
Strategic Evaluation of the Traceless Staudinger Ligation for Radiolabeling with the Tricarbonyl Core



Abstract
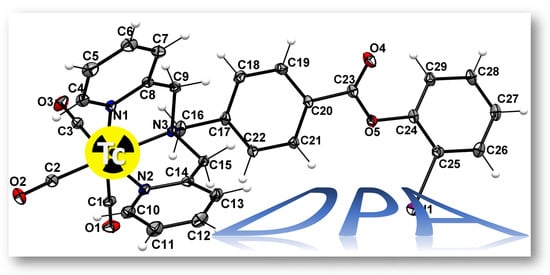
:
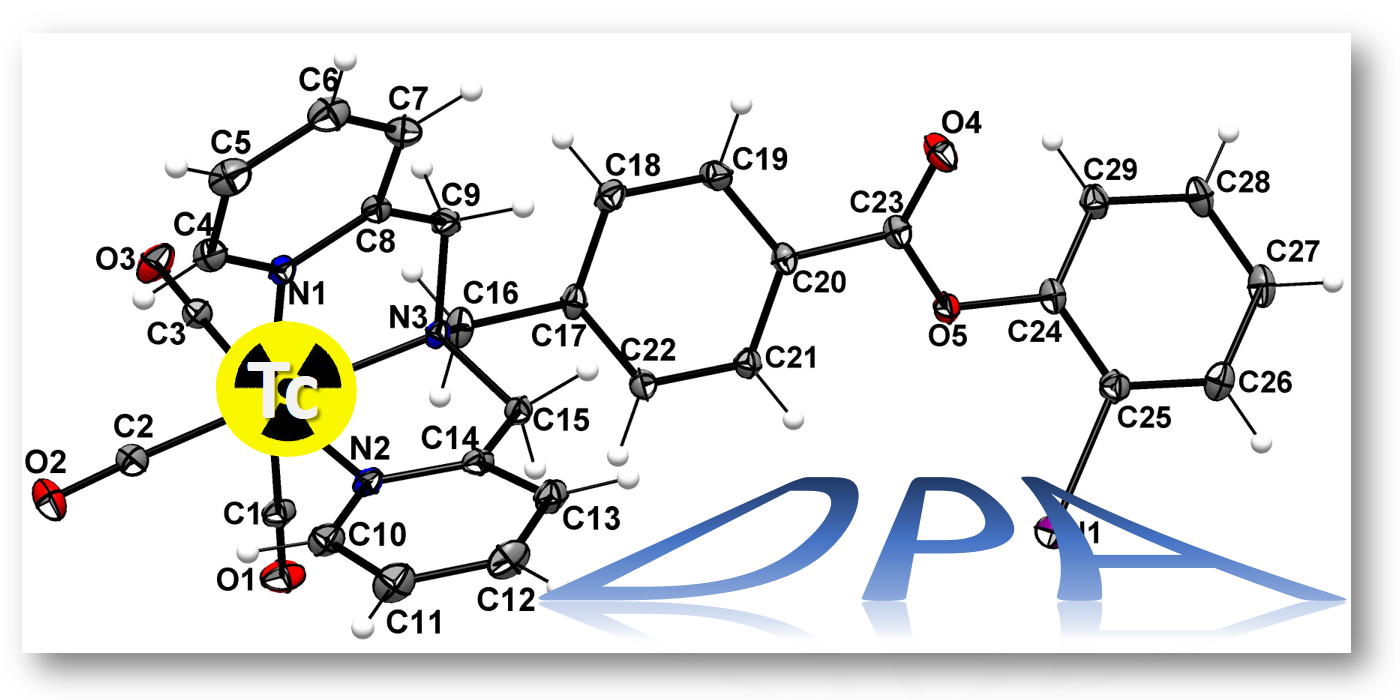{kind=link}
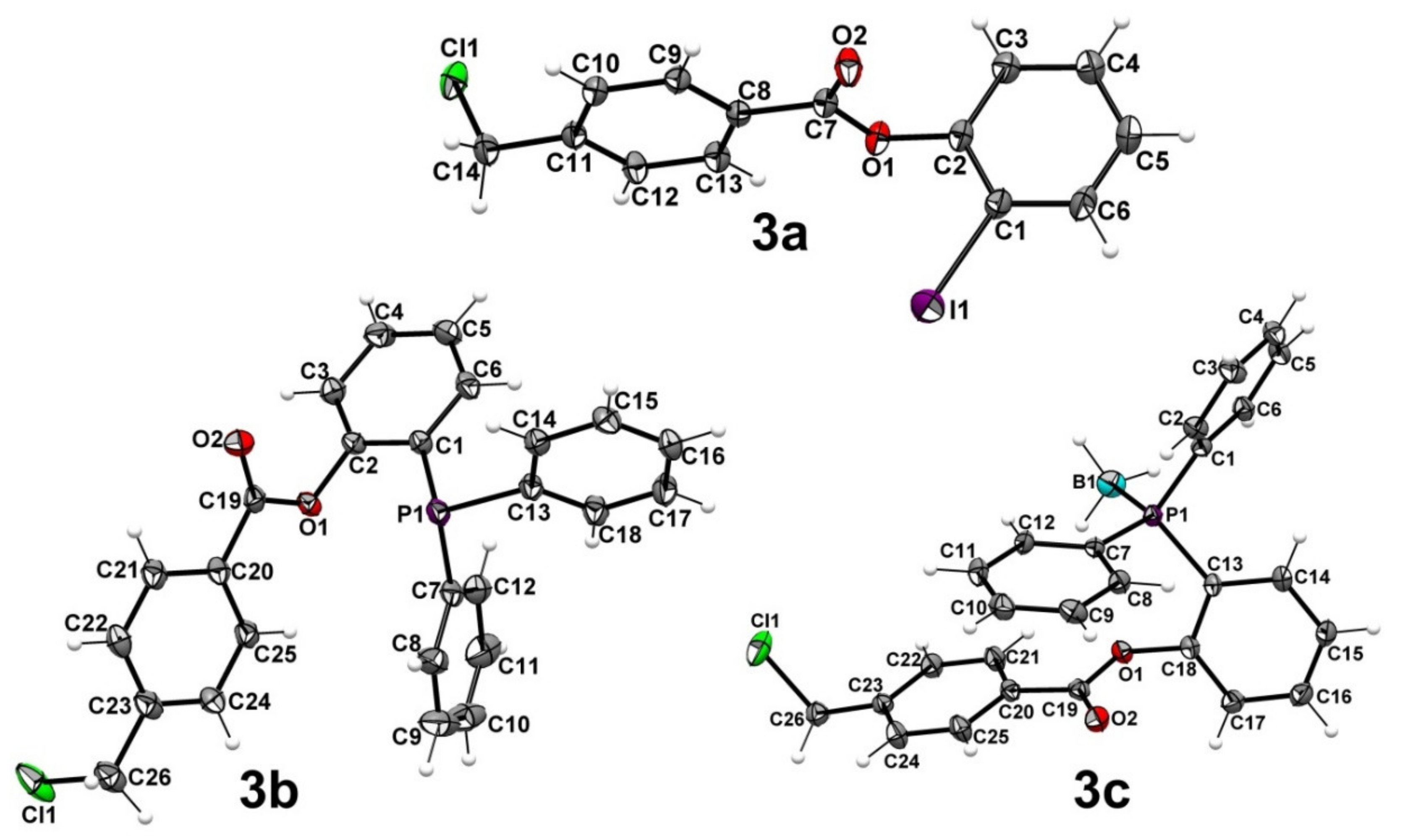{kind=link}
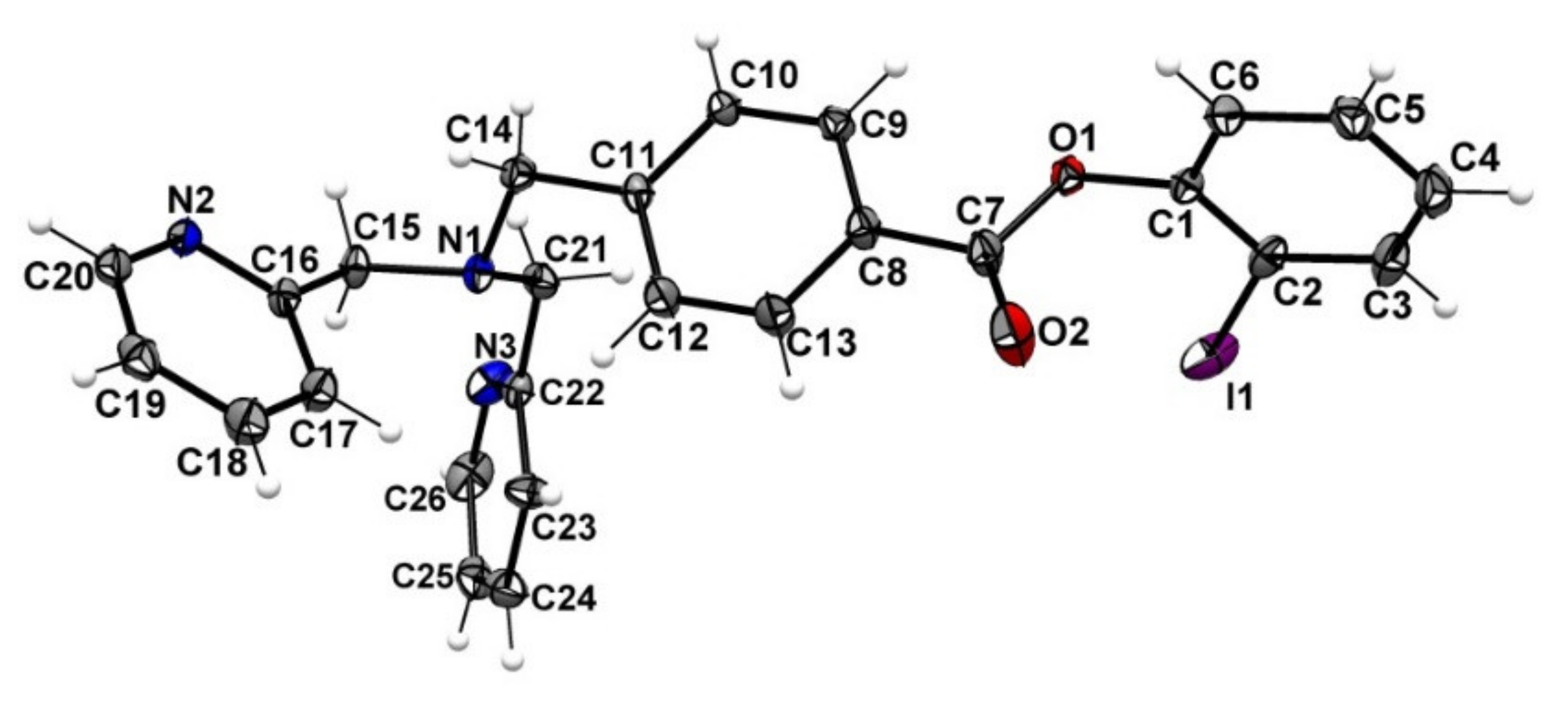{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
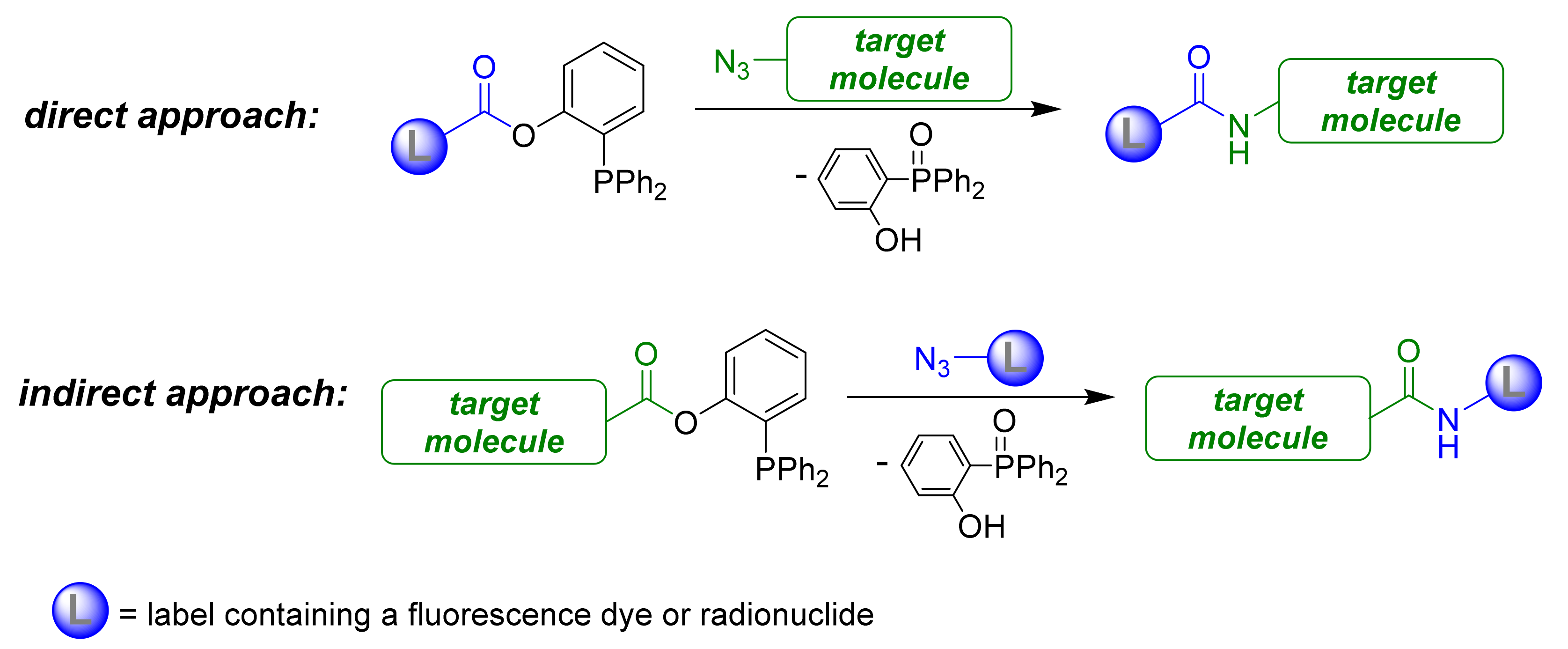
1. Introduction
2. Results and Discussion
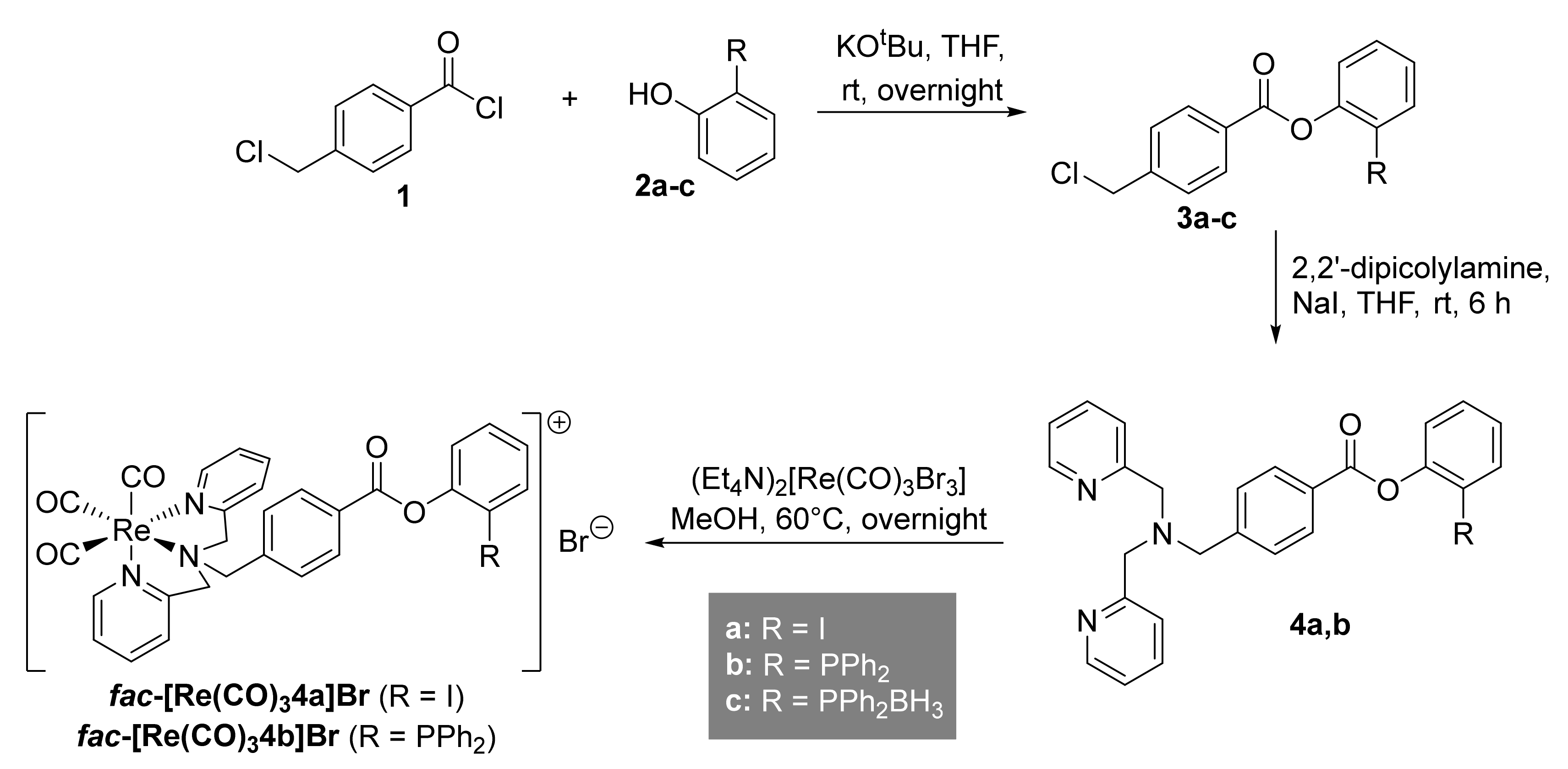
2.1. Preparation of Phosphanes
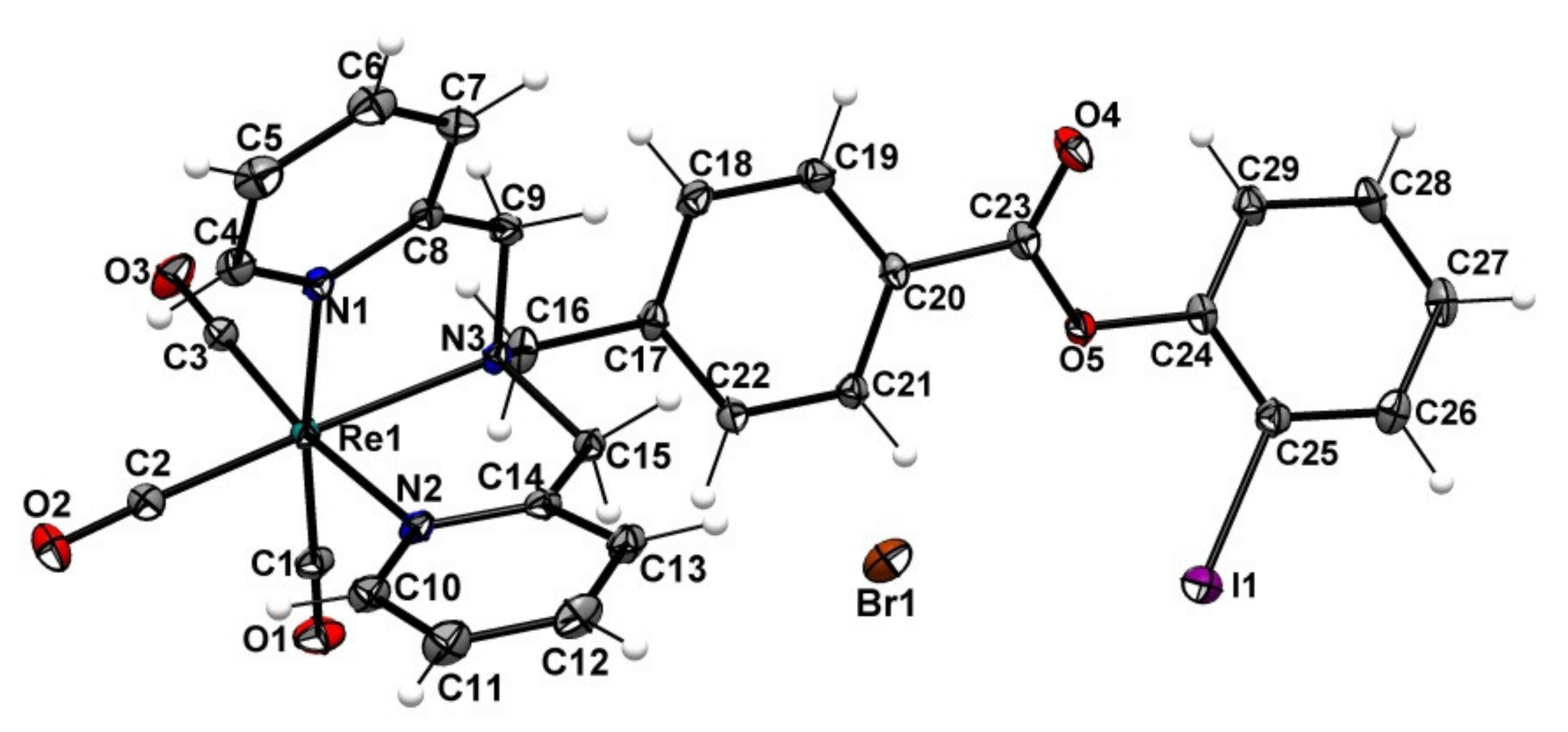
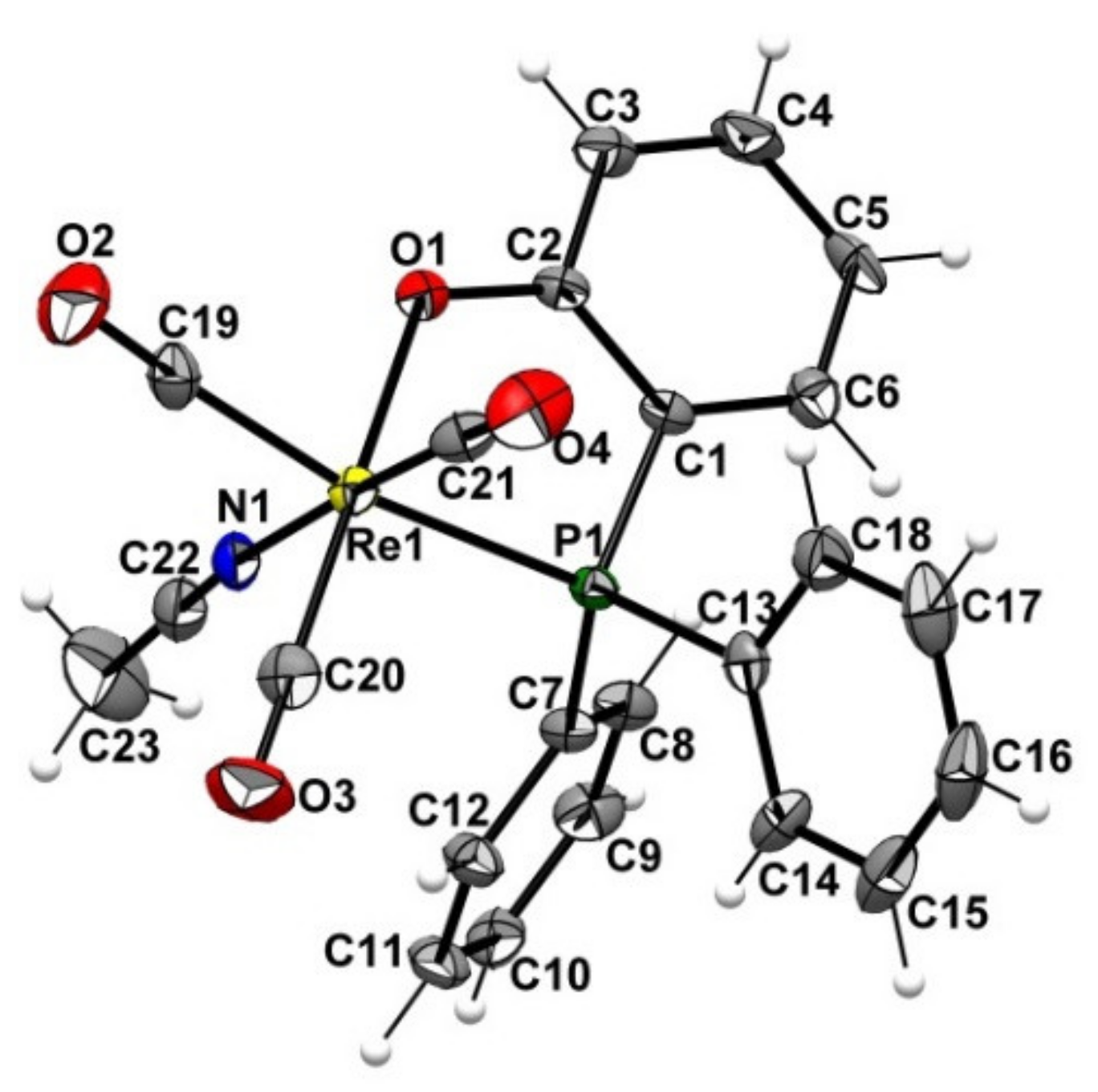
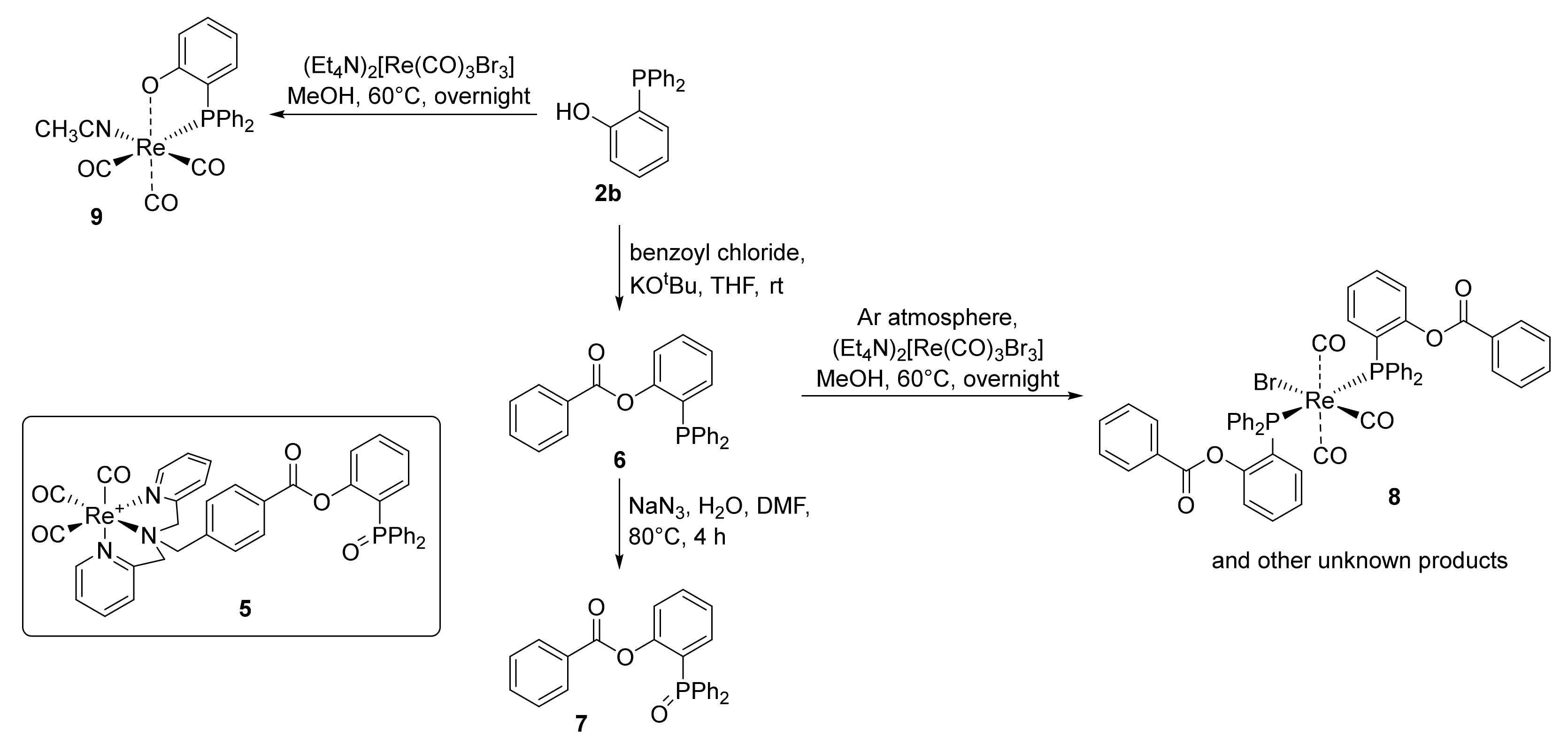
2.2. Preparation of the Non-Radioactive Rhenium-Reference Compounds
2.3. Attempt to Identify the Side Products from Complexation
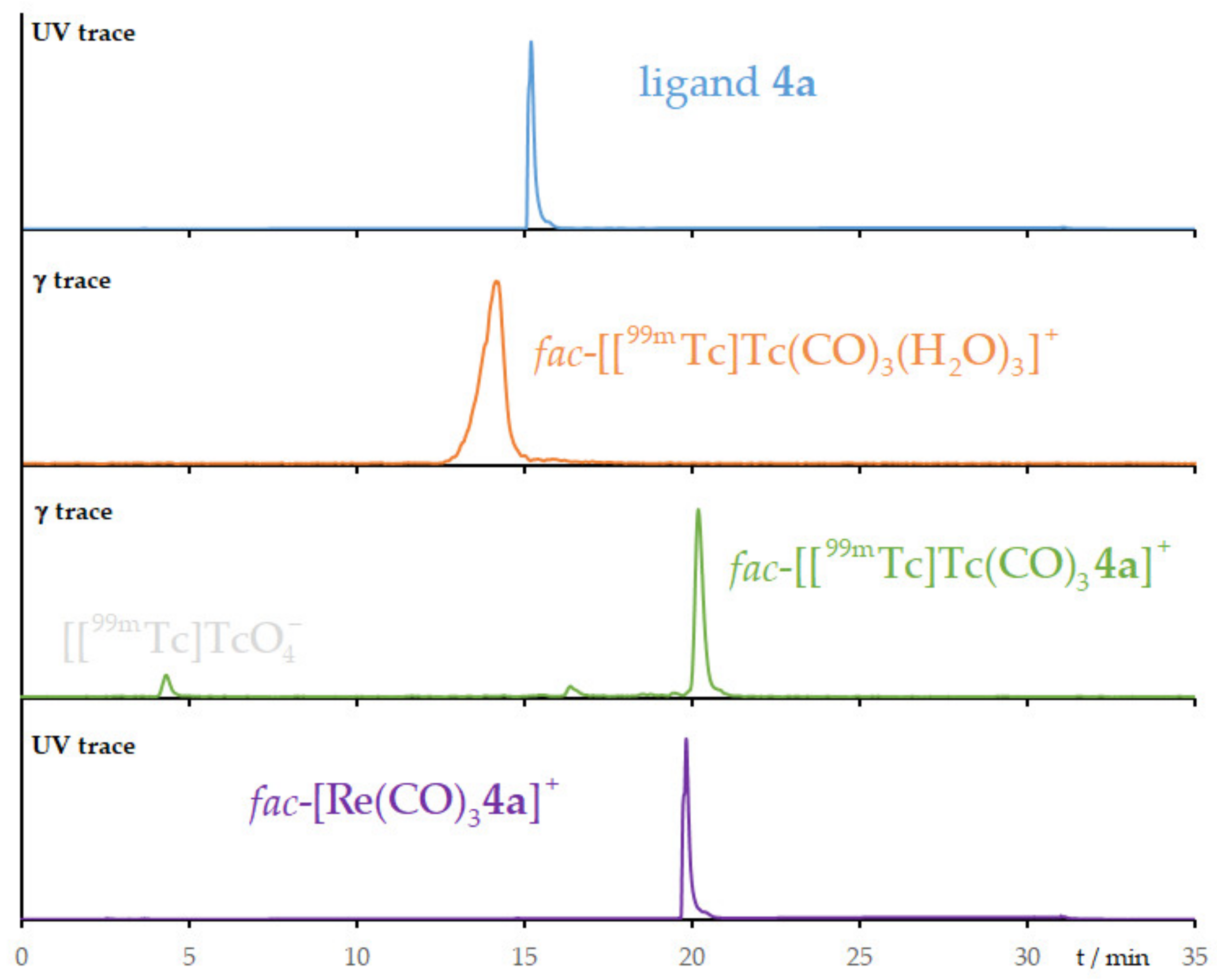
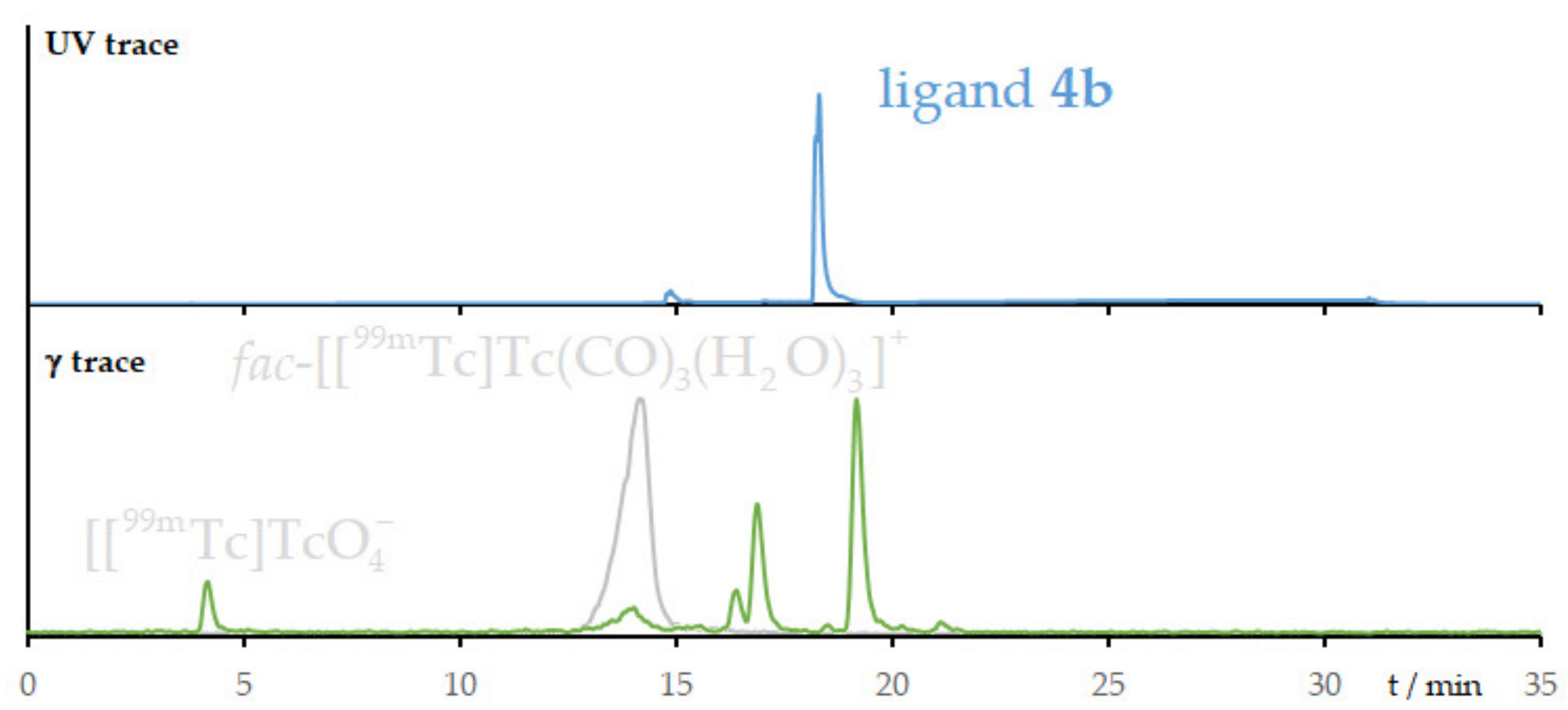
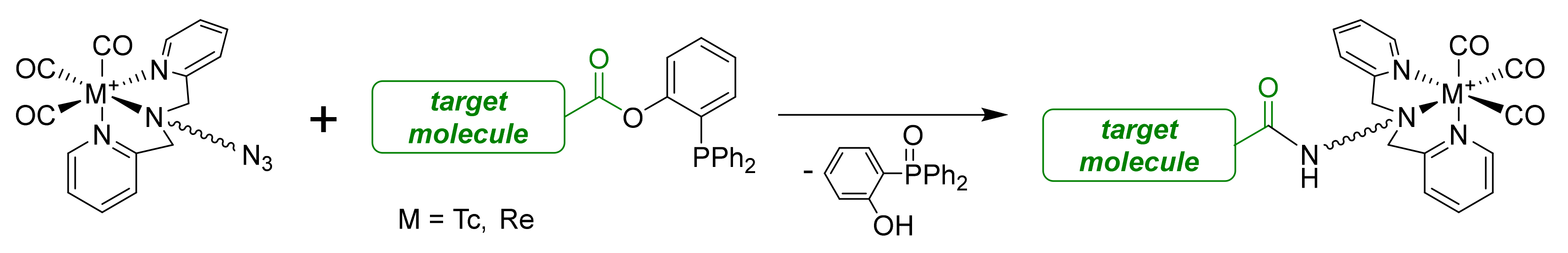
2.4. Radiolabeling with Technetium-99m
3. Materials and Methods
3.1. General
3.2. X-ray Diffraction Studies
3.3. Chemical Syntheses
3.4. Radiolabeling Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Köhn, M.; Breinbauer, R. Die Staudinger-Ligation—Ein Geschenk für die Chemische Biologie. Angew. Chem. 2004, 116, 3168–3178. [Google Scholar] [CrossRef]
- Schilling, C.I.; Jung, N.; Biskup, M.; Schäpers, U.; Bräse, S. Bioconjugation via azide—Staudinger ligation: An overview. Chem. Soc. Rev. 2011, 40, 4840–4871. [Google Scholar] [CrossRef]
