
A Comprehensive Computational Screening of Phytochemicals Derived from Saudi Medicinal Plants against Human CC Chemokine Receptor 7 to Identify Potential Anti-Cancer Therapeutics


Abstract
:1. Introduction
2. Results and Discussion
2.1. Library Filtration
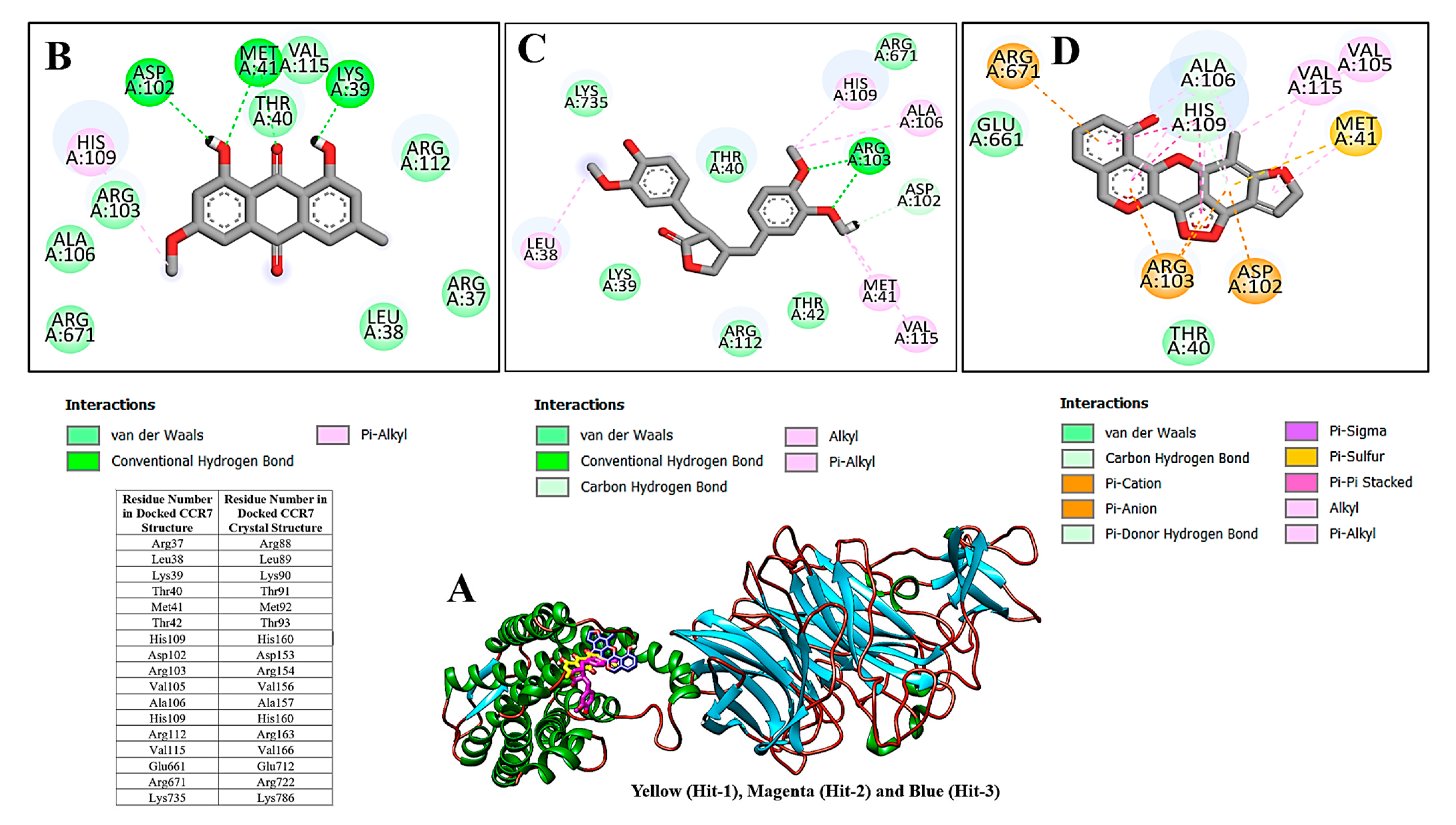
2.2. Molecular Docking Analysis
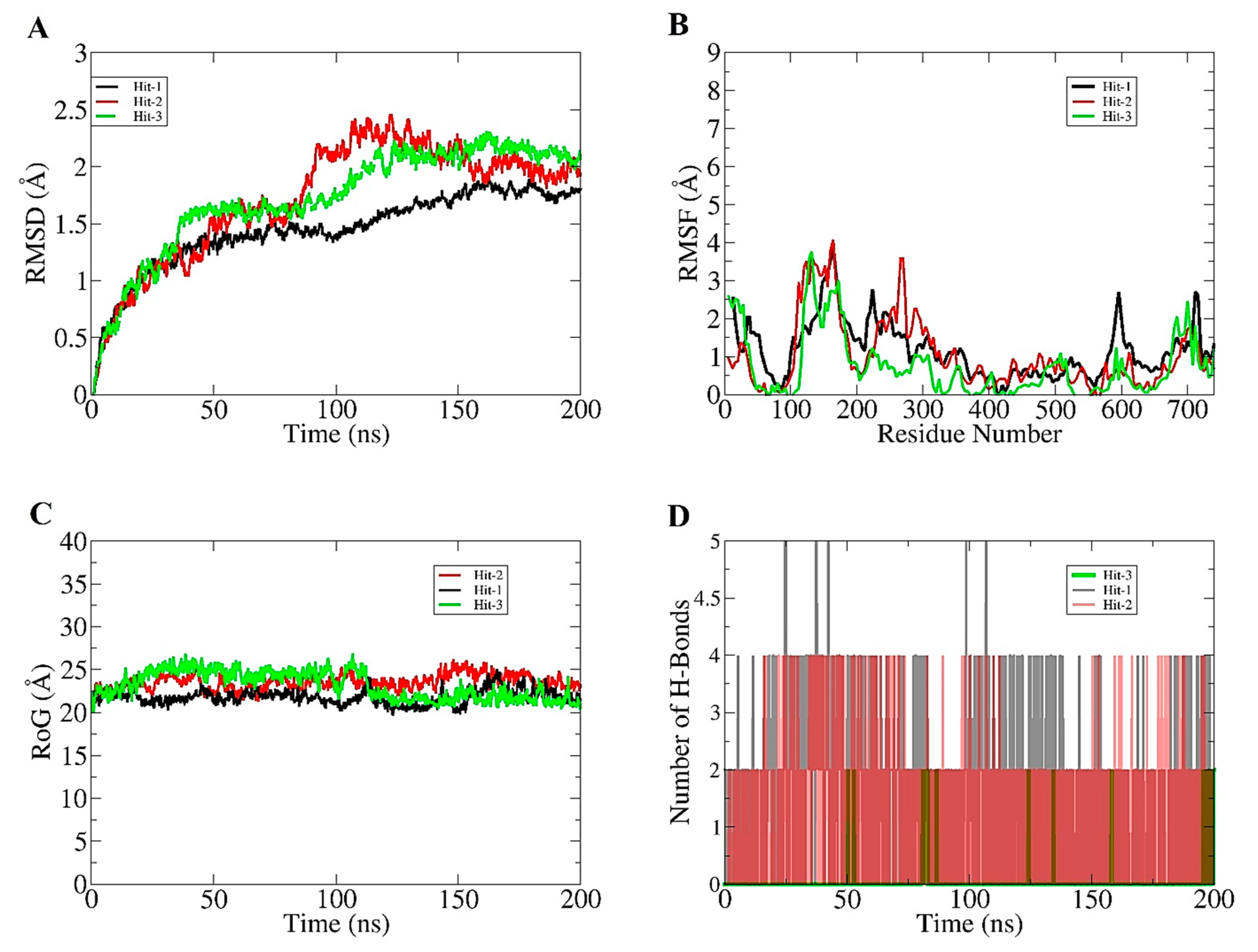
2.3. Molecular Dynamics Simulation
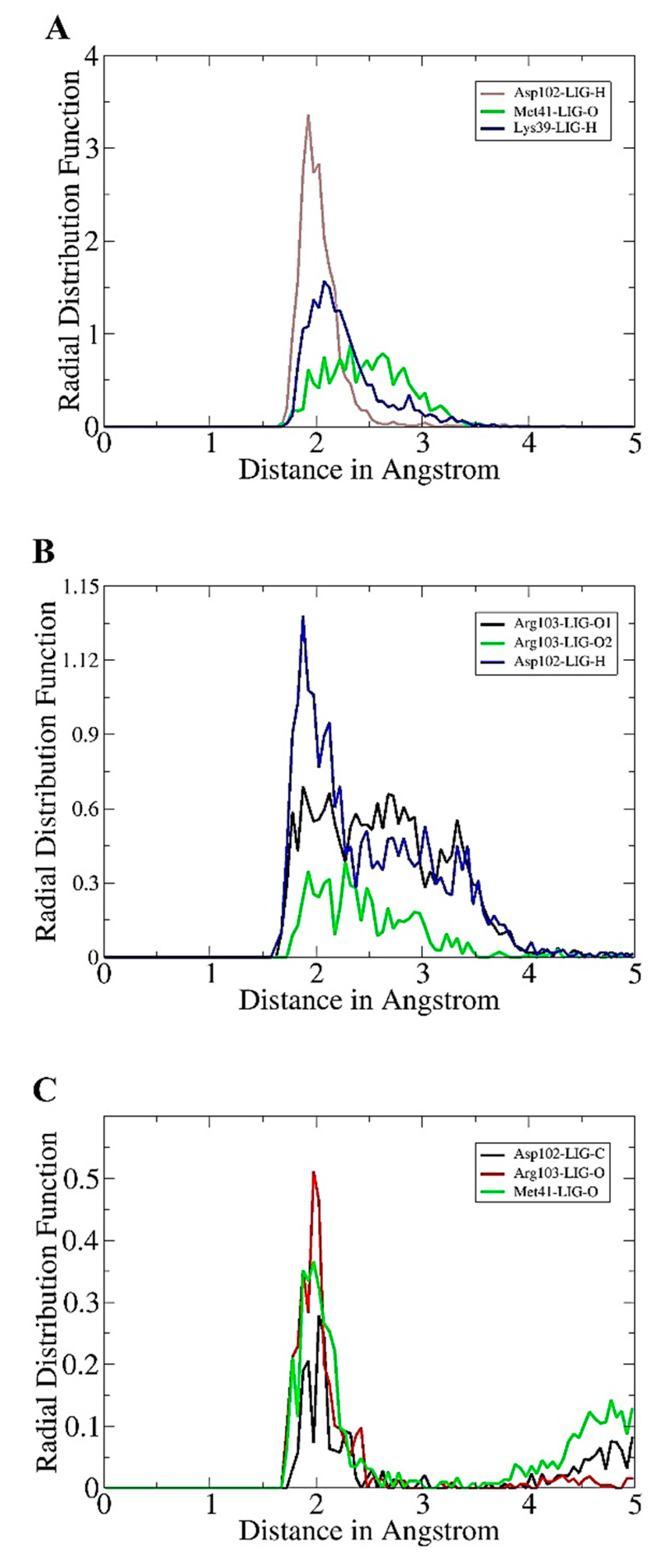
2.4. Radial Distribution Plotting
2.5. MM/GBPBSA Analysis
2.6. Enzyme Hotspot Residues
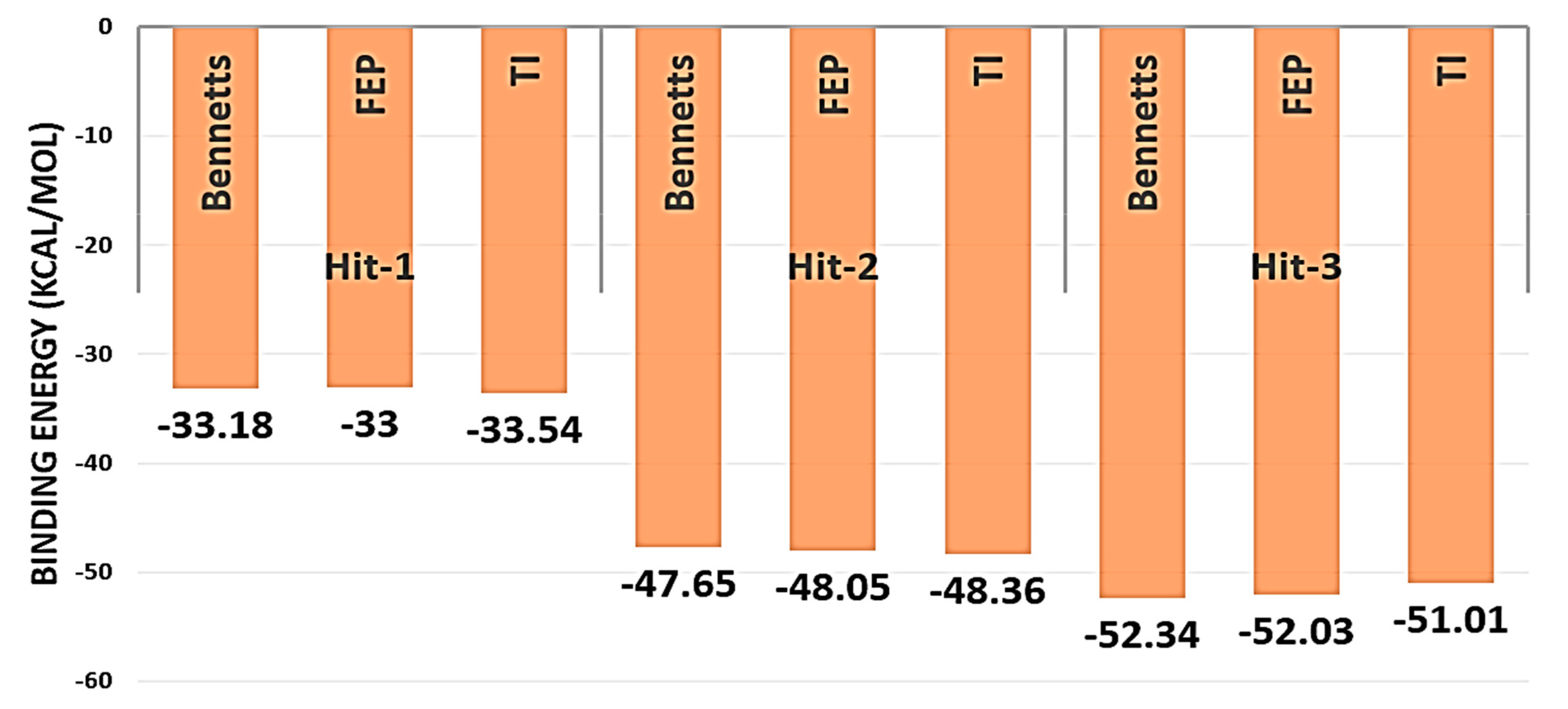
2.7. WaterSwap Analysis
2.8. Entropy Calculations
2.9. Compounds Pharmacokinetics Predictions
2.10. Alanine Scanning
3. Materials and Methods
3.1. Phytochemicals Library Preparation
3.2. Structure Based Virtual Screening
3.3. Molecular Dynamics Simulation
3.4. MM/GBPBSA and Alanine Scanning Analysis
3.5. WaterSwap Analysis
3.6. In Silico Pharmacokinetics, Medicinal Chemistry and Toxicity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Jaeger, K.; Bruenle, S.; Weinert, T.; Guba, W.; Muehle, J.; Miyazaki, T.; Weber, M.; Furrer, A.; Haenggi, N.; Tetaz, T.; et al. Structural basis for allosteric ligand recognition in the human CC chemokine receptor 7. Cell 2019, 178, 1222–1230. [Google Scholar] [CrossRef] [Green Version]
- Jin, T.; Xu, X.; Hereld, D. Chemotaxis, chemokine receptors and human disease. Cytokine 2008, 44, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Förster, R.; Davalos-Misslitz, A.C.; Rot, A. CCR7 and its ligands: Balancing immunity and tolerance. Nat. Rev. Immunol. 