
The Reactions of 6-(Hydroxymethyl)-2,2-dimethyl-1-azaspiro[4.4]nonanes with Methanesulfonyl Chloride or PPh3-CBr4

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
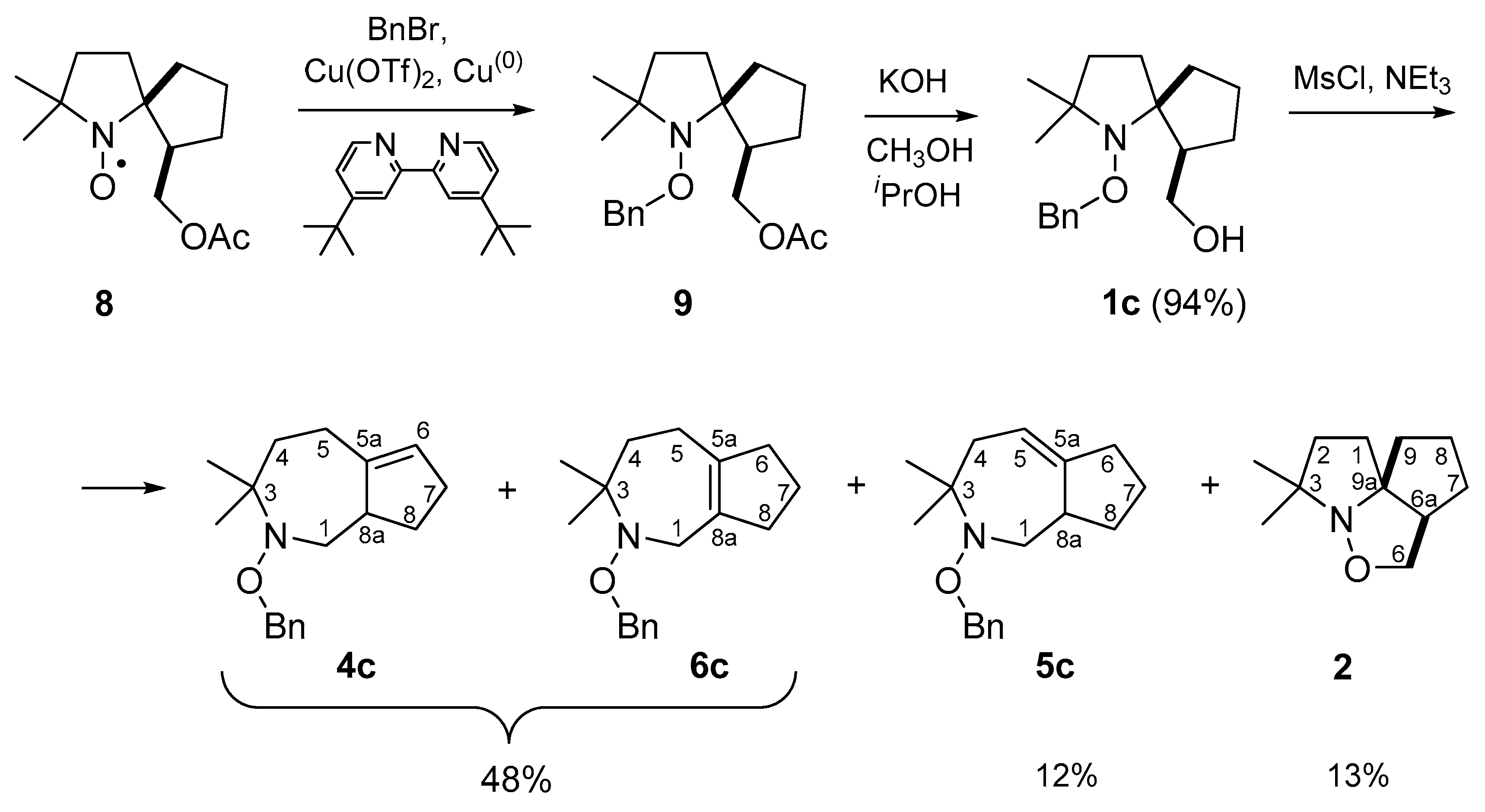
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. Reaction of 6-(Hydroxymethyl)-2,2-dimethyl-1-azaspiro[4.4]nonan-1-oxyl 1a with Methanesulfonyl Chloride
3.2.2. Reaction of 6-(Hydroxymethyl)-2,2-dimethyl-1-azaspiro[4.4]nonan-1-oxyl 1a with PPh3-CBr4
3.2.3. Reaction of (2,2-Dimethyl-1-azaspiro[4.4]nonan-6-yl)methanol 1b with Methanesulfonyl Chloride
3.2.4. Reaction of (5aS,8aR)-3,3-Dimethyloctahydrocyclopenta[2,3]azeto[1,2-a]pyrrole 3 with HBr
3.2.5. Reaction of ((5R(S),6R(S))-2,2-Dimethyl-1-azaspiro[4.4]nonan-6-yl)methanol 1b with PPh3-CBr4
3.2.6. (1-(Benzyloxy)-2,2-dimethyl-1-azaspiro[4.4]nonan-6-yl)-methanol (1c)
3.2.7. Reaction of (1-(Benzyloxy)-2,2-dimethyl-1-azaspiro[4.4]nonan-6-yl)-methanol 1c with Methanesulfonyl Chloride
3.2.8. Reaction of (1-(Benzyloxy)-2,2-dimethyl-1-azaspiro[4.4]nonan-6-yl)-methanol 1c with PPh3-CBr4
3.2.9. 6-(Hydroxymethyl)-2,2-dimethyl-1-azaspiro[4.4]nonan-1-yl benzoate (1d)
3.2.10. Reaction of (1-(Benzoyloxy)-2,2-dimethyl-1-azaspiro[4.4]nonan-6-yl)-methanol 1d with PPh3-CBr4
3.2.11. Reduction of Nitroxide 13 with Zn in CF3COOH for NMR
3.2.12. 3,3,4-Trimethyloctahydro-1H-cyclopenta[2,3]azeto[1,2-a]pyrrol-4-ium iodide (15)
3.2.13. 2,3,3-Trimethyl-1,2,3,4,5,7,8,8a-octahydrocyclopenta[c]azepine (16)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Appendix A
Appendix A.1. 2-(Benzyloxy)-3,3-dimethyl-1,2,3,4,5,7,8,8a-octahydrocyclopenta[c]azepine (4c)
Appendix A.2. 2-(Benzyloxy)-3,3-dimethyl-1,2,3,4,6,7,8,8a-octahydrocyclopenta[c]azepine (5c)
Appendix A.3. 3-Dimethyl-3,4,6,7,8,8a-hexahydrocyclopenta[c]azepin-2(1H)-yl benzoate (5d)
Appendix A.4. (5aS(R),8aR(S))-3,3-Dimethyloctahydro-1H-cyclopenta[2,3]azeto[1,2-a]pyrrol-4-ium bromide (3×HBr) and 2,3,3-Trimethyl-1,2,3,4,5,7,8,8a-octahydrocyclopenta[c]azepine (16)
References
- Nelson, J.D. Aliphatic Nucleophilic Substitution. In Practical Synthetic Organic Chemistry: Reactions, Principles, and Techniques, 2nd ed.; Caron, S., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2020; Chapter 1; pp. 1–71. [Google Scholar]
- Spener, F. Reactions of aliphatic methanesulfonates. Chem. Phys. Lipids 1973, 11, 229–243. [Google Scholar] [CrossRef]
- Appel, R. Tertiary Phosphane/Tetrachloromethane, a Versatile Reagent for Chlorination, Dehydration, and P–N Linkage. Angew. Chem. Int. Ed. 1975, 14, 801–811. [Google Scholar] [CrossRef]
