
Analytical Ultracentrifugation as a Matrix-Free Probe for the Study of Kinase Related Cellular and Bacterial Membrane Proteins and Glycans


Abstract
:1. Introduction
- Once purified, in common with many membrane-associated proteins and glycoproteins they are usually stubborn non-crystallizers making them not amenable to high resolution crystallographic analysis [7], although low resolution shapes of macromolecules, still considered important [8], are still possible
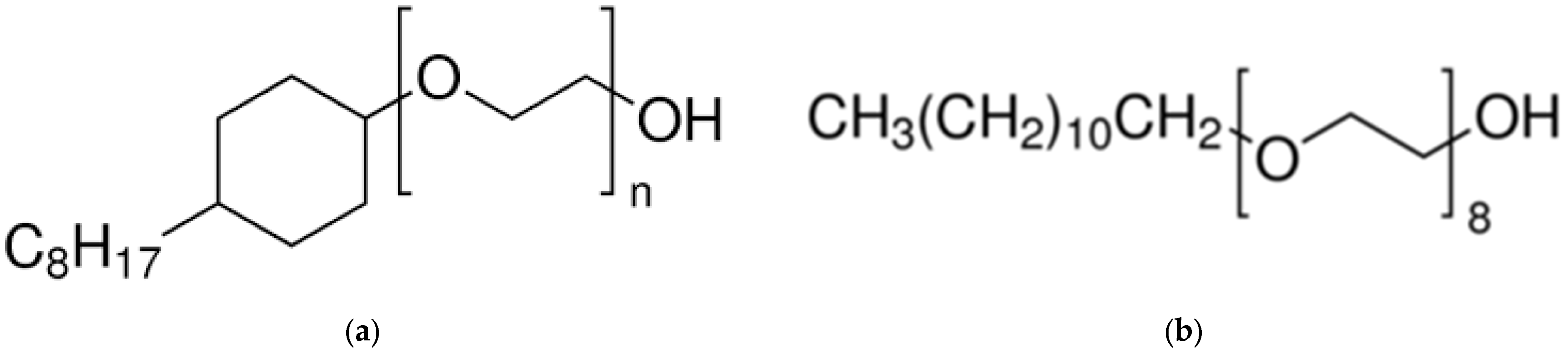
- Solubilisation and stabilisation in an appropriate solvent for solution studies (hydrodynamics, NMR) is not easy [9]. (i) For membrane-associated proteins/glycoproteins (integral or peripheral) solubilisation with detergent is required. Different detergents can disrupt the native structure to different degrees, and for interpretation of measurements the average extent of binding (which could be dynamic) of detergent to the protein is normally required for correct interpretation of the data [1,10]; (ii) For proteins/glycoproteins with a significant cytoplasmic domain (or extracellular region) account needs to be made of the different solvent requirements for the cytoplasmic domain (CD) (aqueous [11]) and transmembrane domain (TMD [10]) (non-aqueous or detergent), before appropriate conclusions can be drawn. Claims on oligomeric states based on either detergent based or aqueous based solvent systems alone need to be treated with caution: in such cases separate studies on the CD and TMD domains are useful [10,11]. Addition of further materials to stabilize the structure such as glycerol can also impact on the solution properties [3].
- Once appropriately solubilised (and stabilised), as before for the TMD domains, the extent of detergent binding is required, taking into account the dynamic nature of the binding process [6]. In addition, the technique(s) chosen to study the oligomeric state and conformation should not disrupt or affect either the intact macromolecule or the CD and TMD domains if being studied individually. This can be difficult with techniques that require a column (e.g., size-exclusion chromatography), separation membrane (e.g., field flow fractionation) or immobilisation onto a surface (atomic force microscopy, surface plasmon resonance).