- Van Berkel, S.S.; van Eldijk, M.B.; van Hest, J.C.M. Staudinger-Ligation als Methode zur Biokonjugation. Angew. Chem. 2011, 123, 8968–8989. [Google Scholar] [CrossRef]
- Wang, Z.-P.A.; Tian, C.-L.; Zheng, J.-S. The recent developments and applications of the traceless - Staudinger reaction in chemical biology study. RSC Adv. 2015, 5, 107192–107199. [Google Scholar] [CrossRef]
- Bednarek, C.; Wehl, I.; Jung, N.; Schepers, U.; Braäse, S. The Staudinger Ligation. Chem. Rev. 2020, 120, 4301–4354. [Google Scholar] [CrossRef] [PubMed]
- Pretze, M.; Pietzsch, D.; Mamat, C. Recent Trends in Bioorthogonal Click-Radiolabeling Reactions Using Fluorine-18. Molecules 2013, 18, 8618–8665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamat, C.; Gott, M.; Steinbach, J. Recent progress using the Staudinger Ligation for radiolableing purposes. J. Label. Compd. Radiopharm. 2018, 61, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Wodtke, R.; König, J.; Pigorsch, A.; Köckerling, M.; Mamat, C. Evaluation of novel fluorescence probes for conjugation purposes using the traceless Staudinger ligation. Dyes Pigm. 2015, 113, 263–273. [Google Scholar] [CrossRef]
- Pretze, M.; Wuest, F.; Peppel, T.; Köckerling, M.; Mamat, C. The traceless Staudinger ligation with fluorine - 18: A novel and versatile labeling technique for the synthesis of PET - radiotracers. Tetrahedron Lett. 2010, 51, 6410–6414. [Google Scholar] [CrossRef]