- Khoroshunova, Y.V.; Morozov, D.A.; Taratayko, A.I.; Gladkikh, P.D.; Glazachev, Y.I.; Kirilyuk, I.A.; Beilstein, J. Synthesis of 1-azaspiro[4.4] nonan-1-oxyls via intramolecular 1,3-dipolar cycloaddition. Org. Chem. 2019, 15, 2036–2042. [Google Scholar] [CrossRef] [Green Version]
- Roser, P.; Schmidt, M.J.; Drescher, M.; Summerer, D. Site-directed spin labeling of proteins for distance measurements in vitro and in cells. Org. Biomol. Chem. 2016, 14, 5468–5476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fielding, J.; Consilio, M.G.; Heaven, G.; Hollas, M.A. New developments in spin labels for pulsed dipolar EPR. Molecules 2014, 19, 16998–17025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denmark, S.E.; Montgomery, J.I. Synthesis of cis,cis,cis,cis-[5.5.5.4]-1-Azafenestrane with Discovery of an Unexpected Dyotropic Rearrangement. Angew. Chem. 2005, 44, 3732–3736. [Google Scholar] [CrossRef]
- Masson, G.; Rioton, S.; Pardo, D.G.; Cossy, J. Synthesis of 2-Fluoroalkyl 4-Substituted Azepanes. Eur. J. Org. Chem. 2019, 2019, 5497–5507. [Google Scholar] [CrossRef]
- Sivaprakasam, M.; Couty, F.; David, O.; Marrot, J.; Sridhar, R.; Srinivas, B.; Rao, K.R. A Straightforward Synthesis of Enantiopure Bicyclic Azetidines. Eur. J. Org. Chem. 2007, 5734–5739. [Google Scholar] [CrossRef]
- Holmes, B.; Ward, A. Meptazinol. A Review of its Pharmacodynamic and Pharmacokinetic Properties and Therapeutic Efficacy. Drugs 1985, 30, 285–312. [Google Scholar] [CrossRef] [PubMed]
- Soderquist, C.J.; Bowers, J.B.; Crosby, D.G. Dissipation of molinate in a rice field. J. Agric. Food Chem. 1977, 25, 940–945. [Google Scholar] [CrossRef]
- Zaimoku, H.; Taniguchi, T. Redox Divergent Synthesis of Fawcettimine-TypeLycopodiumAlkaloids. Chem. A Eur. J. 2014, 20, 9613–9619. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.R.; Fromhold, M.G.; Earley, J.D. Total Synthesis of (−)-Stemospironine. Org. Lett. 2001, 3, 2721–2724. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, S. Recent progress in the synthesis of heterocyclic natural productsby the Staudinger/intramolecular aza-Wittig reaction. ARKIVOC 2005, 2, 98–119. [Google Scholar] [CrossRef] [Green Version]
- Albertini, E.; Barco, A.; Benetti, S.; De Risi, C.; Pollini, G.P.; Zanirato, V. Efficient Synthesis of Chiral N-Tosyl-3,4-Disubstituted Hexahydroazepins from D-(-)-Quinic Acid. Synlett 1996, 1996, 29–30. [Google Scholar] [CrossRef]
- Ducandas, C.; Lasne, M.C.; Moreau, B. Syntheses of piperidine and perhydroazepine derivatives, precursors of two selective antagonists of muscarinic M2 receptors: AF-DX 384 and its perhydroazepine isomer. J. Chem. Soc. Perkin Trans. 1 1996, 2925–2932. [Google Scholar] [CrossRef]