2. Analytical Ultracentrifugation: What It Can Tell Us
3. Sedimentation Velocity Analytical Ultracentrifugation
4. Sedimentation Equilibrium Analytical Ultracentrifugation
5. Band 3 Protein
5.1. Estimation of Detergent Bound
5.2. Partial Specific Volume
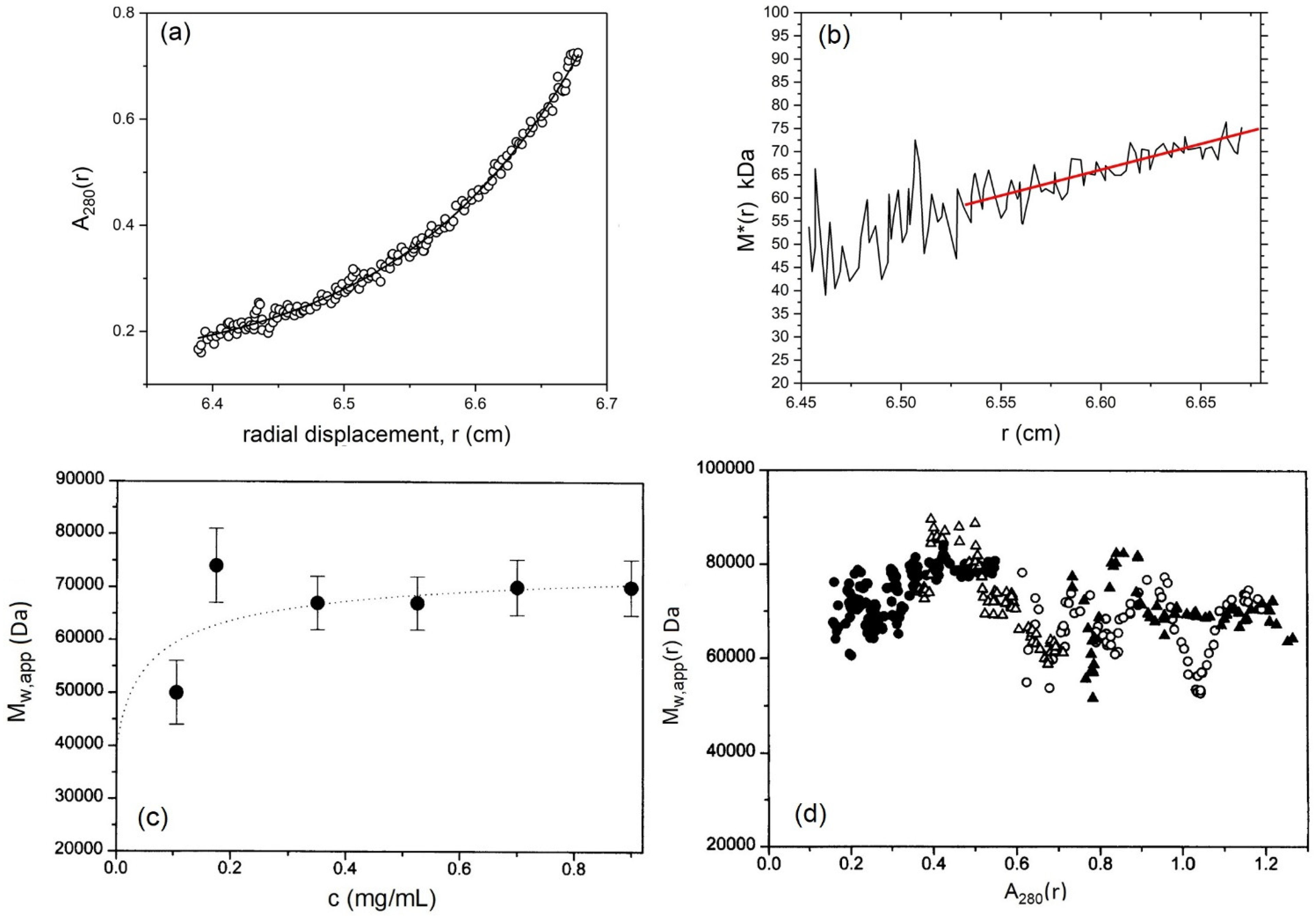
5.3. Weight Average Molecular Mass
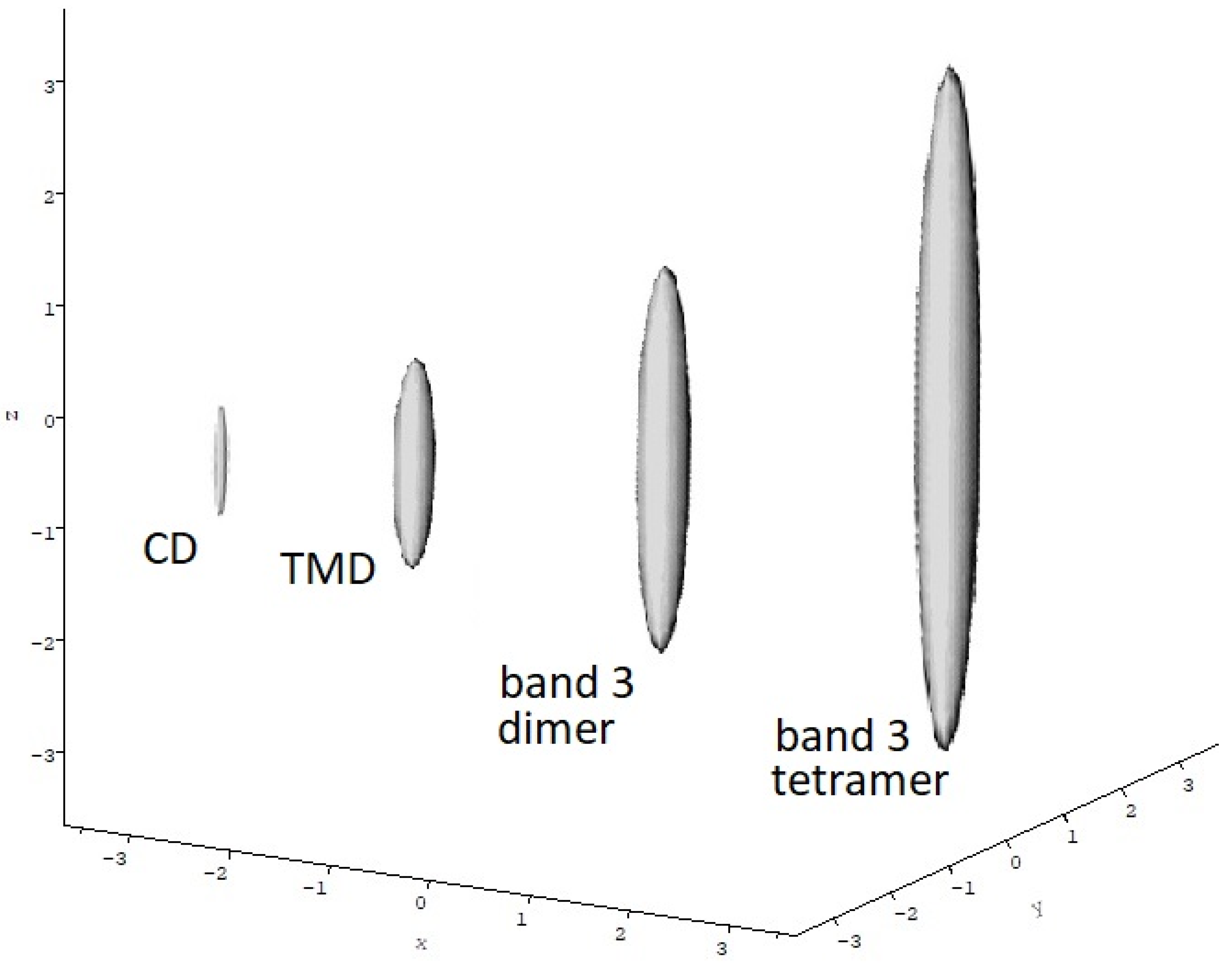
5.4. Sedimentation Coefficient and Low Resolution Conformation
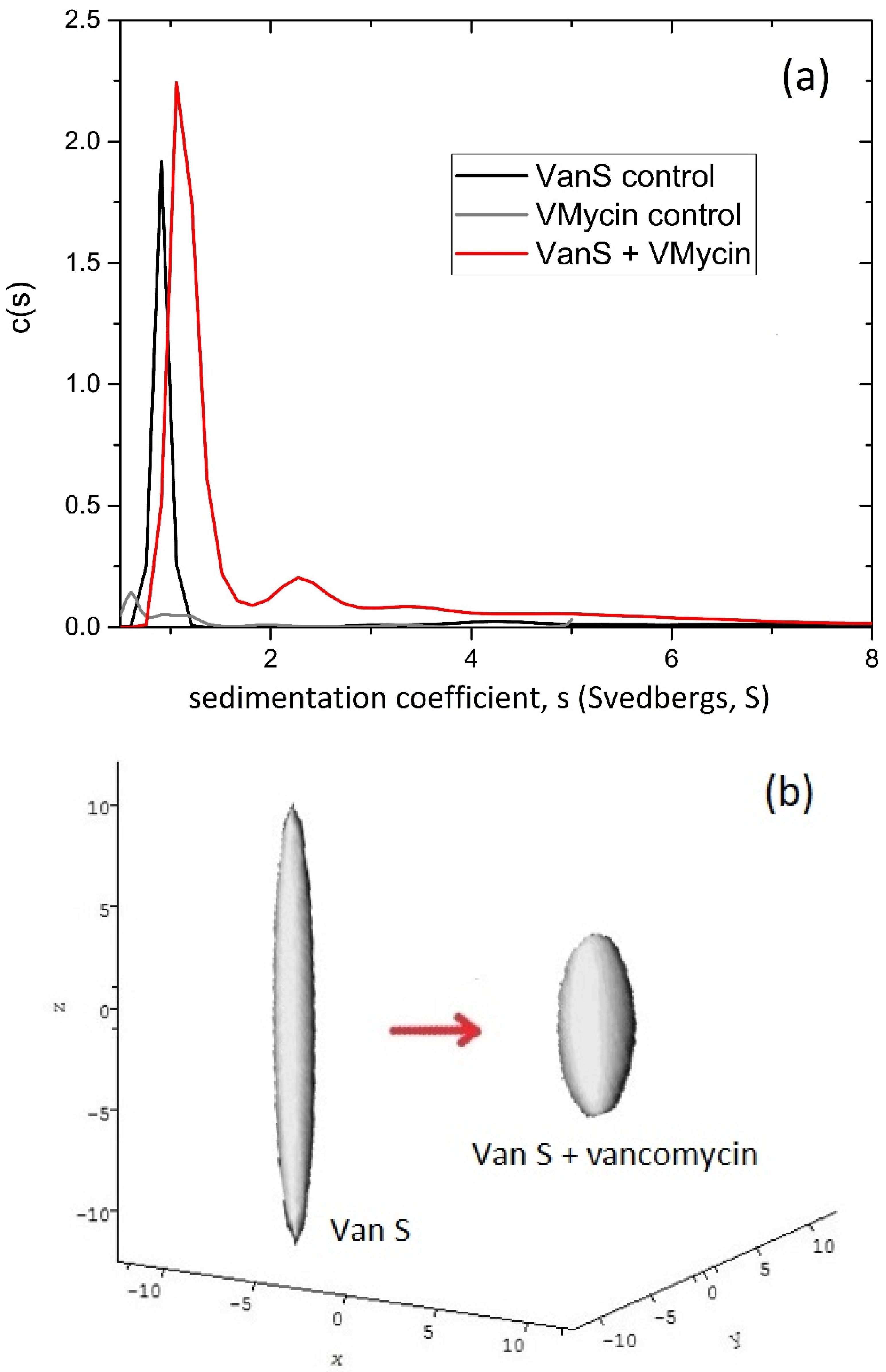
6. Enterococcal VanS—Vancomycin System
Vancomycin Dimerisation
7. Vancomycin-Mucin Interactions
Co-Sedimentation Assay for Mucin-Vancomycin Complexation
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Taylor, A.M.; Boulter, J.; Harding, S.E.; Cölfen, H.; Watts, A. Hydrodynamic properties of human erythrocyte band 3 solubilized in reduced Triton X-100. Biophys. J. 1999, 76, 2043–2055. [Google Scholar] [CrossRef] [Green Version]
- Pantaleo, A.; Ferrule, E.; Pau, M.C.; Khadjavi, A.; Mandili, G.; Mattè, A.; Spano, A.; De Franceschi, L.; Pippia, P.; Turrini, F. Band 3 erythrocyte membrane protein acts as redox stress sensor leading to its phosphorylation by p72 Syk. Oxid. Med. Cell. Longev. 2016, 2016, 6051093. [Google Scholar] [CrossRef] [Green Version]
- Phillips-Jones, M.K.; Channell, G.; Kelsall, C.J.; Hughes, C.S.; Ashcroft, A.E.; Patching, S.G.; Dinu, V.; Gillis, R.B.; Adams, G.G.; Harding, S.E. Hydrodynamics of the VanA-type VanS histidine kinase: An extended solution conformation and first evidence for interactions with vancomycin. Sci. Rep. 2017, 7, 46180. [Google Scholar] [CrossRef]
- Phillips-Jones, M.K.; Lithgo, R.; Dinu., V.; Gillis, R.B.; Harding, J.E.; Adams, G.G.; Harding, S.E. Full hydrodynamic reversibility of the weak dimerization of vancomycin and elucidation of its interaction with VanS monomers at clinical concentration. Sci. Rep. 2017, 7, 12697. [Google Scholar] [CrossRef]
- Phillips-Jones, M.K.; Harding, S.E. Antimicrobial resistance (AMR) nanomachines – mechanisms for fluoroquinolone and glycopeptide recognition, efflux and deactivation. Biophys. Rev. 2018, 10, 347–362. [Google Scholar] [CrossRef] [Green Version]
- Casey, J.R.; Reithmeier. Analysis of the oligomeric state of band 3, the anion transport protein of the erythrocyte membrane, by size exclusion chromatography: Oligomeric stability and origin of heterogeneity. J. Biol. Chem. 1991, 266, 15726–15737. [Google Scholar] [CrossRef]