2008, 8, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 2004, 4, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Pron, B.; Boumaila, C.; Jaubert, F.; Berche, P.; Milon, G.; Geissmann, F.; Gaillard, J.-L. Dendritic cells are early cellular targets of Listeria monocytogenes after intestinal delivery and are involved in bacterial spread in the host. Cell. Microbiol. 2001, 3, 331–340. [Google Scholar] [CrossRef]
- Balkwill, F.R. The chemokine system and cancer. J. Pathol. 2012, 226, 148–157. [Google Scholar] [CrossRef]
- Günther, K.; Leier, J.; Henning, G.; Dimmler, A.; Weißbach, R.; Hohenberger, W.; Förster, R. Prediction of lymph node metastasis in colorectal carcinoma by expressionof chemokine receptor CCR7. Int. J. Cancer 2005, 116, 726–733. [Google Scholar] [CrossRef]
- Müller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef]
- Chang, J.W.; DIng, Y.; Tahir Ul Qamar, M.; Shen, Y.; Gao, J.; Chen, L.L. A deep learning model based on sparse auto-encoder for prioritizing cancer-related genes and drug target combinations. Carcinogenesis 2019, 40, 624–632. [Google Scholar] [CrossRef]
- Zlotnik, A.; Burkhardt, A.M.; Homey, B. Homeostatic chemokine receptors and organ-specific metastasis. Nat. Rev. Immunol. 2011, 11, 597–606. [Google Scholar] [CrossRef]
- Cunningham, H.D.; Shannon, L.A.; Calloway, P.A.; Fassold, B.C.; Dunwiddie, I.; Vielhauer, G.; Zhang, M.; Vines, C.M. Expression of the CC chemokine receptor 7 mediates metastasis of breast cancer to the lymph nodes in mice. Transl. Oncol. 2010, 3, 354–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Mishan, M.A.; Ahmadiankia, N.; Bahrami, A.R. CXCR4 and CCR7: Two eligible targets in targeted cancer therapy. Cell Biol. Int. 2016, 40, 955–967. [Google Scholar] [CrossRef]
