pharm. Biomed. Anal. 2016, 126, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Ip, K.; Shan, A.; Carvalho, M.; Baker, S.; Banov, D. Physicochemical stability of extemporaneously prepared oral suspension of fluconazole 50 mg/mL in SuspendIt™. Pharm. Technol. Hosp. Pharm. 2018, 3, 101–112. [Google Scholar] [CrossRef]
- Center for Drug Evaluation and Research, U.S. Food and Drug Administration. Reviewer Guidance, Validation of Chromatographic Methods; FDA: Rockville, MD, USA, 1994. [Google Scholar]
- Danzer, K.; Currie, L.A. Guidelines for calibration in analytical chemistry—Part, I. Fundamentals and single component calibration (IUPAC Recommendations 1998). Pure Appl. Chem. 1998, 70, 993–1014. [Google Scholar] [CrossRef]
- Raposo, F. Evaluation of analytical calibration based on least-squares linear regression for instrumental techniques: A tutorial review. TrAC Trends Anal. Chem. 2016, 77, 167–185. [Google Scholar] [CrossRef]
- Bressolle, F.; Bromet-Petit, M.; Audran, M. Validation of liquid chromatographic and gas chromatographic methods. Applications to pharmacokinetics. J. Chromatogr. B Biomed. Appl. 1996, 686, 3–10. [Google Scholar] [CrossRef]
- Barwick, V.J. Preparation of calibration curves: A guide to best practice. In LGC/VAM/2003/032; LGC: Teddington, UK, 2003; pp. 1–27. [Google Scholar]
- Ershadi, S.; Shayanfar, A. Are LOD and LOQ reliable parameters for sensitivity evaluation of spectroscopic methods? J. AOAC Int. 2018, 101, 1212–1213. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, A.G.; Herrador, M.A.; Asuero, A.G. Intra-laboratory assessment of method accuracy (trueness and precision) by using validation standards. Talanta 2010, 82, 1995–1998. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Injection No. | Retention Time (min) | Peak Area | USP Tailing Factor | USP Plate Count |
|---|---|---|---|---|
| 1 | 2.423 | 56,048 | 1 | 13,002 |
| 2 | 2.430 | 56,025 | 1 | 12,925 |
| 3 | 2.429 | 55,648 | 1 | 12,866 |
| 4 | 2.428 | 55,929 | 1 | 12,824 |
| 5 | 2.423 | 55,887 | 1 | 12,973 |
| Mean | 2.427 | 55,907 | 1 | 12,918 |
| %CV | 0.14 | 0.28 | 0.00 | 0.57 |
| Stress Condition | Incubation Time (h) | Purity Angle | Purity Threshold | MPA-CUR Remaining (%) | Peak Purity |
|---|---|---|---|---|---|
| Control (untreated) | 0 | 0.190 | 0.435 | 100.00 | Passed |
| Acid hydrolysis (100 µL of 0.1 N HCl), 80 °C | 3 | 0.086 | 0.462 | 77.65 | Passed |
| Basic hydrolysis (100 µL of 0.1 N NaOH), 80 °C | 3 | 0.025 | 12.054 | 3.21 | Passed |
| Oxidation (100 µL of 3% H2O2) | 0 | 0.130 | 0.393 | 94.88 | Passed |
| Oxidation (100 µL of 3% H2O2), 80 °C | 1 | 0.152 | 0.373 | 91.16 | Passed |
| Moisture hydrolysis (100 µL of water), 80 °C | 3 | 0.197 | 0.409 | 96.85 | Passed |
| Moisture hydrolysis (100 µL of water), 80 °C | 6 | 0.196 | 0.440 | 99.60 | Passed |
| Temperature degradation, 80 °C | 3 | 0.212 | 0.475 | 102.20 | Passed |