- Alberto, R.; Ortner, K.; Wheatley, N.; Schibli, R.; Schubiger, P.A. Synthesis and Properties of Boranocarbonate: A Convenient in Situ CO Source for the Aqueous Preparation of [99mTc(OH2)3(CO)3]+. J. Am. Chem. Soc. 2001, 123, 3135–3136. [Google Scholar] [CrossRef]
- Alberto, R.; Schibli, R.; Egli, A.; Schubiger, P.A. A Novel Organometallic Aqua Complex of Technetium for the Labeling of Biomolecules: Synthesis of [99mTc(OH2)3(CO)3]+ from [99mTcO4]− in Aqueous Solution and Its Reaction with a Bifunctional Ligand. J. Am. Chem. Soc. 1998, 120, 7987–7988. [Google Scholar] [CrossRef]
- Alberto, R.; Schibli, R.; Waibel, R.; Abram, U.; Schubiger, P.A. Basic aqueous chemistry of [M(OH2)3(CO)3]+ (M=Re, Tc) directed towards radiopharmaceutical application. Coord. Chem. Rev. 1999, 190-192, 901–919. [Google Scholar] [CrossRef]
- Porchia, M.; Bolzati, C.; Refosco, F.; Vittadini, A. The preparation of substitution-inert 99Tc metal-fragments: Promising candidates for the design of new 99mTc radiopharmaceuticals. Coord. Chem. Rev. 2006, 250, 2034–2045. [Google Scholar]
- Mindt, T.L.; Struthers, H.; Brans, L.; Anguelov, T.; Schweinsberg, C.; Maes, V.; Tourwé, D.; Schibli, R. “Click to Chelate”: Synthesis and Installation of Metal Chelates into Biomolecules in a Single Step. J. Am. Chem. Soc. 2006, 128, 15096–15097. [Google Scholar] [CrossRef] [PubMed]
- Mindt, T.L.; Müller, mC.; Melis, M.; de Jong, M.; Schibli, R. “Click-to-Chelate”: In Vitro and In Vivo Comparison of a 99mTc(CO)3-Labeled N(τ)-Histidine Folate Derivative with Its Isostructural, Clicked 1,2,3-Triazole Analogue. Bioconjug. Chem. 2008, 19, 1689–1695. [Google Scholar] [CrossRef] [PubMed]