- Sussman, J.L.; Lin, D.; Jiang, J.; Manning, N.O.; Prilusky, J.; Ritter, O.; Abola, E.E. Protein Data Bank (PDB): Database of three-dimensional structural information of biological macromolecules. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 1078–1084. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; MacKerell, A.D. Computer-aided drug design methods. In Antibiotics; Springer: Berlin/Heidelberg, Germany, 2017; pp. 85–106. [Google Scholar]
- Altharawi, A.; Ahmad, S.; Alamri, M.A.; ul Qamar, M.T. Structural insight into the binding pattern and interaction mechanism of chemotherapeutic agents with Sorcin by docking and molecular dynamic simulation. Colloids Surf. B Biointerfaces 2021, 208, 112098. [Google Scholar] [CrossRef]
- Arif, R.; Ahmad, S.; Mustafa, G.; Mahrosh, H.S.; Ali, M.; Tahir ul Qamar, M.; Dar, H.R. Molecular Docking and Simulation Studies of Antidiabetic Agents Devised from Hypoglycemic Polypeptide-P of Momordica charantia. Biomed. Res. Int. 2021, 2021. [Google Scholar] [CrossRef] [PubMed]
- Alamri, M.A.; Altharawi, A.; Alabbas, A.B.; Alossaimi, M.A.; Alqahtani, S.M. Structure-based virtual screening and molecular dynamics of phytochemicals derived from Saudi medicinal plants to identify potential COVID-19 therapeutics. Arab. J. Chem. 2020, 13, 7224–7234. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Bensky, D.; Clavey, S.; Stõger, E. Materia medica. Chin. Herb. Med. 2004, 1, 3–6. [Google Scholar]