- Chayen, N.E.; Saridakis, E. Protein crystallization: From purified protein to diffraction-quality crystal. Nat Methods. 2008, 5, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.R.; Thornton, J.M.; Turnell, W.G. An ellipsoidal approximation of protein shape. J. Mol. Graph. 1983, 1, 30–38. [Google Scholar] [CrossRef]
- Roy, A. Membrane preparation and solubilization. Meth. Enzymol. 2015, 557, 45–56. [Google Scholar]
- Cölfen, H.; Boulter, J.M.; Harding, S.E.; Watts, A. Ultracentrifugation studies on the transmembrane domain of the human erythrocyte anion transporter Band 3 in the detergent C12E8. Eur. Biophys. J. 1998, 27, 651–655. [Google Scholar]
- Cölfen, H.; Harding, S.E.; Boulter, J.M.; Watts, A. Hydrodynamic examination of the dimeric cytoplasmic domain of the human erythrocyte anion transporter, band 3. Biophys. J. 1996, 71, 1611–1615. [Google Scholar] [CrossRef] [Green Version]
- Ebel, C. Sedimentation velocity to characterize surfactants and solubilized membrane proteins. Methods 2011, 54, 56–66. [Google Scholar] [CrossRef]
- le Maire, M.; Arnou, B.; Olesen, C.; Georgin, D.; Ebel, C.; Møller, J.V. Gel chromatography and analytical ultracentrifugation to determine the extent of detergent binding and aggregation, and Stokes radius of membrane proteins using sarcoplasmic reticulum Ca2+ -ATPase as an example. Nat. Protoc. 2008, 3, 1782–1795. [Google Scholar] [CrossRef] [PubMed]
- Doran, J.D.; Mohanty, A.K.; Fox, T. Resolving the challenge of measuring ligand binding to membrane proteins by combining analytical ultracentrifugation and light scattering photometry. J. Pharm. Sci. 2012, 101, 92–101. [Google Scholar] [CrossRef]
- Maslennikov, I.; Krupa, M.; Dickson, C.; Esquivies, L.; Blain, K.; Kefala, G.; Choe, S.; Kwiatkowski, W. Characterization of protein detergent complexes by NMR, light scattering, and analytical ultracentrifugation. J. Struct. Fluid Genom. 2009, 10, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Roy, A.; Wang, K.; Schaack, B.; Schuck, P.; Breyton, C.; Ebel, C. AUC and small-angle scattering for membrane proteins. Meth. Enzymol. 2015, 562, 257–286. [Google Scholar]