- Kluba, C.A.; Mindt, T.L. Click-to-Chelate: Development of Technetium and Rhenium-Tricarbonyl Labeled Radiopharmaceuticals. Molecules 2013, 18, 3206–3226. [Google Scholar] [CrossRef]
- Mamat, C.; Ramenda, T.; Wuest, F.R. Recent Applications of Click Chemistry for the Synthesis of Radiotracers for Molecular Imaging. Mini-Rev. Org. Chem. 2009, 6, 21–34. [Google Scholar] [CrossRef]
- Choi, J.Y.; Lee, B.C. Click Reaction: An Applicable Radiolabeling Method for Molecular Imaging. Nucl. Med. Mol. Imaging 2015, 49, 258–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, J.P.; Adumeau, P.; Lewis, J.S.; Zeglis, B.M. Click Chemistry and Radiochemistry: The First 10 Years. Bioconjug. Chem. 2016, 27, 2791–2807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, S.; Maresca, K.P.; Allis, D.G.; Valliant, J.F.; Eckelman, W.; Babich, J.W.; Zubieta, J. Extension of the Single Amino Acid Chelate Concept (SAAC) to Bifunctional Biotin Analogues for Complexation of the M(CO)3+1 Core (M = Tc and Re): Syntheses, Characterization, Biotinidase Stability, and Avidin Binding. Bioconjug. Chem. 2006, 17, 579–589. [Google Scholar] [CrossRef]
- Maresca, K.P.; Hillier, S.M.; Femia, F.J.; Zimmerman, C.N.; Levadala, M.K.; Banerjee, S.R.; Hicks, J.; Sundararajan, C.; Valliant, J.; Zubieta, J.; et al. Comprehensive Radiolabeling, Stability, and Tissue Distribution Studies of Technetium-99m Single Amino Acid Chelates (SAAC). Bioconjug. Chem. 2009, 20, 1625–1633. [Google Scholar] [CrossRef] [PubMed]