| Temperature degradation, 80 °C | 6 | 0.222 | 0.452 | 101.20 | Passed |
| Photolysis (UV and Fluorescence) | 5 days | 1.077 | 5.290 | 0.03 | Passed |
| Standard (μg/mL) | Peak Area | Peak Area Ratio | s2 | Fcal | Ftable | |
|---|---|---|---|---|---|---|
| 0.100 | 643 | 1.000 | 643 | 11.533 | 219,113.642 | 99.000 |
| 620 | 1.000 | 620 | ||||
| 633 | 1.000 | 633 | ||||
| 24.942 | 177,307 | 1.000 | 177,307 | 5398 | ||
| 173,466 | 1.000 | 173,466 | ||||
| 184,125 | 1.000 | 184,125 | ||||
| Replication | Model | Weighting Factor | Slope | Intercept | r | r2 | ∑|%RE| | Minimum | Result |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 1 | 7096.149 | −112.938 | 0.9999 | 0.9990 | 23.727 | 20.43 | 1/x2 |
| 2 | 1/x | 7093.347 | −107.486 | 0.9998 | 0.9997 | 22.790 | |||
| 3 | 1/x2 | 7003.813 | −61.143 | 0.9985 | 0.9970 | 20.430 | |||
| 2 | 1 | 1 | 6973.427 | −441.224 | 0.9999 | 0.9990 | 57.482 | 10.59 | 1/x2 |
| 2 | 1/x | 6928.311 | −123.907 | 0.9999 | 0.9998 | 17.914 | |||
| 3 | 1/x2 | 6809.330 | −62.322 | 0.9996 | 0.9993 | 10.593 | |||
| 3 | 1 | 1 | 7368.774 | −885.046 | 0.9999 | 0.9998 | 114.5429 | 13.63 | 1/x2 |
| 2 | 1/x | 7269.613 | −173.504 | 0.9998 | 0.9996 | 25.753 | |||
| 3 | 1/x2 | 7084.538 | −77.709 | 0.9994 | 0.9989 | 13.625 |
| Compound | Nominal Conc. (µg/mL) | Back-Calculated Concentration (µg/mL) | Mean Back-Calculated Concentration (µg/mL) | %RE | %CV | ||
|---|---|---|---|---|---|---|---|
| Day 1 | Day 2 | Day 3 | |||||
| MPA-CUR | 0.100 | 0.101 | 0.100 | 0.100 | 0.100 ± 0.001 | 0.250 | 0.50 |
| 1.000 | 0.898 | 0.953 | 0.945 | 0.949 ± 0.03 | −5.100 | 4.40 | |
| 3.000 | 3.175 | 2.986 | 2.942 | 3.026 ± 0.124 | 0.858 | 3.39 | |
| 8.000 | 8.071 | 7.927 | 7.996 | 7.996 ± 0.072 | −0.053 | 0.74 | |
| 15.000 | 15.035 | 15.383 | 15.282 | 15.175 ± 0.179 | 1.167 | 1.23 | |
| 25.000 | 25.325 | 25.484 | 26.001 | 25.453 ± 0.353 | 1.810 | 1.64 | |
| r2 | 0.9997 | ||||||
| Fcal | 52,968.10878 | ||||||
| Ftable | 1.35816 × 10−29 | ||||||
| p-value of slope | 1.35816 × 10−29 | ||||||
| p-value of intercept | 0.1596 | ||||||
| Injection No. | LOD | Sample No. | LOQ | ||||
|---|---|---|---|---|---|---|---|
| Peak Area | S/N | Added Conc. (µg/mL) | Found Conc. (µg/mL) | %Recovery | S/N | ||
| 1 | 178 | 4 | 1 | 0.100 | 0.094 | 93.9 | 9 |
| 2 | 171 | 3 | 2 | 0.100 | 0.091 | 90.8 | 11 |
| 3 | 184 | 3 | 3 | 0.100 | 0.091 | 90.5 | 11 |
| 4 | 193 | 3 | 4 | 0.100 | 0.094 | 94.1 | 14 |
| 5 | 191 | 4 | 5 | 0.100 | 0.093 | 93.3 | 14 |
| Mean | 183 | 3 | 0.093 | 92.5 | 12 | ||
| %CV | 5.0 | 1.6 | |||||
| Nominal Conc. (µg/mL) | Intra-Day (n = 3) | Inter-Day (n = 9) | ||||||
|---|---|---|---|---|---|---|---|---|
| Added Conc. (µg/mL) | Found Conc. (µg/mL) | %Recovery | %CV | Added Conc. (µg/mL) | Found Conc. (µg/mL) | %Recovery | %CV | |
| 0.10 | 0.100 | 0.098 ± 0.001 | 98.4 | 0.52 | 0.10 | 0.099 ± 0.001 | 98.5 | 0.77 |