- He, Y.; Fan, Q.; Cai, T.; Huang, W.; Xie, X.; Wen, Y.; Shi, Z. Molecular mechanisms of the action of Arctigenin in cancer. Biomed. Pharmacother. 2018, 108, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Kuzmanic, A.; Zagrovic, B. Determination of ensemble-average pairwise root mean-square deviation from experimental B-factors. Biophys. J. 2010, 98, 861–871. [Google Scholar] [CrossRef] [Green Version]
- Haq, F.U.; Abro, A.; Raza, S.; Liedl, K.R.; Azam, S.S. Molecular dynamics simulation studies of novel β-lactamase inhibitor. J. Mol. Graph. Model. 2017, 74, 143–152. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Abro, A.; Azam, S.S. Binding free energy based analysis of arsenic (+3 oxidation state) methyltransferase with S-adenosylmethionine. J. Mol. Liq. 2016, 220, 375–382. [Google Scholar] [CrossRef]
- Woods, C.J.; Malaisree, M.; Michel, J.; Long, B.; McIntosh-Smith, S.; Mulholland, A.J. Rapid decomposition and visualisation of protein-ligand binding free energies by residue and by water. Faraday Discuss. 2014, 169, 477–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, W.; Ashfaq, U.A.; Aslam, S.; Saif, S.; Aslam, T.; Tusleem, K.; Maryam, A.; ul Qamar, M.T. Anticancer screening of medicinal plant phytochemicals against Cyclin-Dependent Kinase-2 (CDK2): An in-silico approach. Adv. Life Sci. 2017, 4, 113–119. [Google Scholar]
- Rehan Khalid, R.; ul Qamar, M.; Maryam, A.; Ashique, A.; Anwar, F.; Geesi, M.H.; Siddiqi, A.R. Comparative Studies of the Dynamics Effects of BAY60-2770 and BAY58-2667 Binding with Human and Bacterial H-NOX Domains. Molecules 2018, 23, 2141. [Google Scholar] [CrossRef] [Green Version]