- Hayes, T.R.; Lyon, P.A.; Silva-Lopez, E.; Twamley, B.; Benny, P.D. Photo-initiated Thiol-ene Click Reactions as a Potential Strategy for Incorporation of [MI(CO)3]+ (M = Re, 99mTc) Complexes. Inorg. Chem. 2013, 52, 3259–3267. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.R.; Levadala, M.K.; Lazarova, N.; Wei, L.; Valliant, J.F.; Stephenson, K.A.; Babich, J.W.; Maresca, K.P.; Zubieta, J. Bifunctional Single Amino Acid Chelates for Labeling of Biomolecules with the {Tc(CO)3}+ and {Re(CO)3}+ Cores. Crystal and Molecular Structures of [ReBr(CO)3(H2NCH2C5H4N)], [Re(CO)3{(C5H4NCH2)2NH}]Br, [Re(CO)3{(C5H4NCH2)2NCH2CO2H}]Br, [Re(CO)3{X(Y)NCH2CO2CH2CH3}]Br(X = Y = 2-pyridylmethyl; X = 2-pyridylmethyl, Y = 2-(1-methylimidazolyl)methyl; X = Y = 2-(1-methylimidazolyl)methyl), [ReBr(CO)3{(C5H4NCH2)NH(CH2C4H3S)}], and [Re(CO)3{(C5H4NCH2)N(CH2C4H3S)(CH2CO2)}]. Inorg. Chem. 2002, 41, 6417–6425. [Google Scholar] [PubMed]
- Mamat, C.; Flemming, A.; Köckerling, M.; Steinbach, J.; Wuest, F.R. Synthesis of benzoate - functionalized phosphanes as novel building blocks for the traceless Staudinger ligation. Synthesis 2009, 3311–3321. [Google Scholar] [CrossRef]
- Mamat, C.; Köckerling, M. Preparation of 4-halobenzoate-containing phosphane-based building blocks for labeling reactions using the traceless Staudinger ligation. Synthesis 2015, 45, 387–394. [Google Scholar] [CrossRef]