- Fleming, K.G. Determination of membrane protein molecular weight using sedimentation equilibrium analytical ultracentrifugation. Curr. Protoc. Prot. Sci. 2008, 53, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Burgess, N.K.; Stanley, A.M.; Fleming, K.G. Determination of membrane protein molecular weights and association equilibrium constants using sedimentation equilibrium and sedimentation velocity. Meth. Cell Biol. 2008, 84, 181–211. [Google Scholar]
- Surya, W.; Torres, J. Sedimentation equilibrium of a small oligomer-forming membrane protein: Effect of histidine protonation on pentameric stability. J. Vis. Exp. 2015, e52404. [Google Scholar] [CrossRef] [Green Version]
- Howlett, G.J. Sedimentation analysis of membrane proteins. In Analytical Ultracentrifugation in Biochemistry and Polymer Science; Harding, S.E., Rowe, A.J., Horton, J.C., Eds.; Royal Society of Chemistry: Cambridge, UK, 1992; pp. 470–483. [Google Scholar]
- Harding, S.E. The Svedberg Lecture 2017. From nano to micro: The huge dynamic range of the analytical ultracentrifuge for characterising the sizes, shapes and interactions of molecules and assemblies in Biochemistry and Polymer Science. Eur. Biophys. J. 2018, 47, 697–707. [Google Scholar] [CrossRef] [Green Version]
- Bordin, L.; Brunati, A.M.; Donella-Deana, A.; Baggio, B.; Toninello, A.; Clari, G. Band 3 is an anchor protein and a target for SHP-2 tyrosine phosphatase in human erythrocytes. Blood 2002, 100, 276–282. [Google Scholar] [CrossRef] [Green Version]
- Harrison, M.L.; Isaacson, C.C.; Burg, D.L.; Geahlen, R.L.; Low, P.S. Phosphorylation of human erythrocyte band 3 by endogenous p72syk. J. Biol. Chem. 1994, 269, 955–959. [Google Scholar] [CrossRef]
- Pantaleo, A.; De Franchesci, L.; Ferru, E.; Vono, R.; Turrini, F. Current knowledge about the functional roles of phosphorylative changes of membrane proteins in normal and diseased red cells. J. Proteom. 2010, 73, 445–455. [Google Scholar] [CrossRef]
- Dinu, V.; Lu, Y.; Weston, N.; Lithgo, R.; Coupe, H.; Channell, G.; Adams, G.G.; Torcello Gómez, A.; Sabater, C.; Mackie, A.; et al. The antibiotic vancomycin induces complexation and aggregation of gastrointestinal and submaxillary mucins. Sci. Rep. 2020, 10, 960. [Google Scholar] [CrossRef]
- Harding, S.E.; Rowe, A.J.; Horton, J.C. (Eds.) Analytical Ultracentrifugation in Biochemistry and Polymer Science; Royal Society of Chemistry: Cambridge, UK, 1992. [Google Scholar]