- Riaz, M.; Ashfaq, U.A.; Qasim, M.; Yasmeen, E.; Qamar, M.T.U.; Anwar, F. Screening of medicinal plant phytochemicals as natural antagonists of p53--MDM2 interaction to reactivate p53 functioning. Anticancer Drugs 2017, 28, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. In Chemical Biology; Springer: Berlin/Heidelberg, Germany, 2015; pp. 243–250. [Google Scholar]
- Suleman, M.; ul Qamar, M.T.; Shoaib Saleem, S.A.; Ali, S.S.; Khan, H.; Akbar, F.; Khan, W.; Alblihy, A.; Alrumaihi, F.; Waseem, M. Mutational Landscape of Pirin and Elucidation of the Impact of Most Detrimental Missense Variants That Accelerate the Breast Cancer Pathways: A Computational Modelling Study. Front. Mol. Biosci. 2021, 8, 692835. [Google Scholar] [CrossRef]
- Case, D.A.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.; Cheatham, T.; Cruzeiro, V.W.D.; Darden, T.; Duke, R.E.; Giambasu, G.; et al. Amber 2020; University of California Press: Auckland, CA, USA, 2020. [Google Scholar]
- Dickson, C.J.; Rosso, L.; Betz, R.M.; Walker, R.C.; Gould, I.R. GAFFlipid: A General Amber Force Field for the accurate molecular dynamics simulation of phospholipid. Soft Matter 2012, 8, 9617–9627. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham III, T.E.; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. The FF14SB force field. Amber 2014, 14, 29–31. [Google Scholar]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Petersen, H.G. Accuracy and efficiency of the particle mesh Ewald method. J. Chem. Phys. 1995, 103, 3668–3679. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham III, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Donohue, J. Radial Distribution Functions of Some Structures of the Polypeptide Chain. Proc. Natl. Acad. Sci. USA 1954, 40, 377–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Woods, C.J.; Malaisree, M.; Hannongbua, S.; Mulholland, A.J. A water-swap reaction coordinate for the calculation of absolute protein-ligand binding free energies. J. Chem. Phys. 2011, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.; Raza, S.; Abro, A.; Liedl, K.R.; Azam, S.S. Toward novel inhibitors against KdsB: A highly specific and selective broad-spectrum bacterial enzyme. J. Biomol. Struct. Dyn. 2019, 37, 1326–1345. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Autodock Vina Binding Free Energy (kcal/mol) |
|---|---|
 | –10.97 |
 | –9.71 |
 | –7.00 |
| Compound | MM/GBSA | ||||||
|---|---|---|---|---|---|---|---|
| ΔG Binding | ΔG Electrostatic | ΔG Bind Van Der Waals | ΔG Bind Gas Phase | ΔG Polar Solvation | ΔG Non Polar Solvation | ΔG Solvation | |
| Hit-1 | −41.36 | −30.58 | −25.67 | −56.25 | 25.17 | −10.28 | 14.89 |
| Hit-2 | −44.91 | −25.67 | −30.82 | −56.49 | 25.63 | −14.05 | 11.58 |
| Hit-3 | −19.58 | −14.58 | −18.45 | 33.03 | 22.39 | −8.94 | 13.45 |
| MM/PBSA | |||||||
| Hit-1 | −41.25 | −30.58 | −25.67 | −56.25 | 24.64 | −9.64 | 15 |
| Hit-2 | −43.66 | −25.67 | −30.82 | −56.49 | 26.47 | −13.64 | 12.83 |
| Hit-3 | −23.06 | −14.58 | −18.45 | 33.03 | 22.31 | −12.34 | 9.97 |
| Ligand/Residue | Hit-1 | Hit-2 | Hit-3 |
|---|---|---|---|
| Arg37 (Arg88) | −0.58 | −0.88 | 0.94 |
| Leu38 (Leu89) | −0.61 | −0.71 | −0.77 |
| Lys39 (Lys90) | −3.69 | −1.86 | −1.00 |
| Thr40 (Thr91) | −1.67 | −2.38 | −2.54 |
| Met41 (Met92) | −4.21 | 1.49 | −1.01 |
| Thr42 (Thr93) | 0.48 | −1.63 | −0.98 |