- Alberto, R.; Egli, A.; Abram, U.; Hegetschweiler, K.; Gramlich, V.; Schubiger, P.A. Synthesis and reactivity of [NEt4]2[ReBr3(CO)3]. Formation and structural characterization of the clusters [NEt4][Re3(µ3-OH)(µ-OH)3(CO)9] and [NEt4][Re2(µ-OH)3(CO)6] by alkaline titration. J. Chem. Soc., Dalton Trans. 1994, 2815–2820. [Google Scholar] [CrossRef]
- Braband, H.; Abram, U. Tricarbonyl complexes of rhenium(I) and technetium(I) thiourea complexes. J. Organomet. Chem. 2004, 689, 2066–2072. [Google Scholar] [CrossRef]
- Abram, U.; Abram, S.; Alberto, R.; Schibli, R. Ligand exchange reactions starting from [Re(CO)3Br3]2−. Synthesis, characterization and structures of rhenium(I) tricarbonyl complexes with thiourea and thiourea derivatives. Inorg. Chim. Acta 1996, 248, 193–202. [Google Scholar] [CrossRef]
- Lepareur, N.; Lacœuille, F.; Bouvry, C.; Hindré, F.; Garcion, E.; Chérel, M.; Noiret, N.; Garin, E.; Knapp, F.F.R. Rhenium-188 Labeled Radiopharmaceuticals: Current Clinical Applications in Oncology and Promising Perspectives. Front. Med. 2019, 6, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilworth, J.R.; Parrott, S.J. The biomedical chemistry of technetium and rhenium. Chem. Soc. Rev. 1998, 27, 43–55. [Google Scholar] [CrossRef]
- Alberto, R. Metal-Based Radiopharmaceuticals. In Bioinorganic Medicinal Chemistry; Alessio, E., Ed.; Wiley-VCH: Weinheim, Germany, 2011; pp. 253–283. [Google Scholar]