- Harding, S.E. Determination of absolute molecular weights using sedimentation equilibrium analytical ultracentrifugation. Meth. Mol. Biol. 1994, 22, 75–84. [Google Scholar]
- Schuck, P.; Zhao, H.; Brautigam, C.A.; Ghirlando, R. Basic Principles of Analytical Ultracentrifugation; CRC Press: Boca Raton, FL, USA, 2020. [Google Scholar]
- Harding, S.E.; Rowe, A.J. Insight into protein–protein interactions from analytical ultracentrifugation. Biochem. Soc. Trans. 2010, 38, 901–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, S.E.; Winzor, D.J. Sedimentation velocity analytical ultracentrifugation. In Protein–Ligand Interactions: Hydrodynamics and Calorimetry; Harding, S.E., Chowdhry, B.Z., Eds.; Oxford University Press: Oxford, UK, 2001; pp. 75–103. [Google Scholar]
- Garcıa de la Torre, J.; Harding, S.E. Hydrodynamic modelling of protein conformation in solution: ELLIPS and HYDRO. Biophys. Rev. 2013, 5, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casey, J.R.; Lieberman, D.M.; Reithmeier, R.A.F. Purification and characterization of Band 3 protein. Meth. Enzymol. 1989, 173, 494–512. [Google Scholar]
- Chaptal, V.; Delolme, F.; Kilburg, A.; Magnard, S.; Montigny, C.; Picard, M.; Prier, C.; Monticelli, L.; Bornert, O.; Agez, M.; et al. Quantification of detergents complexed with membrane proteins. Sci. Rep. 2017, 7, 41751. [Google Scholar] [CrossRef]
- Butler, P.J.P.; Tate, C.G. Correcting for the buoyancy of macromolecules; density increments and apparent partial specific volumes with particular reference to the study of membrane proteins. In Modern Analytical Ultracentrifugation: Techniques and Methods; Scott, D.J., Harding, S.E., Rowe, A.J., Eds.; Royal Society of Chemistry: Cambridge, UK, 2005; pp. 133–151. [Google Scholar]
- Dam, J.; Schuck, P. Determination of sedimentation coefficient distributions by direct modeling of the sedimentation boundary with Lamm equation solutions. Meth. Enzymol. 2004, 384, 185–212. [Google Scholar]