| 12.5 | 12.47 | 12.71 ± 0.01 | 101.6 | 0.04 | 12.47 | 12.64 ± 0.16 | 101.2 | 1.23 |
| 25.0 | 24.942 | 25.15 ± 0.20 | 100.6 | 0.81 | 24.942 | 24.96 ± 0.63 | 99.8 | 2.53 |
| Chromatographic Parameters | Retention Time | Peak Area | Tailing Factor (T) | Theoretical Plate (N) |
|---|---|---|---|---|
| %CV | %CV | |||
| Concentrations of formic acid solution | ||||
| 0.09% | 0.08 | 0.84 | 1.0 | 12,615 |
| 0.10% | 0.23 | 0.44 | 1.0 | 13,182 |
| 0.11% | 0.13 | 0.51 | 1.0 | 12,587 |
| Analytical columns from different batches | ||||
| Column # 1 Batch no. 0293370651 | 0.23 | 0.44 | 1.0 | 13,182 |
| Column # 2 Batch no. 0318381361 | 0.08 | 0.59 | 1.1 | 12,609 |
| Time (h) | Added Conc. (µg/mL) | Found Conc. (µg/mL) | %Recovery |
|---|---|---|---|
| 0 | 7.922 | 7.894 | 99.66 |
| 6 | 7.922 | 8.146 | 102.83 |
| 9 | 7.922 | 8.099 | 102.24 |
| 12 | 7.922 | 8.092 | 102.15 |
| 24 | 7.922 | 8.177 | 103.23 |
| pH of Buffer Solutions | r2 | ||
|---|---|---|---|
| Zero Order | Pseudo-First Order | Second Order | |
| 1.2 | 0.964 | 0.978 | 0.862 |
| 4.5 | 0.949 | 0.982 | 0.934 |
| 6.8 | 0.966 | 0.972 | 0.883 |
| 7.4 | 0.964 | 0.977 | 0.804 |
| pH of Buffer Solutions | Kinetic Parameters | |
|---|---|---|
| kobs (h−1) | t1/2 (h) | |
| 1.2 | 0.045 ± 0.003 | 15.67 ± 1.2 |
| 4.5 | 0.041 ± 0.010 | 18.59 ± 7.5 |
| 6.8 | 0.036 ± 0.036 | 19.73 ± 4.4 |
| 7.4 | 0.044 ± 0.002 | 15.94 ± 0.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuyun, Y.; Jithavech, P.; Thitikornpong, W.; Vajragupta, O.; Rojsitthisak, P. A Stability-Indicating Ultra Performance Liquid Chromato-Graphic (UPLC) Method for the Determination of a Mycophenolic Acid-Curcumin Conjugate and Its Applications to Chemical Kinetic Studies. Molecules 2021, 26, 5398. https://doi.org/10.3390/molecules26175398
Yuyun Y, Jithavech P, Thitikornpong W, Vajragupta O, Rojsitthisak P. A Stability-Indicating Ultra Performance Liquid Chromato-Graphic (UPLC) Method for the Determination of a Mycophenolic Acid-Curcumin Conjugate and Its Applications to Chemical Kinetic Studies. Molecules. 2021; 26(17):5398. https://doi.org/10.3390/molecules26175398
Chicago/Turabian StyleYuyun, Yonelian, Ponsiree Jithavech, Worathat Thitikornpong, Opa Vajragupta, and Pornchai Rojsitthisak. 2021. "A Stability-Indicating Ultra Performance Liquid Chromato-Graphic (UPLC) Method for the Determination of a Mycophenolic Acid-Curcumin Conjugate and Its Applications to Chemical Kinetic Studies" Molecules 26, no. 17: 5398. https://doi.org/10.3390/molecules26175398
APA StyleYuyun, Y., Jithavech, P., Thitikornpong, W., Vajragupta, O., & Rojsitthisak, P. (2021). A Stability-Indicating Ultra Performance Liquid Chromato-Graphic (UPLC) Method for the Determination of a Mycophenolic Acid-Curcumin Conjugate and Its Applications to Chemical Kinetic Studies. Molecules, 26(17), 5398. https://doi.org/10.3390/molecules26175398