- Gomes Marin, J.F.; Nunes, R.F.; Coutinho, A.M.; Zaniboni, E.C.; Costa, L.B.; Barbosa, F.G.; Queiroz, M.A.; Cerri, G.G.; Buchpiguel, C.A. Theranostics in Nuclear Medicine: Emerging and Re-emerging Integrated Imaging and Therapies in the Era of Precision Oncology. Radiographics 2020, 40, 1715–1740. [Google Scholar] [CrossRef] [PubMed]
- Yordanova, A.; Eppard, E.; Kürpig, S.; Bundschuh, R.A.; Schönberger, S.; Gonzalez-Carmona, M.; Feldmann, G.; Ahmadzadehfar, H.; Essler, M. Theranostics in nuclear medicine practice. OncoTargets Ther. 2017, 10, 4821–4828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, E.; Onai, S. Crystal structure of cis-bis(triphenylphosphine)-fac-(tricarbonyl)-rhenium(I) bromide, Re(CO)3(C18H15P)2Br. Z. Krist. NCS 2001, 216, 454–456. [Google Scholar] [CrossRef] [Green Version]
- Shegani, A.; Triantis, C.; Nock, B.A.; Maina, T.; Kiritsis, C.; Psycharis, V.; Raptopoulou, C.; Pirmettis, I.; Tisato, F.; Papadopoulos, M.S. Rhenium(I) Tricarbonyl Complexes with (2-Hydroxyphenyl)diphenylphosphine as PO Bidentate Ligand. Inorg. Chem. 2017, 56, 8175–8186. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Sihver, W.; Bergmann, R.; Belter, B.; Bolzati, C.; Salvarese, N.; Steinbach, J.; Pietzsch, J.; Pietzsch, H.-J. Synthesis, Characterization, and Initial Biological Evaluation of [99mTc]Tc-Tricarbonyl-labeled DPA-α-MSH Peptide Derivatives for Potential Melanoma Imaging. ChemMedChem 2018, 13, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Grandjean, C.; Boutonnier, A.; Guerreiro, C.; Fournier, J.