- Harding, S.E.; Schuck, P.; Abdelhammed, A.S.; Adams, G.; Kök, M.S.; Morris, G.A. Extended Fujita approach to the molecular weight distribution of polysaccharides and other polymer systems. Methods 2011, 54, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuck, P.; Gillis, R.B.; Besong, D.; Almutairi, F.; Adams, G.G.; Rowe, A.J.; Harding, S.E. SEDFIT-MSTAR: Molecular weight and molecular weight distribution analysis of polymers by sedimentation equilibrium in the ultracentrifuge. Analyst 2014, 139, 79–92. [Google Scholar] [CrossRef] [Green Version]
- Gillis, R.B.; Adams, G.G.; Heinze, T.; Nikolajski, M.; Harding, S.E.; Rowe, A.J. MultiSig: A new high-precision approach to the analysis of complex biomolecular systems. Eur. Biophys. J. 2013, 42, 777–786. [Google Scholar] [CrossRef] [Green Version]
- Dam, J.; Schuck, P. Sedimentation velocity analysis of heterogeneous protein-protein interactions: Sedimentation coefficient distributions c(s) and asymptotic boundary profiles from Gilbert-Jenkins theory. Biophys. J. 2005, 89, 651–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rallison, J.M.; Harding, S.E. Excluded volume for pairs of triaxial ellipsoids at dominant Brownian motion. J. Coll. Int. Sci. 1985, 103, 284–289. [Google Scholar] [CrossRef]
- Harding, S.E.; Horton, J.C.; Winzor, D.J. COVOL: An answer to your thermodynamic non-ideality problems? Biochem. Soc. Trans. 1998, 26, 737–740. [Google Scholar] [CrossRef] [Green Version]
- Harding, S.E.; Horton, J.C.; Jones, S.; Thornton, J.M.; Winzor, D.J. COVOL: An interactive program for evaluating second virial coefficients from the triaxial shape or dimensions of rigid macromolecules. Biophys. J. 1999, 76, 2432–2438. [Google Scholar] [CrossRef] [Green Version]
- Creeth, J.M.; Harding, S.E. Some observations on a new type of point average molecular weight. J. Biochem. Biophys. Methods 1982, 7, 25–34. [Google Scholar] [CrossRef]
- Harding, S.E.; Horton, J.C.; Morgan, P.J. MSTAR: A FORTRAN algorithm for the model independent molecular weight analysis of macromolecules using low speed or high speed sedimentation equilibrium. In Analytical Ultracentrifugation in Biochemistry and Polymer Science; Harding, S.E., Rowe, A.J., Horton, J.C., Eds.; Royal Society of Chemistry: Cambridge, UK, 1992; pp. 275–294. [Google Scholar]
- Cölfen, H.; Harding, S.E. MSTARA and MSTARI: Interactive PC algorithms for simple, model independent evaluation of sedimentation equilibrium data. Eur. Biophys. J. 2006, 333–346. [Google Scholar] [CrossRef]