| His109 (His160) | 1.23 | −0.54 | −2.37 |
| Asp102(Asp153) | −5.01 | −2.74 | −0.88 |
| Arg103 (Arg154) | −2.67 | −2.51 | −2.35 |
| Val105 (Val156) | 0.21 | −0.21 | −0.62 |
| Ala106 (Ala157) | 0.42 | −0.78 | −0.55 |
| Arg112 (Arg163) | −1.62 | −0.54 | 0.24 |
| Val115 (Val166) | 0.21 | 0.41 | 0.62 |
| Glu661 (Glu712) | 1.25 | −0.58 | −3.24 |
| Arg671 (Arg722) | −1.0 | −1.50 | −1.56 |
| Lys735 (Lys786) | 2.25 | −1.36 | −0.87 |
| Property | Compounds | ||
|---|---|---|---|
| Physicochemical Properties | Hit-1 | Hit-2 | Hit-3 |
| Formula | C16H14O5 | C21H24O6 | C19H12O6 |
| Molecular weight | 286.28 g/mol | 372.41 g/mol | 336.29 g/mol |
| Num. heavy atoms | 21 | 27 | 25 |
| Num. arom. heavy atoms | 6 | 12 | 18 |
| Fraction Csp3 | 0.19 | 0.38 | 0.16 |
| Num. rotatable bonds | 1 | 7 | 0 |
| Num. H-bond acceptors | 5 | 6 | 6 |
| Num. H-bond donors | 3 | 1 | 1 |
| Molar Refractivity | 76.80 | 100.60 | 86.94 |
| TPSA | 86.99 A² | 74.22 A² | 78.11 A² |
| Lipophilicity | |||
| Consensus Log Po/w | 1.94 | 3.10 | 3.00 |
| Water Solubility | Soluble | Moderately soluble | Soluble |
| Pharmacokinetics | |||
| GI absorption | High | High | High |
| BBB permeant | No | Yes | Yes |
| P-gp substrate | No | No | Yes |
| CYP1A2 inhibitor | Yes | No | Yes |
| CYP2C19 inhibitor | No | No | No |
| CYP2C9 inhibitor | Yes | Yes | Yes |
| CYP2D6 inhibitor | No | Yes | No |
| CYP3A4 inhibitor | Yes | Yes | Yes |
| Log Kp (skin permeation) | –6.59 cm/s | –6.02 cm/s | –6.65 cm/s |
| Drug-likeness | |||
| Lipinski | Yes | Yes | Yes |
| Medicinal chemistry | |||
| PAINS | 0 alert | 0 alert | 0 alert |
| Synthetic accessibility | 3.50 | 3.43 | 4.12 |