-M.; Mulard, L.A. On the Preparation of Carbohydrate−Protein Conjugates Using the Traceless Staudinger Ligation. J. Org. Chem. 2005, 70, 7123–7132. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Reissig, F.; Mamat, C.; Steinbach, J.; Pietzsch, H.-J.; Freudenberg, R.; Navarro-Retamal, C.; Caballero, J.; Kotzerke, J.; Wunderlich, G. Direct and Auger electron-induced, single- and double-strand breaks on plasmid DNA caused by 99mTc-labeled pyrene derivatives and the effect of bonding distance. PLoS ONE 2016, 11, e0161973. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamat, C.; Jentschel, C.; Köckerling, M.; Steinbach, J. Strategic Evaluation of the Traceless Staudinger Ligation for Radiolabeling with the Tricarbonyl Core. Molecules 2021, 26, 6629. https://doi.org/10.3390/molecules26216629
Mamat C, Jentschel C, Köckerling M, Steinbach J. Strategic Evaluation of the Traceless Staudinger Ligation for Radiolabeling with the Tricarbonyl Core. Molecules. 2021; 26(21):6629. https://doi.org/10.3390/molecules26216629
Chicago/Turabian StyleMamat, Constantin, Christian Jentschel, Martin Köckerling, and Jörg Steinbach. 2021. "Strategic Evaluation of the Traceless Staudinger Ligation for Radiolabeling with the Tricarbonyl Core" Molecules 26, no. 21: 6629. https://doi.org/10.3390/molecules26216629
APA StyleMamat, C., Jentschel, C., Köckerling, M., & Steinbach, J. (2021). Strategic Evaluation of the Traceless Staudinger Ligation for Radiolabeling with the Tricarbonyl Core. Molecules, 26(21), 6629. https://doi.org/10.3390/molecules26216629