- Úr, G.; Kálai, T.; Balog, M.; Bognár, B.; Gulyás-Fekete, G.; Hideg, K. Synthesis of New Pyrroline Nitroxides with Ethynyl Functional Group. Synth. Commun. 2015, 45, 2122–2129. [Google Scholar] [CrossRef] [Green Version]
- Bushmakina, N.G.; Misharin, A.Y. A Simple Synthesis of 4-Amino-2,2,6,6-tetramethyl-1-piperidinyloxy Radical. Synthesis 1986, 1986, 966. [Google Scholar] [CrossRef]
- Dobrynin, S.; Kutseikin, S.; Morozov, D.; Krumkacheva, O.; Spitsyna, A.; Gatilov, Y.; Silnikov, V.; Angelovski, G.; Bowman, M.K.; Kirilyuk, I.; et al. Human Serum Albumin Labelled with Sterically-Hindered Nitroxides as Potential MRI Contrast Agents. Molecules 2020, 25, 1709. [Google Scholar] [CrossRef] [Green Version]
- Kálai, T.; Balog, M.; Jekö, J.; Hideg, K. Synthesis and Reactions of a Symmetric Paramagnetic Pyrrolidine Diene. Synthesis 1999, 973–980. [Google Scholar] [CrossRef]
- Yasui, S.; Shioji, K.; Tsujimoto, M.; Ohno, A. Reactivity of a trivalent phosphorus radical cation as an electrophile toward pyridine derivatives. Heteroat. Chem. 2000, 11, 152–157. [Google Scholar] [CrossRef]
- Hu, J.; Wang, J.; Nguyen, T.H.; Zheng, N. The chemistry of amine radical cations produced by visible light photoredox catalysis. Beilstein J. Org. Chem. 2013, 9, 1977–2001. [Google Scholar] [CrossRef]
- Chalmers, B.; Morris, J.C.; Fairfull-Smith, K.; Grainger, R.; Bottle, S.E. A novel protecting group methodology for syntheses using nitroxides. Chem. Commun. 2013, 49, 10382–10384. [Google Scholar] [CrossRef] [Green Version]
- Cardona, F.; Goti, A.; Picasso, S.; Vogel, P.; Brandi, A. Polyhydroxypyrrolidine Glycosidase Inhibitors Related to (+)-Lentiginosine. J. Carbohydr. Chem. 2000, 19, 585–601. [Google Scholar] [CrossRef]
- Tufariello, J.J.; Tegeler, J.J. Nitrone cycloaddition. A route to the lupin class of alkaloids. Tetrahedron Lett. 1976, 17, 4037–4040. [Google Scholar] [CrossRef]
- McCaig, A.E.; Meldrum, K.P.; Wightman, R.H. Synthesis of Trihydroxylated Pyrrolizidines and Indolizidines using Cycloaddition Reactions of Functionalized Cyclic Nitrones, and the Synthesis of (+)- and (-)-Lentiginosine. Tetrahedron. 1998, 54, 9429–9446. [Google Scholar] [CrossRef]
- Cope, A.C.; Trumbull, E.R. Olefins from Amines: The Hofmann Elimination Reaction and Amine Oxide Pyrolysis. Org. React. 1960, 11, 317–493. [Google Scholar] [CrossRef]
- Ebnőter, A.; Jucker, E. Ringerweiterungen bei Reaktionen von 2-[N-Methyl-pyrrolidinyl-(2)]-1chlor-äthan und 2-[N-Methyl-piperidyI-(2)]-l-chlor-athan mit nucleophilen Reagenzien. Untersuchungen über synthetische Arzneimittel, 13. Mitteilung. Helv. Chim. Acta. 1964, 47, 745–756. [Google Scholar] [CrossRef]
- Braslau, R.; Anderson, M.O.; Rivera, F.; Jimenez, A.; Haddad, T.; Axon, J.R. Acyl hydrazines as precursors to acyl radicals. Tetrahedron 2002, 58, 5513–5523. [Google Scholar] [CrossRef]