- Reithmeier, R.A.F.; Casey, J.R.; Kalli, A.C.; Sansom, M.S.P.; Alguel, Y.; Iwata, S. Band 3, the human red cell chloride/bicarbonate anion exchanger (AE1, SLC4A1), in a structural context. Biochim. Biophys. Acta-Biomembr. 2016, 1858, 1507–1532. [Google Scholar] [CrossRef]
- Durchschlag, H.; Zipper, P. Calculation of the partial volume of organic compounds and polymers. Prog. Colloid Polym. Sci. 1994, 94, 20–39. [Google Scholar]
- Kratky, O.; Leopold, H.; Stabinger, H. The determination of the partial specific volume of proteins by the mechanical oscillator technique. Meth. Enzymol. 1973, 27, 98–110. [Google Scholar]
- Roark, D.E.; Yphantis, D.A. Studies of self-associating systems by equilibrium ultracentrifugation. Ann. N. Y. Acad. Sci. 1969, 164, 245–278. [Google Scholar] [CrossRef]
- Nikolajski, M.; Adams, G.G.; Gillis, R.B.; Besong, D.T.; Rowe, A.J.; Heinze, T.; Harding, S.E. Protein–like fully reversible tetramerisation and super-association of an aminocellulose. Sci. Rep. 2014, 4, 3861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, S.E.; Horton, J.C.; Cölfen, H. The ELLIPS suite of macromolecular conformation algorithms. Eur. Biophys. J. 1997, 25, 347–359. [Google Scholar] [CrossRef]
- Ma, P.; Phillips-Jones, M.K. Membrane sensor histidine kinases: Insights from structural, ligand and inhibitor studies of full-length proteins and sSignalling domains for antibiotic discovery. Molecules 2021, 26, 5110. [Google Scholar] [CrossRef] [PubMed]
- Kegeles, G.; Rao, M.S.N. Ultracentrifugation of chemically reacting systems. J. Am. Chem. Soc. 1958, 80, 5721–5724. [Google Scholar] [CrossRef]
- Kim, H.; Deonier, R.C.; Williams, J.W. The investigation of self-association reactions by equilibrium ultracentrifugation. Chem. Rev. 1977, 77, 659–690. [Google Scholar] [CrossRef]
- Chun, T.; Corfield, A.P.; Dinu, V.; Harding, S.E. Oligomerisation of teicoplanin and its interactions with gasterointestinal mucin studied by analytical ultracentrifugation. 2021; Manuscript in preparation. [Google Scholar]
- Hussain, R.; Harding, S.E.; Hughes, C.S.; Ma, P.; Patching, S.G.; Edara, S.; Siligardi, G.; Henderson, P.J.; Phillips-Jones, M.K. Purification of bacterial membrane sensor kinases and biophysical methods for determination of their ligand and inhibitor interactions. Biochem. Soc. Trans. 2016, 44, 810–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciunas, L.J.; Porter, N.; Lee, P.J.; Gupta, K.; Loll, P.J. Structures of full-length VanR from Streptomyces coelicolor in both the inactive and activated states. Acta Crystallogr. Sect. D. Struct. Biol. 2021, 77, 1027–1039. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CD Domain | TMD Domain | Intact Band 3 | |