| Toxicity | |||
| Hepatotoxicity | No | No | No |
| Skin Sensitisation | No | No | No |
| T. pyriformis toxicity | 0.316 (log ug/L) | 0.469 (log ug/L) | 0.29 (log ug/L) |
| AMES toxicity | Yes | No | Yes |
| Minnow toxicity | 1.836 (log mM) | 0.482 (log mM) | –0.137 (log mM) |
| Carcino mouse | No | No | No |
| Excretion | |||
| Total Clearance | 0.44 (log mL/min/kg) | 0.25 (log mL/min/kg) | 0.093 (log mL/min/kg) |
| Renal OCT2 substrate | No | No | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alrumaihi, F. A Comprehensive Computational Screening of Phytochemicals Derived from Saudi Medicinal Plants against Human CC Chemokine Receptor 7 to Identify Potential Anti-Cancer Therapeutics. Molecules 2021, 26, 6354. https://doi.org/10.3390/molecules26216354
Alrumaihi F. A Comprehensive Computational Screening of Phytochemicals Derived from Saudi Medicinal Plants against Human CC Chemokine Receptor 7 to Identify Potential Anti-Cancer Therapeutics. Molecules. 2021; 26(21):6354. https://doi.org/10.3390/molecules26216354
Chicago/Turabian StyleAlrumaihi, Faris. 2021. "A Comprehensive Computational Screening of Phytochemicals Derived from Saudi Medicinal Plants against Human CC Chemokine Receptor 7 to Identify Potential Anti-Cancer Therapeutics" Molecules 26, no. 21: 6354. https://doi.org/10.3390/molecules26216354
APA StyleAlrumaihi, F. (2021). A Comprehensive Computational Screening of Phytochemicals Derived from Saudi Medicinal Plants against Human CC Chemokine Receptor 7 to Identify Potential Anti-Cancer Therapeutics. Molecules, 26(21), 6354. https://doi.org/10.3390/molecules26216354