- Shelrick, G.M. SADABS, v. 2008/1; Bruker AXS Inc.: Madison, WI, USA, 2008. [Google Scholar]
- Sheldrick, G.M. SHELX-97 Programs for Crystal Structure Analysis (Release 97-2). University of Göttingen, Germany, 1997. Sheldrick, G.M. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.; Schweiger, A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Morozov, D.A.; Kirilyuk, I.A.; Komarov, D.A.; Goti, A.; Bagryanskaya, I.Y.; Kuratieva, N.V.; Grigorev, I.A.J. Synthesis of a Chiral C2-Symmetric Sterically Hindered Pyrrolidine Nitroxide Radical via Combined Iterative Nucleophilic Additions and Intramolecular 1,3-Dipolar Cycloadditions to Cyclic Nitrones. J. Org. Chem. 2012, 77, 10688–10698. [Google Scholar] [CrossRef] [PubMed]
- Edeleva, M.V.; Parkhomenko, D.A.; Morozov, D.A.; Dobrynin, S.A.; Trofimov, D.G.; Kanagatov, B.; Kirilyuk, I.A.; Bagryanskaya, E.G. Controlled/living polymerization of methyl methacrylate using new sterically hindered imidazoline nitroxides prepared via intramolecular 1,3-dipolar cycloaddition reaction. J. Polym. Sci. Part A Polym. Chem. 2014, 52, 929–943. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khoroshunova, Y.V.; Morozov, D.A.; Taratayko, A.I.; Dobrynin, S.A.; Eltsov, I.V.; Rybalova, T.V.; Sotnikova, Y.S.; Polovyanenko, D.N.; Asanbaeva, N.B.; Kirilyuk, I.A. The Reactions of 6-(Hydroxymethyl)-2,2-dimethyl-1-azaspiro[4.4]nonanes with Methanesulfonyl Chloride or PPh3-CBr4. Molecules 2021, 26, 6000. https://doi.org/10.3390/molecules26196000
Khoroshunova YV, Morozov DA, Taratayko AI, Dobrynin SA, Eltsov IV, Rybalova TV, Sotnikova YS, Polovyanenko DN, Asanbaeva NB, Kirilyuk IA. The Reactions of 6-(Hydroxymethyl)-2,2-dimethyl-1-azaspiro[4.4]nonanes with Methanesulfonyl Chloride or PPh3-CBr4. Molecules. 2021; 26(19):6000. https://doi.org/10.3390/molecules26196000
Chicago/Turabian StyleKhoroshunova, Yulia V., Denis A. Morozov, Andrey I. Taratayko, Sergey A. Dobrynin, Ilia V. Eltsov, Tatyana V. Rybalova, Yulia S. Sotnikova, Dmitriy N. Polovyanenko, Nargiz B. Asanbaeva, and Igor A. Kirilyuk. 2021. "The Reactions of 6-(Hydroxymethyl)-2,2-dimethyl-1-azaspiro[4.4]nonanes with Methanesulfonyl Chloride or PPh3-CBr4" Molecules 26, no. 19: 6000. https://doi.org/10.3390/molecules26196000
APA StyleKhoroshunova, Y. V., Morozov, D. A., Taratayko, A. I., Dobrynin, S. A., Eltsov, I. V., Rybalova, T. V., Sotnikova, Y. S., Polovyanenko, D. N., Asanbaeva, N. B., & Kirilyuk, I. A. (2021). The Reactions of 6-(Hydroxymethyl)-2,2-dimethyl-1-azaspiro[4.4]nonanes with Methanesulfonyl Chloride or PPh3-CBr4. Molecules, 26(19), 6000. https://doi.org/10.3390/molecules26196000