|---|---|---|---|
| Solvent: | pH8 Tris Buffer | C12E8 | Reduced Triton |
| detergent binding 1 | - | 0.945 g/g | 0.77 g/g |
| partial specific volumes 2: protein detergent complex | 0.740 mL/g | 0.7642 mL/g (0.9732 ± 0.0003) mL/g 0.866 mL/g | (0.740 ± 0.007) mL/g (0.9732 ± 0.0003) mL/g (0.842 ± 0.004) mL/g |
| monomer molecular mass M1 (Da) | 40,000 | 122,800 | 200,000 |
| molecular mass Mw in solution (Da) | 80,000 (dimer) | 250,000 (dimer) | 400,000–800,000 (dimer-tetramer) + some hexamer/higher order associations |
| dissociation constant Kd (μM) | 2.8 ± 0.5 | <1 | |
| sedimentation coefficient so20,w (S) | 3.74 ± 0.07 | 4.94 ± 0.07 | dimer = (6.9 ± 0.1) S tetramer = (10.6 ± 0.7) S |
| translational frictional ratio f/fo | 1.7 ± 0.2 | 1.29 ± 0.02 | dimer = (1.55 ± 0.080) tetramer = (1.68 ± 0.28) |
| Perrin P shape parameter (based on hydration = 0.2) | 1.5 | 1.15 ± 0.05 | Pdimer = 1.44 ± 0.08 Ptetramer = 1.49 ± 0.21 |
| axial ratio a/b | ~10 | ~3.5 | dimer a/b ~7 tetramer a/b ~10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harding, S.E. Analytical Ultracentrifugation as a Matrix-Free Probe for the Study of Kinase Related Cellular and Bacterial Membrane Proteins and Glycans. Molecules 2021, 26, 6080. https://doi.org/10.3390/molecules26196080
Harding SE. Analytical Ultracentrifugation as a Matrix-Free Probe for the Study of Kinase Related Cellular and Bacterial Membrane Proteins and Glycans. Molecules. 2021; 26(19):6080. https://doi.org/10.3390/molecules26196080
Chicago/Turabian StyleHarding, Stephen E. 2021. "Analytical Ultracentrifugation as a Matrix-Free Probe for the Study of Kinase Related Cellular and Bacterial Membrane Proteins and Glycans" Molecules 26, no. 19: 6080. https://doi.org/10.3390/molecules26196080
APA StyleHarding, S. E. (2021). Analytical Ultracentrifugation as a Matrix-Free Probe for the Study of Kinase Related Cellular and Bacterial Membrane Proteins and Glycans. Molecules, 26(19), 6080. https://doi.org/10.3390/molecules26196080